
Abstract
Atherosclerosis, a disease of blood vessels, is driven by cholesterol accumulation and inflammation. Gene therapy that removes cholesterol from blood vessels and decreases inflammation is a promising approach for prevention and treatment of atherosclerosis. In previous work, we reported that helper-dependent adenoviral (HDAd) overexpression of apolipoprotein A-I (apoAI) in endothelial cells (ECs) increases cholesterol efflux in vitro and reduces atherosclerosis in vivo. However, the effect of HDAdApoAI on atherosclerosis is partial. To improve this therapy, we considered concurrent overexpression of ATP-binding cassette subfamily A, member 1 (ABCA1), a protein that is required for apoAI-mediated cholesterol efflux. Before attempting combined apoAI/ABCA1 gene therapy, we tested whether an HDAd that expresses ABCA1 (HDAdABCA1) increases EC cholesterol efflux, whether increased cholesterol efflux alters normal EC physiology, and whether ABCA1 overexpression in ECs has anti-inflammatory effects. HDAdABCA1 increased EC ABCA1 protein (∼3-fold; p < 0.001) and apoAI-mediated cholesterol efflux (2.3-fold; p = 0.007). Under basal culture conditions, ABCA1 overexpression did not alter EC proliferation, metabolism, migration, apoptosis, nitric oxide production, or inflammatory gene expression. However, in serum-starved, apoAI-treated EC, ABCA1 overexpression had anti-inflammatory effects: decreased inflammatory gene expression (∼50%; p ≤ 0.02 for interleukin [IL]-6, tumor necrosis factor [TNF]-α, and vascular cell adhesion protein-1); reduced lipid-raft Toll-like receptor 4 (80%; p = 0.001); and a trend towards increased nitric oxide production (∼55%; p = 0.1). In ECs stimulated with lipopolysaccharide, ABCA1 overexpression markedly decreased inflammatory gene expression (∼90% for IL-6 and TNF-α; p < 0.001). Therefore, EC ABCA1 overexpression has no toxic effects and counteracts the two key drivers of atherosclerosis: cholesterol accumulation and inflammation. In vivo testing of HDAdABCA1 is warranted.
Introduction
Cardiovascular disease, largely caused by atherosclerosis, is responsible for up to one-third of deaths worldwide. 1 Accordingly, improved strategies to prevent and treat atherosclerosis would likely reduce mortality on a global scale. Pharmacologic therapies for atherosclerosis are aimed primarily at decreasing plasma low-density lipoprotein cholesterol (LDL-C), for example, with statin drugs. Statins and other LDL-C–lowering agents have significantly reduced atherosclerosis-related morbidity and mortality 2 ; however, despite achievement of median LDL-C levels as low as 30 mg/dL, major adverse cardiovascular events are far from eliminated. 3 In addition, statin therapy is life-long and is associated with compliance problems due in part to patient-reported side effects. 4 Other therapies aimed at modifying plasma lipids to prevent atherosclerosis include fibrates and drugs that increase high-density lipoprotein (HDL) cholesterol (e.g., niacin and inhibitors of cholesterol-ester transfer protein). However, neither fibrates nor HDL-C–raising drugs were effective when added to statins in large clinical trials. 5 –8
An alternative approach to atherosclerosis prevention and treatment is enhancement of cholesterol removal from the blood vessel wall. This approach, which aims to accelerate an endogenous process known as reverse cholesterol transport (RCT), 9 would likely work synergistically with LDL-C–lowering drug therapy. 10 RCT begins with cholesterol efflux from vascular cells via the cell-surface cholesterol transporter ATP-binding cassette subfamily A, member 1 (ABCA1). 11 Effluxed cholesterol is bound by plasma-derived apolipoprotein (apo) AI, forming HDL-C, which transports cholesterol to the liver for excretion. Several groups have developed therapeutic approaches aimed at acceleration of RCT, including infusions of apoAI protein, apoAI-mimetic peptides, or synthetically reconstituted HDL. 12 –14 These strategies have shown promise in animal models and small clinical studies, but none has yielded positive results in a large clinical trial. Moreover, life-long protein or peptide infusions are impractical and expensive.
Gene therapy is an attractive strategy for increasing apoAI and ABCA1 in the blood vessel wall, thereby increasing RCT and preventing or reversing atherosclerosis. We initially pursued this approach by using a helper-dependent adenoviral (HDAd) vector to express apoAI in endothelial cells (ECs) in carotid arteries of fat-fed rabbits. EC-expressed apoAI increased cholesterol efflux when added to lipid-loaded cells in vitro and significantly reduced lesion mass as well as lipid and inflammatory cell accumulation in vivo. 15 –17 These therapeutic effects were substantial (30–40% reductions); however, they were only partial. Because apoAI-mediated cholesterol efflux depends on cell-surface expression of ABCA1, 18 we considered that co-expression of apoAI and ABCA1 in ECs might act synergistically to prevent and reverse atherosclerosis. Others showed that modest (<2-fold) transgenic overexpression of ABCA1 in ECs of fat-fed mice can reduce aortic atherosclerosis; however, a concurrent 40% increase in plasma HDL-C precluded attribution of reduced atherosclerosis to a local effect of ABCA1 overexpression in ECs. 19
As a prelude to experiments in which apoAI and ABCA1 are co-expressed in ECs in vivo, we tested the hypothesis that EC ABCA1 expression and cholesterol efflux could be increased by transduction of ECs with an ABCA1-expressing HDAd. Because cholesterol depletion could have negative consequences on ECs, including impaired cell migration, 20 altered transmembrane signaling, 21 –23 decreased endothelial nitric oxide synthase (eNOS) activity, 24,25 and increased apoptosis, 26 –28 we also tested whether overexpression of ABCA1 altered several important aspects of EC physiology. Finally, based on data suggesting an anti-inflammatory role for EC ABCA1, 29 we tested whether ECs that overexpress ABCA1 also express lower levels of pro-inflammatory genes.
Materials and Methods
Cell culture
Bovine aortic ECs (BAECs) (Cell Applications, San Diego, CA), 293-Cre cells (Microbix, Toronto, ON, Canada), 30 and baby hamster kidney (BHK) cells (ATCC, Manassas, VA) were cultured in standard growth medium containing high-glucose Dulbecco's modified Eagle's medium (DMEM; GIBCO, Grand Island, NY), 10% fetal bovine serum (FBS; VWR Life Science Seradigm, Radnor, PA), and 1% penicillin/streptomycin (GIBCO) at 37°C and 5% CO2. We used BHK cells that contain a mifepristone-inducible human ABCA1 gene. 31 Medium for 293-Cre cells was supplemented with a final concentration of 400 μg/mL G418 (Thermo Fisher Scientific, Waltham, MA). BAECs were used between passages 3 and 7. Human apolipoprotein AI (apoAI) was purified from plasma as described. 32
Cloning of rabbit ABCA1 and HDAd vector construction, production, and characterization
A rabbit ABCA1 cDNA (NCBI Reference Sequence: XM_008257475.1) containing an inserted Kozak sequence, 33 an XhoI restriction site at the 5′ end, and a NotI restriction site at the 3′ end was synthesized and inserted into pUC57 (GenBank: Y14837.1; GenScript, Piscataway, NJ). We synthesized rabbit ABCA1 (instead of using mouse or human ABCA1 clones) because we anticipate testing an HDAd that overexpresses ABCA1 in our rabbit atherosclerosis models. 15,17,34 Use of a native (i.e., rabbit) protein in our rabbit models eliminates the risk of a foreign protein-targeted immune response and ensures physiologic interactions with other rabbit proteins. 35 The ABCA1 cDNA was removed with an XhoI/NotI digest and ligated into XhoI/NotI-digested pCI (Promega, Madison, WI) to yield pCIcABCA1 (Supplementary Fig. S1). pCI contains a CMV major immediate-early enhancer/promoter, a chimeric intron, and a late SV40 polyadenylation signal. pCIcABCA1 was digested with SnaBI and HpaI, removing a fragment containing part of the promoter sequence, the rabbit ABCA1 cDNA transgene, and part of the SV40 polyadenylation signal. This fragment was ligated into SnaBI/HpaI-digested pBsCMVgApoAI, 36 restoring the CMV promoter and SV40 sequences and yielding pBsCMVcABCA1. pBsCMVcABCA1 was digested with KpnI and SacII, releasing the ABCA1 expression cassette flanked by sequences homologous to stuffer DNA flanking the AscI site in the HDAd backbone plasmid pC4HSU. 37 To accommodate the large ABCA1 expression cassette (∼8.1 kb) and to enable better separation of HDAdABCA1 from the larger helper virus by density-gradient ultracentrifugation, we modified pC4HSU by removing a ∼7.8-kb fragment of stuffer DNA (by digestion with PspXI and re-ligation). The re-ligated plasmid, pC4HSUt, was then linearized by digestion with AscI. AscI-digested pC4HSUt and the KpnI/SacII fragment of pBsCMVcABCA1 (containing the ABCA1 expression cassette flanked by homology arms) were electroporated into recombination-permissive BJ5183 cells (Stratagene, La Jolla, CA). Ampicillin-resistant cells were screened by colony PCR, and plasmid DNA from positive cells was transformed into DH10B cells (Invitrogen, Waltham, MA). Plasmid DNA (pC4HSUtCMVcABCA1) from the DH10B cells was mapped with restriction digests and sequenced to verify presence of the cABCA1 expression cassette. We used PmeI-digested pC4HSUtCMVcABCA1 along with AdNG163 helper virus and Cre-expressing 116 cells 38 to generate and amplify HDAdABCA1. HDAdNull, which contains the same expression cassette as HDAdABCA1, but without the cABCA1 transgene, 39 was used as a control. HDAd virions were purified by CsCl ultracentrifugation and quantified by spectrophotometry at 260 nm. 40 Helper virus and E1A contamination were measured as described. 39 All HDAd stocks contained <1% helper virus genomes and <1 in 106 E1A-containing genomes.
In vitro transduction
For all in vitro experiments, BAECs were plated and incubated overnight in growth medium. HDAd stocks were diluted to 1 × 1010 vp/mL in growth medium and added to BAECs. After a 6-h transduction period, medium was removed and BAECs were rinsed with Dulbecco's phosphate-buffered saline (DPBS) and fed with fresh medium. Phenol red-free medium was used for all experiments involving colorimetric assays. During all serum-starved conditions, cells were provided fatty acid-free albumin (FAFA; 1 mg/mL; Sigma-Aldrich, St. Louis, MO). Human apoAI was added at 5 μg/mL for cholesterol-efflux experiments and at 50 μg/mL for other experiments. All protein quantification was performed with the BCA assay (Thermo Scientific Pierce, Waltham, MA).
ABCA1 protein expression
To detect expression of ABCA1 protein, we prepared lysates of transduced BAECs with a Roche cOmplete Lysis-M kit (Penzberg, Germany). BHK cells with mifepristone-inducible ABCA1 expression 18 were used as technical controls. Equal amounts of protein were separated on 4%–12% Invitrogen NuPAGE Bis-Tris gels, transferred to Immobilon-P membranes (Millipore, Billerica, MA), and membranes blocked with 5% (w/v) nonfat dry milk. Blots were probed with murine anti-ABCA1 (1:750 dilution; sc-58219, Santa Cruz Biotechnology, Dallas, TX) and goat anti-GAPDH (1:1,000 dilution; sc-20357, Santa Cruz). Bound antibodies were detected with horseradish peroxidase-conjugated goat anti-mouse IgG (1:7,500 dilution; 1706516, Bio-Rad) or donkey anti-goat IgG (1:7,500 dilution; sc-2033, Santa Cruz). Peroxidase activity was detected with Bio-Rad Clarity Western ECL Substrate. Blots were imaged (Bio-Rad ChemiDoc MP Imaging System, Hercules, CA) and signal was quantified using ImageJ software (National Institutes of Health, Bethesda, MD). ABCA1 signal was normalized to GAPDH signal, as a loading control.
Measurement of cholesterol efflux
BAECs were plated in 48-well plates at 2 × 104 cells per well and transduced with either HDAdNull or HDAdABCA1. Twenty-four hours after addition of HDAd, BAECs were rinsed with DPBS and loaded with [ 3 H]cholesterol by addition of DMEM and [ 3 H]cholesterol (1 μCi/mL; PerkinElmer Life Sciences, Waltham, MA). Twenty-four hours later, the cells were rinsed with DPBS and fed with DMEM, with or without apoAI for 24 h. Conditioned medium was collected and then filtered and centrifuged (2500 × g for 10 min) to remove detached cells. [ 3 H]Cholesterol in the supernatant was measured with a scintillation counter. Cells were lysed by adding 250 μL of NaOH (200 mM) per well, gently shaking cells at room temperature for 1 h, and freeze-thawing the plates once. [ 3 H]Cholesterol in cell lysates was measured with a scintillation counter. BHK cells (with and without mifepristone treatment) were used as technical controls. ApoAI-mediated cholesterol efflux was calculated by dividing [ 3 H] in the medium by total [ 3 H] (medium + cells), after subtracting background cholesterol efflux (measured in medium harvested from BAECs not treated with apoAI).
Bromodeoxyuridine (BrdU) incorporation
BAECs were plated in 96-well plates at 2 × 103 cells per well and transduced with HDAdNull or HDAdABCA1. Cell proliferation was measured with a BrdU ELISA kit (Abcam, Cambridge, United Kingdom), after exposure to BrdU for 18 h, beginning 54 h after addition of HDAd. Cells were fixed and DNA denatured according to the manufacturer's protocol and were then incubated with anti-BrdU for 1 h at room temperature, followed by incubation with peroxidase-conjugated goat anti-mouse IgG for 30 min at room temperature. The peroxidase substrate was added and the plates were wrapped in aluminum foil for 30 min at room temperature. Stop solution was added and absorbance at 450 nm was measured with a microplate reader (Molecular Devices SoftMax Pro 5, Sunnyvale, CA). As a technical control, we treated BAECs with the antiproliferative agent hydroxyurea (2 mM final concentration) for 24 h before incubating these cells with BrdU.
Cellular metabolic activity
BAECs were plated in 96-well plates at 2 × 103 cells per well and transduced with HDAdNull or HDAdABCA1. We used an MTT assay kit (Vybrant, Thermo Fisher Scientific) to measure metabolic activity. We added MTT at 6, 30, or 54 h after addition of HDAd, and incubated the cells for 4 h. We then added SDS-HCl solution to each well, incubated the cells for 14 h at 37°C, and read absorbance at 562 nm with a microplate reader. As a technical control, we incubated BAECs with hydroxyurea (added 6 h before addition of MTT).
Cell migration
We measured BAEC migration with a monolayer wound-healing assay. BAECs were seeded at 6 × 104 cells per well in 24-well plates and transduced with HDAdNull or HDAdABCA1. Twenty-four hours later, monolayers were wounded by scraping with a pipette tip. The wound was photographed immediately post-scraping and then again 3, 6, and 9 h later. Wound area was measured with ImageJ software. We also treated BAECs with hydroxyurea for 24 h before wounding, to test whether wound healing detected by our assay was because of cell proliferation.
Flow cytometry
We used the Vybrant Apoptosis Kit #6 (Thermo Fisher Scientific) and flow cytometry to measure apoptosis. We used an LSRII flow cytometer (BD Biosciences, San Jose, CA) equipped with a UV laser (350 nm; detected with a 450/50 filter) and a yellow-green laser (561 nm; detected with a 610/20 filter) for flow cytometry measurements and we used BD FACSDiva 8.0.1 software for collection. Data were also analyzed with FCS Express (De Novo Software, Glendale, CA). BAECs were seeded at 2 × 105 cells per well in six-well plates and transduced with HDAdNull or HDAdABCA1. Cells were trypsinized 24, 48, and 72 h after transduction, washed in cold DPBS, and resuspended in 100 μL of 1X annexin-binding buffer. We then added 5 μL of biotin-X annexin V to each sample and incubated the cells for 15 min at room temperature. The cells were then washed in 1X annexin-binding buffer and resuspended in 100 μL of 1X annexin-binding buffer. We added 1 μL of Alexa Fluor 350 streptavidin solution, and placed the cells on ice for 30 min. After incubation, cells were resuspended in 500 μL of 1X annexin-binding buffer, 1 μL of propidium iodide solution was added, and the cells were incubated on ice for 10 min. As a technical control, we treated BAECs with camptothecin (50 μM) for 24 hours, to trigger apoptosis. We used a gating strategy to identify apoptotic and dead cells. Annexin V-positive and propidium iodide-negative cells were considered to be apoptotic. We recorded 10,000 events for each sample.
Measurement of nitric oxide
We used electron paramagnetic resonance (EPR) spin-trapping to measure nitric oxide (NO). To measure NO under basal conditions, we seeded BAECs at 1.5 × 106 cells per 100-mm plate and transduced the cells with HDAdNull or HDAdABCA1. Twenty-four hours after addition of HDAd, the cells were rinsed with Krebs-HEPES buffer and incubated for 1 h in a Krebs-HEPES buffer-based trap solution (deoxygenated using argon gas) containing iron sulfate heptahydrate (62 μM; Sigma-Aldrich) and sodium diethyldithiocarbamate trihydrate (798 μM; Sigma-Aldrich). The cells were rinsed in Krebs-HEPES buffer, removed with a cell scraper, and resuspended in 110 μL of Krebs-HEPES buffer. We used 10 μL to quantify total protein and the remaining cell suspension was immediately frozen in liquid nitrogen. The EPR signals were measured with a Miniscope MS 200 EPR spectrometer and EPR signal amplitude was normalized to total protein in each sample by dividing EPR signal by total protein. As a technical control, BAECs were treated with the NO synthase inhibitor N(ω)-nitro-L-arginine methyl ester (1 mM; Sigma-Aldrich) during the 1-h trap medium incubation period. To measure NO under serum-starved conditions and after treatment with apoAI, we waited until 48 h after addition of HDAd, and then rinsed the cells with DPBS and incubated them in serum-free DMEM with added apoAI for 24 h. The cells were then washed with Krebs-HEPES buffer and processed for measurement of NO, as described above.
Measurement of mRNA
To measure mRNA under basal culture conditions, BAECs were seeded (2 × 105 cells per well in six-well plates), transduced with HDAdNull or HDAdABCA1, and maintained in serum-containing medium. At 24, 48, or 72 h after addition of HDAd, cells were lysed with TRIzol (Thermo Fisher Scientific), and total RNA was extracted (RNeasy kit; Qiagen, Hilden, Germany). As a positive control for stimulation of inflammatory gene expression, BAECs were treated with lipopolysaccharide (LPS) (100 ng/mL; Sigma-Aldrich) for 24 h. We also measured mRNA in serum-starved cells treated with apoAI; some of these cells were also stimulated with LPS. For these experiments, BAECs were seeded and transduced with HDAd as described above, and then fed with serum-containing medium. Forty-eight hours after addition of HDAd, cells were rinsed with DPBS and incubated in serum-free DMEM with added apoAI for 24 h. For experiments involving LPS treatment, BAECs were incubated in serum-free DMEM with added apoAI for 18 h, and then LPS (10 ng/mL) was added for 6 h. Total RNA was extracted as described above and quantified using NanoDrop (Thermo Fisher Scientific). We treated RNA samples with DNase I (1 U/μL; Thermo Fisher Scientific) at 37°C for 30 min to remove genomic DNA. We used 100 ng of sample RNA and the qScript XLT 1-Step RT-PCR Kit (Quantabio, Beverly, MA) for both cDNA synthesis and amplification. Primers and probes are listed in the Supplementary Table. mRNA was measured using the ΔΔCT method, 41 with normalization to GADPH mRNA, measured in the same samples.
Lipid raft isolation and immunoblotting
BAECs were seeded at 1.5 × 106 cells in 100-mm plates and transduced with HDAdNull or HDAdABCA1. Forty-eight hours after addition of HDAd, cells were rinsed with DPBS and incubated for 18 h in DMEM with added apoAI. The cells were then rinsed with DPBS and incubated in DMEM containing LPS (10 ng/mL) for 6 h. We adapted a detergent-free, gradient ultracentrifugation protocol 42 to isolate BAEC lipid-raft and non–lipid-raft fractions. This protocol includes centrifugation of cell lysates at 288,244 g in an SW41 rotor for 12 h at 4°C, and then collection of nine equal (∼1.2 mL) fractions, with no. 9 as the top fraction and no. 1 as the bottom fraction. We pooled equal amounts of protein from three non-raft fractions (nos. 2, 3, and 4) and three lipid-raft fractions (nos. 6, 7, and 8), separated proteins within the pooled samples by SDS-PAGE (4%–15% Mini-PROTEAN TGX gels; Bio-Rad), and transferred proteins onto Immobilon-P membranes. After blocking with nonfat dry milk, proteins were detected with a mouse antibody to Toll-like-receptor-4 (TLR4; 1:750 dilution; 1706516, Santa Cruz) and a rabbit antibody to caveolin-1 (1:1,500 dilution; 3267, Cell Signaling Technology, Danvers, MA). Secondary antibodies were horseradish peroxidase-linked goat anti-mouse IgG (1:7,500 dilution; 1706516, Bio-Rad) or horseradish peroxidase-linked goat anti-rabbit IgG (1:7,500 dilution; 7074, Cell Signaling). We detected peroxidase activity with either Amersham ECL Select for TLR4 blots (GE Life Sciences, Chicago, IL) or Bio-Rad Clarity Western ECL Substrate for caveolin-1 blots.
Statistics
Statistical analyses were performed using SigmaPlot (Systat Software Inc, San Jose, CA). We tested normality using a Shapiro-Wilk test and equal variance using a Brown-Forsythe test. When conditions of normality and equal variance were met, we performed a t-test. Otherwise, we used the nonparametric Mann-Whitney rank-sum test. For experiments in which the control group values were assigned an arbitrary value of 1 and the experimental group values were calculated in relation to the control group value of 1, we performed a one sample t-test.
Results
HDAdABCA1 increases EC ABCA1 protein and apoAI-mediated cholesterol efflux without altering metabolic activity or cell proliferation
In an earlier study, 39 we showed that—compared to mock-transduced ECs—transduction of ECs with HDAdNull does not alter metabolic activity, proliferation, migration, apoptosis, NO production, cytokine expression, or adhesion molecule expression. Therefore, in the present study, we used only HDAdNull-transduced ECs as controls. Transduction of ECs with HDAdABCA1 increased ABCA1 protein by ∼3-fold compared to cells transduced with HDAdNull (p < 0.001; Fig. 1A and 1B). This level of ABCA1 overexpression increased apoAI-mediated cholesterol efflux by 2.3-fold (p = 0.007; Fig. 1C). Because cellular cholesterol is essential for membrane biosynthesis as well as transmembrane signaling that regulates cell growth, viability, and proliferation, 21 –23 we tested whether overexpression of ABCA1 reduces EC metabolic activity (with the MTT assay) or proliferation (with a BrdU incorporation assay). Compared to ECs transduced with HDAdNull, ABCA1-overexpressing ECs had equivalent levels of metabolic activity and proliferation (p > 0.1 for both; Fig. 2A and 2B).
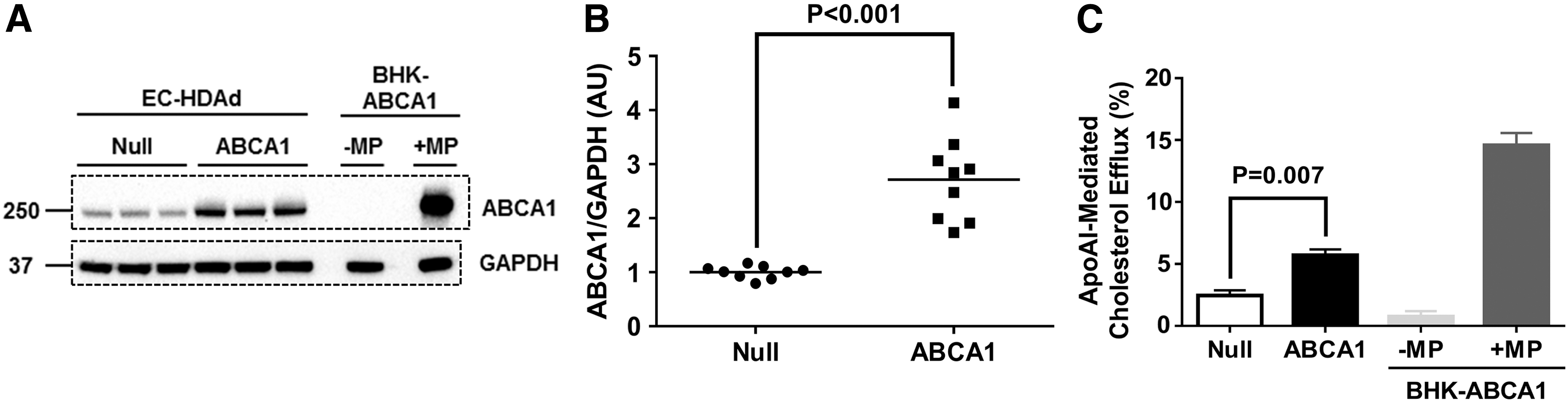
Expression and function of rabbit ABCA1 in endothelial cells (ECs).
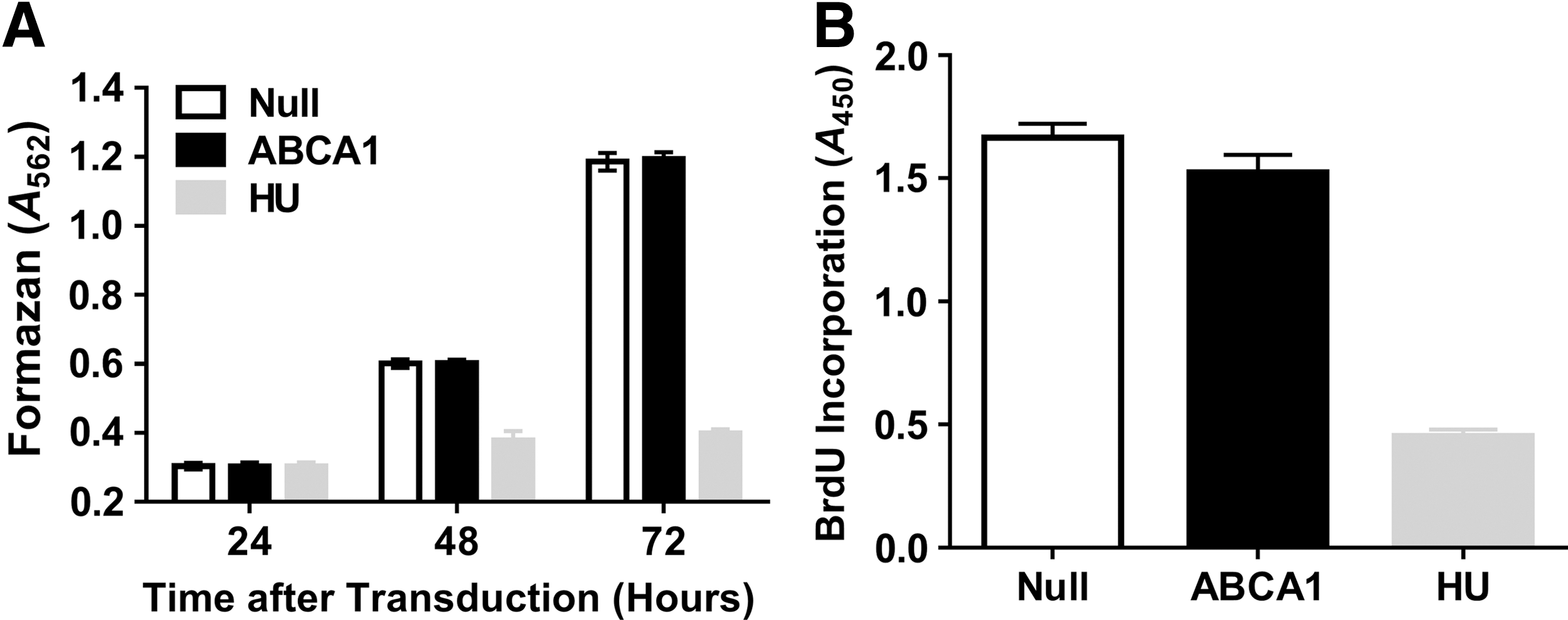
Proliferation and metabolic activity in ECs overexpressing ABCA1. ECs were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1).
ABCA1 overexpression does not alter cell migration or trigger apoptosis
Lateral migration is a critical EC function because it allows rapid coverage of areas of the blood vessel luminal surface from which ECs are denuded; for example, during interventional procedures such as stent placement or catheter-mediated intraluminal gene transfer. 43,44 EC lateral migration could also prevent acute coronary syndromes associated with plaque erosion, in which loss of luminal endothelial cells precipitates mural thrombosis. 45 Cholesterol depletion can inhibit actin cytoskeleton assembly, interfering with cell migration. 20 We used a monolayer wound-healing assay to measure lateral migration in ECs transduced with HDAdNull or HDAdABCA1. ABCA1 overexpression did not alter EC migration at any time point (p > 0.1 for all comparisons of HDAdNull and HDAdABCA1-transduced EC; Fig. 3). Equivalent lateral migration in ECs treated with the antiproliferative agent hydroxyurea compared with untreated ECs confirmed that cell proliferation did not contribute to wound coverage in this assay, validating it as a specific test of cell migration. Excessive cellular cholesterol depletion can cause both cell lysis and apoptosis. 26 –28 Therefore, we used flow cytometry to measure EC apoptosis after transduction with either HDAdNull or HDAdABCA1 (Fig. 4A and 4B). As a positive control, we treated ECs with camptothecin (Fig. 4C). Compared to control HDAdNull-transduced ECs, overexpression of ABCA1 did not increase EC apoptosis (p > 0.1 for all comparisons of HDAdNull and HDAdABCA1-transduced ECs; Fig. 4D).
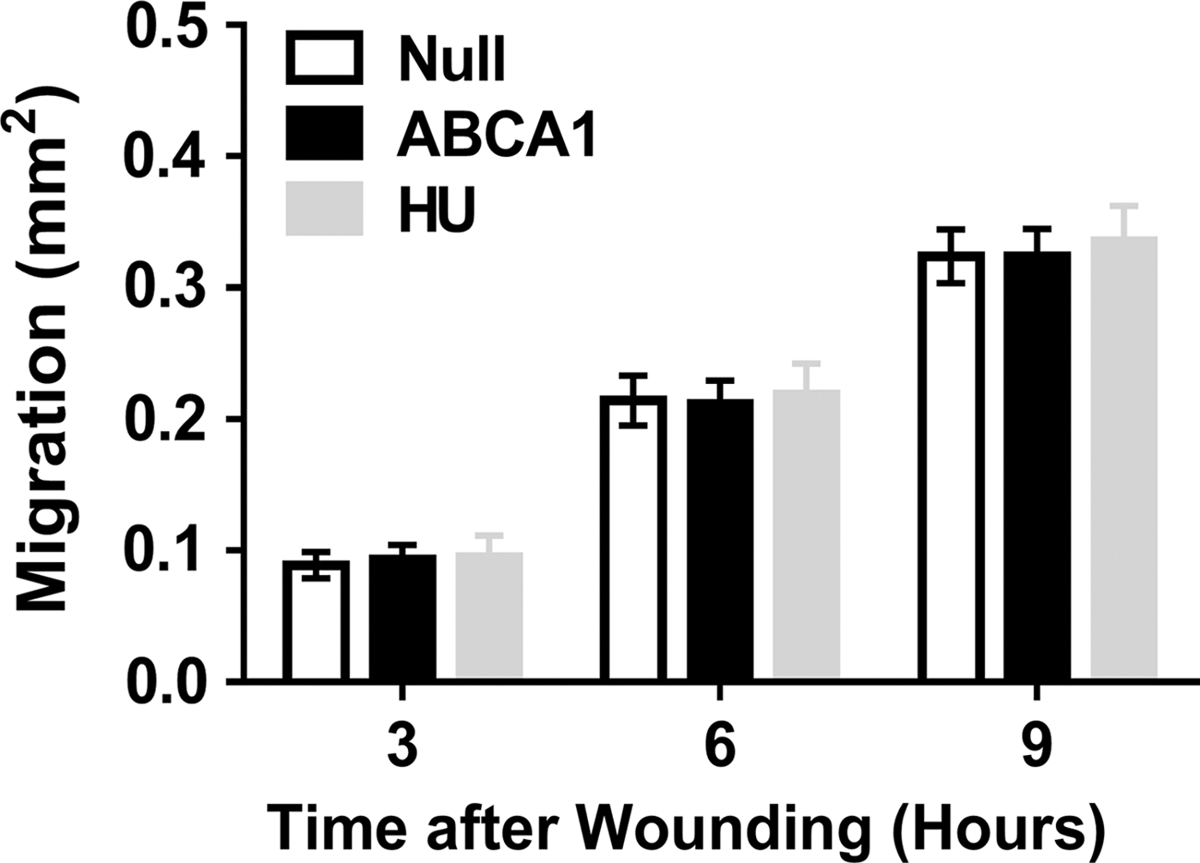
Migration in ECs overexpressing ABCA1. ECs were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1). Confluent EC monolayers were wounded with a pipette tip and imaged immediately after wounding and 3, 6, and 9 h later. Migration is the difference in wound area between each time point and t = 0. HU-treated ECs (untransduced) were included to determine whether wound coverage resulted from EC proliferation. Three independent transductions; each with three wells per condition. Data are mean ± SEM.

Overexpression of ABCA1 in ECs does not trigger apoptosis. ECs were transduced with either HDAdNull (Null) or HDAdABCA1 (ABCA1). ECs treated with camptothecin (CPT) were a technical control.
Overexpression of ABCA1 does not affect baseline NO production or increase inflammatory markers
Synthesis and release of NO is a critical EC function because EC-derived NO contributes to normal vasomotor function, blood pressure regulation, EC survival, and angiogenesis. In addition, physiologic levels of EC-derived NO inhibit platelet aggregation, leukocyte adhesion, and smooth muscle cell proliferation. 46 Depletion of cholesterol in ECs and their caveolae by treatment with either β-cyclodextrin or cyclosporin A displaces eNOS from caveolae and inhibits eNOS activation. 24,25 By increasing cholesterol efflux and depleting EC cholesterol, overexpression of ABCA1 in ECs could potentially reduce eNOS activity and NO synthesis. Therefore, we compared NO production in ECs transduced with either HDAdABCA1 or HDAdNull. Overexpression of ABCA1 had no effect on EC NO production (p > 0.1; Supplementary Fig. S2), providing additional evidence that increasing EC cholesterol efflux via ABCA1 overexpression does not cause EC dysfunction. Dysfunctional ECs are characterized by increased expression of the cytokine interleukin (IL)-6 and the adhesion molecules intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion protein (VCAM)-1, molecules that contribute to the pathogenesis of inflammatory vascular diseases such as atherosclerosis. 47,48 Accordingly, we tested whether EC overexpression of ABCA1 altered baseline expression of these inflammatory mediators. We found no differences in expression of IL-6, ICAM-1, or VCAM-1 between ABCA1-overexpressing ECs and controls (p > 0.1; Supplementary Fig. S3A-C).
ABCA1 overexpression decreases nuclear factor (NF)-κB target gene expression and lipid raft TLR4, and increases NO in serum-starved, apoAI-treated ECs
After initiation of this project, others reported that inhibition of cholesterol efflux in ECs of hyperlipidemic mice (by EC-specific deletion of both ABCA1 and ATP-binding cassette transporter subfamily G1, member 1 [ABCG1]) increased expression of NF-κB target genes (including both cytokines and adhesion molecules) in ECs stimulated ex vivo with LPS. 29 LPS activates NF-κB, 49 which stimulates pro-inflammatory target gene expression. Accordingly, we designed experiments to investigate whether overexpression of ABCA1 in ECs would decrease expression of NF-κB–stimulated cytokines and adhesion molecules, either without or with addition of LPS. To increase the sensitivity of our assay system to detect effects both of overexpressed ABCA1 and of LPS, we cultured HDAdABCA1- and HDAdNull-transduced ECs in serum-free medium and supplied exogenous apoAI protein. This experimental design differed from the experiments described above, in which we measured expression of NO, IL-6, ICAM-1, and VCAM-1 in ECs grown in serum-containing medium without added apoAI. When the transduced ECs were cultured in serum-free medium supplemented with the ABCA1 ligand apoAI (Fig. 5A), expression of several NF-κB target genes (IL-6, TNF-α, ICAM-1, and VCAM-1) was reduced by ≥50% in HDAdABCA1-transduced ECs compared to HDAdNull-transduced ECs (p ≤ 0.06 for all; Fig. 5B–5E). We repeated this experiment with LPS-treated ECs (Fig. 5F). In the presence of LPS, IL-6 and TNF-α expression was reduced by ∼90% in HDAdABCA1-transduced ECs (p < 0.001 for both; Fig 5G and 5H) and expression of ICAM-1 and VCAM-1 was reduced by ∼50% (p < 0.001 for both; Fig. 5I and 5J).

Overexpression of ABCA1 attenuates nuclear factor (NF)-κB target-gene expression in apoAI-treated ECs.
LPS stimulates NF-κB target gene expression by binding to TLR4, which drives TLR4 translocation into plasma-membrane lipid rafts, initiating intracellular signals that lead to activation of NF-κB target genes. 42 Because increased ABCA1/apoAI-mediated cholesterol efflux decreases LPS-mediated inflammatory signals by disrupting lipid rafts, 50,51 we hypothesized that decreased NF-κB target gene expression in apoAI-treated HDAdABCA1-transduced ECs was associated with a decrease in lipid-raft TLR4. To test this hypothesis, we isolated lipid-raft and non–lipid-raft fractions from ECs that were transduced with either HDAdABCA1 or HDAdNull, and then treated with apoAI. Caveolin-1, a marker of lipid rafts, 51 was present only in the lipid-raft fractions, confirming effective lipid-raft isolation (Fig. 6A). Lipid-raft TLR4 protein was significantly decreased in ABCA1-overexpressing ECs compared to HDAdNull-transduced ECs (∼80% decrease; p = 0.001; Fig. 6A and 6B).

Overexpression of ABCA1 decreases LPS-stimulated Toll-like receptor 4 (TLR4) translocation into lipid rafts and causes a trend towards increased nitric oxide production in apoAI-treated ECs.
As already mentioned, others recently showed that impairment of EC cholesterol efflux by combined EC-specific deletion of ABCA1 and ABCG1 in mice decreases aortic eNOS activity and NO production. 29 Moreover, an earlier study reported modestly increased eNOS mRNA in aortas of transgenic mice with EC-targeted ABCA1 overexpression. 19 We therefore used our serum-free culture system (with added apoAI; Fig. 5A) to test the hypothesis that EC overexpression of ABCA1 increases NO production. Under these conditions, we observed a trend towards increased NO production in HDAdABCA1-transduced ECs (∼55% increase compared to HDAdNull-transduced ECs; p = 0.1; Fig. 6C).
Discussion
We tested whether overexpression of ABCA1 in ECs in vitro enhances apoAI-mediated cholesterol efflux without negatively impacting EC physiology, and we tested the hypothesis that ABCA1 overexpression in ECs has anti-inflammatory effects. Our major findings are (1) transduction of ECs with ABCA1 significantly and substantially increases ABCA1 protein and apoAI-mediated cholesterol efflux; (2) a ∼3-fold overexpression of ABCA1 does not alter EC proliferation, metabolic activity, migration, apoptosis, NO production, or expression of inflammatory markers under basal culture conditions; (3) when ECs are maintained in serum-free medium and treated with apoAI, ABCA1 overexpression is accompanied by decreased expression of inflammatory markers both in the presence and absence of added LPS; and (4) apoAI-treated ABCA1-overexpressing ECs have a large (∼80%) decrease in lipid-raft TLR4, and a trend towards increased NO production. These results strongly support further testing of ABCA1 overexpression in ECs as a gene-therapy strategy for atherosclerosis.
RCT is an attractive target for atheroprotective gene therapy because RCT removes excess cholesterol from peripheral cells—including lipid-laden cells within the artery wall that contribute to atherosclerosis—and transports cholesterol to the liver for excretion. 9 Moreover, the key molecules (and genes) that control RCT are known, 52 suggesting that RCT could be enhanced and atherosclerosis could be reduced by gene therapy that increases levels of these molecules in the vessel wall. In earlier work, we attempted to increase RCT from blood vessels by transducing ECs in hyperlipidemic rabbits with vectors expressing apoAI, a plasma protein that initiates RCT by accepting cholesterol from ABCA1. 53 Despite secretion of only nanogram amounts per day of apoAI by the transduced EC 16 (insufficient to increase plasma apoAI levels), 15,17 this approach substantially reduced atherosclerosis progression (∼30%) and accelerated regression of small- to moderate-sized (but not large) atherosclerotic lesions. 15,17 EC-targeted apoAI gene therapy is likely successful because it delivers lipid-poor apoAI protein precisely where it is needed to initiate RCT: adjacent to lipid-laden artery-wall cells.
Because EC-targeted apoAI gene therapy is only partially effective in preventing and reversing atherosclerosis, 15,17 we considered how to increase its efficacy. We noted that EC ABCA1 can increase transport of apoAI across ECs to the subendothelium, 54 where apoAI could more effectively remove lipid from intimal macrophages and smooth muscle cells. Moreover, ABCA1 is a rate-controlling protein in the RCT pathway 55 and ABCA1 overexpression in ECs is atheroprotective in transgenic mice. 19 Accordingly, overexpression of ABCA1 in ECs along with apoAI seemed a promising approach. However, before attempting combination gene therapy (apoAI + ABCA1), we first tested whether we could overexpress ABCA1 in ECs. Unlike apoAI, ABCA1 is expressed in ECs, 56 necessitating an increase above endogenous ABCA1 expression levels. In addition, because ABCA1 mRNA and protein levels correlate poorly in many cell types, 57 it was uncertain that overexpression of ABCA1 mRNA would also increase ABCA1 protein. We also considered that unregulated ABCA1 overexpression in ECs might have deleterious consequences due to cellular cholesterol depletion. We found that under typical cell culture conditions, HDAdABCA1 increases EC ABCA1 protein and cholesterol efflux by ∼3-fold and does not alter critical EC functions. These results bode well for EC-targeted ABCA1 gene therapy.
Despite several reports of toxicity in ECs and other cell types subjected to cholesterol depletion, 20 –28 we found no evidence of toxicity in ABCA1-overexpressing ECs. Absence of toxicity might be explained by the ability of ECs (as with most cell types) to both synthesize cholesterol and to absorb extracellular cholesterol, primarily via cell surface receptors. 58 Toxicity associated with increased ABCA1-mediated cholesterol efflux might also be prevented by upregulation of EC miR33a, consequent to cellular cholesterol depletion that activates SREBP2, 59 the host gene for miR33a. 60 Elevated miR33a would downregulate endogenous ABCA1 and ABCG1, preserving cellular cholesterol. 60 However, miR33a cannot downregulate HDAdABCA1-expressed ABCA1 because the miR33a target sequence in the ABCA1 3′ untranslated region 60 is not present in HDAdABCA1. Toxicity of EC ABCA1 overexpression could be a more important concern in the clinical setting because candidates for HDAdABCA1 gene therapy would likely be taking statin drugs, which both inhibit cholesterol synthesis and lower plasma cholesterol. Future studies, performed in animal models, are needed to continue to assess whether constitutive overexpression of ABCA1 is toxic to ECs.
Our gene therapy approach envisions an atheroprotective role for EC ABCA1. Roles for ABCA1 in atherosclerosis are most thoroughly studied in other cell types: specifically, hepatocytes and macrophages. These studies have yielded conflicting results with regard to cell-type–specific atheroprotective effects of ABCA1. 61 –63 Nevertheless, several themes have emerged. First, hepatocyte ABCA1 plays a major role in synthesis of plasma HDL 62,64 –66 and also increases plasma levels of atherogenic plasma apoB-containing lipoproteins. 62,66 Second, manipulation of hepatocyte ABCA1 expression via knockout or overexpression yields variable effects on atherosclerosis. Whether hepatocyte ABCA1 expression increases or reduces atherosclerosis seems to depend on model-specific variability in effects of altered hepatocyte ABCA1 expression on relative levels of plasma HDL and apoB-containing lipoproteins. 62,66,67 In one study, adenovirus-mediated overexpression of ABCA1 in hepatocytes resulted in elevated, but dysfunctional plasma HDL, and increased atherosclerosis. 68 Third, macrophage ABCA1 expression is atheroprotective in most animal models. 65,69,70 However, macrophage ABCA1-mediated atheroprotection was absent in one model. This outcome was explained by a surprising role discovered for myeloid cell ABCA1 in increasing in hepatic VLDL secretion and plasma levels of atherogenic apoB-containing lipoproteins. 63
In contrast to these variable results obtained in mice with altered hepatic and macrophage ABCA1 expression, EC ABCA1 is consistently atheroprotective in mouse models. Terasaka et al. 71 reported that EC-expressed ABCA1 protects against high-fat diet–induced EC dysfunction. More recently, Vaisman et al. 19 showed that germ line EC-targeted overexpression of ABCA1 in mice reduced atherosclerosis, while also increasing plasma HDL-C. After initiation of the present study, Westerterp et al. 29 reported accelerated atherosclerosis in mice with EC-specific loss of ABCA1. Our results are congruent with all of these studies in suggesting that increased EC ABCA1 expression will be atheroprotective. Moreover, our results are more clinically applicable because—unlike germline EC-targeted transgenesis 19 —our somatic gene-transfer approach can be applied clinically. In addition, our approach of focal EC gene transfer, performed in small vessel segments, 15,17 is highly unlikely to alter plasma HDL levels. Therefore, in contrast to the Vaisman study in which pan-endothelial ABCA1 overexpression increased atheroprotective plasma HDL as well as EC ABCA1 levels, our approach will allow a specific test of whether increased EC ABCA1 expression per se is atheroprotective.
Our study reveals three mechanisms through which elevated EC ABCA1 could reduce atherosclerosis (Supplementary Fig. S4): (1) enhanced EC cholesterol efflux, which could reduce or eliminate atherogenic cholesterol crystal formation both within and adjacent to luminal EC 72 ; (2) decreased EC inflammatory signaling, via depletion of lipid rafts and reduction of lipid-raft TLR4 73 ; and (3) increased eNOS activity and NO production, likely mediated by HDL formation and binding of HDL to EC scavenger receptor B1. 74 Another potential atheroprotective effect of increased EC ABCA1, not investigated here, is activation of JAK2/STAT3-dependent anti-inflammatory signals. Activation of this pathway by binding of apoAI to ABCA1 reduced LPS-mediated induction of inflammatory cytokines in macrophages and was independent of the lipid-transport function of ABCA1. 32 Whether JAK2/STAT3 signaling is also activated in ABCA1-overexpressing ECs will be the subject of future work.
In summary, in vitro studies suggest that EC-targeted ABCA1 overexpression could diminish both lipid-related and inflammation-related contributions to atherosclerosis. Although several groups have attempted gene therapy with apoAI, we are aware of only one report of gene therapy with ABCA1 (mentioned above, in which liver-targeted ABCA1 gene therapy in mice increased atherosclerosis). 68 The paucity of ABCA1 gene therapy studies might be due—in part—to the large size of the ABCA1 cDNA (∼8 kB), which exceeds the capacity of many viral vectors. Because HDAd can accommodate inserts >30 kB, it is possible to construct a single HDAd that expresses both ABCA1 and apoAI. Future studies, performed in our rabbit models, 15,17,34 will determine whether EC ABCA1 overexpression is atheroprotective either alone or in combination with apoAI overexpression.
Footnotes
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grant R01HL114541 and the John L. Locke Jr. Charitable Trust. Dr. Stamatikos was supported by NIH grant T32HL007828 and by a postdoctoral fellowship from the American Heart Association 17POST33650120. We thank AdVec, Inc. for permission to use the HDAd reagents, Genscript for molecular cloning services, Sanjay Kubsad for technical assistance, the University of Washington Department of Pathology Flow Cytometry Core Facility for FACS analysis assistance and Julia Feyk for excellent administrative assistance.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.