
Abstract
Peripheral artery disease (PAD) is a debilitating and prevalent condition characterized by blockage of the arteries, leading to limb amputation in more severe cases. Mesenchymal stem/stromal cells (MSC) are known to have intrinsic regenerative properties that can be potentiated by the introduction of pro-angiogenic genes such as the vascular endothelial growth factor (VEGF). Herein, the use of human bone marrow MSC transiently transfected with minicircles encoding for VEGF is proposed as an ex vivo gene therapy strategy to enhance angiogenesis in PAD patients. The VEGF gene was cloned in minicircle and conventional plasmid vectors and used to transfect bone marrow–derived MSC ex vivo. VEGF expression was evaluated both by quantitative polymerase chain reaction and enzyme-linked immunosorbent assay. The number of VEGF transcripts following MSC transfection with minicircles increased 130-fold relative to the expression in non-transfected MSC, whereas for the plasmid (pVAX1)-based transfection, the increase was 50-fold. Compared to the VEGF basal levels secreted by MSC (11.1 ± 3.4 pg/1,000 cells/day), significantly higher values were detected by enzyme-linked immunosorbent assay after both minicircle and pVAX1 transfection (644.8 ± 82.5 and 508.3 ± 164.0 pg/1,000 cells/day, respectively). The VEGF overexpression improved the angiogenic potential of MSC in vitro, as confirmed by endothelial cell tube formation and cell migration assays, without affecting the expansion potential ex vivo, as well as multilineage differentiation capacity or immunophenotype of MSC. Although preclinical in vivo studies are required, these results suggest that minicircle-mediated VEGF gene delivery, combined with the unique properties of human MSC, could represent a promising ex vivo gene therapy approach for an improved angiogenesis in the context of PAD.
Introduction
Peripheral arterial disease (PAD) is a chronic disease caused by obstruction of the arteries, leading to decreased blood flow to the lower extremities. PAD affects 3–10% of the world population, and about 30% of these patients are faced with the possibility of limb amputation. In order to recover tissue oxygenation, those patients are dependent on the adaptation of pre-existing vessels or on the formation of new vessels. 1 Therapeutic delivery of angiogenic factors and/or stem/progenitor cells to promote revascularization of the ischemic regions represents a potential therapeutic approach to regenerate damaged tissue and prevent amputations in patients with PAD. 2,3
Human mesenchymal stem/stromal cells (MSC) are multipotent cells known to set up a regenerative environment and modulate anti-inflammatory responses. 4 Though the mechanisms underlying the immunomodulatory capacity of MSC are not totally understood, some studies have suggested that it is not only dependent on cell–cell contact but also most likely relies on the secretion of soluble factors that act through a paracrine way on other cells. 5 –7 These paracrine effects of MSC include immunomodulation, anti-apoptosis, angiogenesis, anti-scarring, chemoattraction, and support of the growth and differentiation of other stem/progenitor cells. 6 While it is not known if potential rejection of donor MSC (described as immunoevasive) may influence the efficacy of allogeneic MSC therapies, 8 several clinical studies have advocated for an allogeneic setting. This would make it possible to use an off-the-shelf product, allowing immediate access for acute interventions. Furthermore, a large number of cells from young and healthy donors would be available that could overcome the age-related loss of the regenerative capacity of patient cells. 9
The action of MSC in the context of an angiogenic therapy has been attributed to the secretion of signalling factors such as vascular endothelial growth factor (VEGF), among others. VEGF is a key regulator of physiological angiogenesis, known to promote endothelial cell growth and survival and one of the most important and powerful promoters of vascular regeneration. 10 Several studies have suggested the use of MSC-based cell therapy to enhance angiogenesis and induce tissue repair in PAD. 11,12 Nevertheless, although the therapeutic benefit of MSC administration has been observed in several studies, clinical trials have produced conflicting data or shown only modest benefits. 13 Thus, strategies to improve MSC therapeutic properties in a robust and reliable way are required. In that regard, genetic engineering of MSC with VEGF-encoding vectors can improve the intrinsic secretion of VEGF by cultured MSC, thus improving the angiogenic potential of these cells in the context of ex vivo gene therapy. 14 Ex vivo gene therapy involves genetic engineering of cells outside the body and their subsequent transplantation back into patients. 15
The genetic modification of human MSC would require vectors that efficiently and safely transfer the target therapeutic gene. Plasmids are especially adequate for this purpose due to their low immunogenicity, non-integrative nature (low risk of insertional mutagenesis), and ease of manufacturing when compared to viral vectors. 16 The key disadvantages of plasmids in the context of ex vivo gene therapy of PAD are the low transfection efficiency and the short duration of transgene expression. 17 However, this can be partially circumvented by using small plasmid derivatives called minicircles that carry only the transgene expression cassette and thus improve transfection on account of their lower size. 18 The lack of bacterial sequences in minicircles (e.g., origin of replication, antibiotic resistant marker) further contributes to improve safety, reduce CpG-mediated immunogenicity, and minimize silencing of transgene expression. A number of studies have shown that minicircle-based cell engineering is able to sustain higher and longer transgene expression, as well as stem-cell survival 19 –23 when compared to conventional plasmid vectors. Furthermore, minicircles have been shown to be superior to plasmids in what concerns angiogenic VEGF expression. 24 –26 In spite of their advantages, the technology for minicircle manufacturing in suitable amounts and with adequate purity to run preclinical and clinical trials is still incipient. In this context, a production strain system and a purification process for optimal minicircle manufacture have been recently established in the authors' laboratory. 27,28
Based on previous studies focused on the optimization of non-viral gene delivery to MSC of different human tissue sources using a reporter fluorescent protein, green fluorescent protein (GFP), 29,30 herein a strategy was developed to improve the angiogenic potential of cultured human bone marrow (BM) MSC by transiently modifying the cells with minicircles encoding VEGF. Engineered MSC were characterized to confirm the maintenance of their intrinsic properties and to evaluate transgene expression and in vitro angiogenic potential. This represents, to the best of the authors' knowledge, the first study where MSC were modified with VEGF-encoding minicircles.
Methods
Plasmid construction, production, and purification
The control plasmid (pVAX-VEGF; 3,531 bp) and parental plasmid (pMINILi-CV; 3,821 bp) expressing the VEGF (165a) gene were obtained from pVAX-VEGF-GFP and pMINILi-CVG, respectively, by removing the GFP gene. Those vectors were constructed as described elsewhere and transformed by heat shock into Escherichia coli strains. 27,28,31 pMINILi-CV contains an expression cassette with VEGF and the human cytomegalovirus (CMV) immediate-early promoter, two multimer resolution sites (MRS) flanking the expression cassette, pMB1 origin of replication, kanamycin resistance gene, and BGH polyadenylation sequence (Fig. 1A). Plasmids coding for VEGF (pVAX-VEGF) were produced in E. coli DH5α strain and purified using an endotoxin-free plasmid DNA purification kit (Macherey-Nagel), as previously described. 29 The concentration of purified pDNA solutions was assayed by spectrophotometry at 260 nm (NanoDrop; Thermo Fisher Scientific), and DNA integrity was confirmed by DNA agarose gels stained with ethidium bromide.

Minicircle production and purification by Escherichia coli BW2P and hydrophobic interaction chromatography (HIC).
Minicircles were produced in a BW2P E. coli strain harboring the parental plasmid pMINLI-CV, according to previously established methods. 27,28 E. coli BW2P was grown until the late exponential phase, and then recombination was induced for 2 h by the addition of 0.01% (w/v) L-(+)arabinose (Merck). As a result of intramolecular recombination between the two MRS, both a minicircle with the expression cassette and a miniplasmid (MP) with the prokaryotic backbone sequences were obtained. Next, all plasmid DNA species were recovered and purified from the producer cells using an endotoxin-free plasmid DNA purification kit (Macherey-Nagel). Then, the minicircle was separated from other DNA forms by digestion with nicking endonuclease (Nb.BbvCI) followed by hydrophobic interaction chromatography (HIC), as described and optimized by the authors' group. 28,32
Isolation and culture of human BM-derived MSC
Human BM-derived MSC were isolated and expanded from healthy donors after informed consent, as described elsewhere, 33 and maintained cryopreserved in liquid/vapor phase nitrogen containers. Upon thawing, cells were cultured for three to five passages under xenogeneic(xeno)-free culture conditions, as previously established. 34,35 Cells were plated at a cell density between 3,000 and 6,000 cells/cm 2 on CELLstart™ CTS™ (Invitrogen) precoated T-flasks using StemPro® MSC SFM XenoFree (Invitrogen) supplemented with 1% GlutaMAX™-I CTS™ (Invitrogen) and 1% antibiotic-antimycotic (Invitrogen). Cells were maintained at 37°C and 5% CO2 in a humidified atmosphere, and the culture medium was changed every 3–4 days. At 70% cell confluence, MSC were detached from the flasks by adding TrypLE™ Select CTS™ (Invitrogen) solution 1 × in phosphate-buffered saline (PBS; Gibco). Cell number and viability were determined using the Trypan Blue (Gibco) exclusion method. BM MSC from between three and five passages were used from four independent donors.
Microporation of BM MSC with pVAX-VEGF and MC-VEGF
Microporation of BM MSC was performed, as previously optimized. 29 For each condition, 1.5 × 106 cells were re-suspended in 100 μL of re-suspension buffer (buffer R; Invitrogen) and incubated with 10 μg of pVAX-VEGF (or the equivalent number of molecules of MC-VEGF). Electroporation was performed using the Neon® Transfection System and a Microporator MP-100 (Digital Bio; Invitrogen) using one pulse (1,000 V; 40 ms width). After microporation, the cell suspension was incubated with 900 μL of Opti-MEM™ I Reduced Serum Medium (Gibco) for 20–30 min. Next, cells were plated at a density of 7,000–8,000 cells/cm 2 in pre-warmed StemPro® XenoFree culture medium. At each time point (days 2, 5, and 7), the number of cells was estimated using the Trypan Blue dye exclusion method. Cell recovery for each microporated sample was determined after 48 h by calculating the ratio between viable cells in the condition where cells were microporated and viable cells in the control condition (non-electroporated), as described elsewhere. 29 Two controls were also prepared: a control for microporation process corresponding to MSC microporated without DNA, and non-microporated cells, referred to herein as “control,” which will be used for comparison in subsequent studies.
In vitro multilineage differentiation potential and immunophenotype characterization of transfected cells
At 7 days after transfection, the osteogenic and adipogenic differentiation potential of cells transfected with both pVAX-VEGF and MC-VEGF were assessed, as previously described, using StemPro® osteogenesis/adipogenesis differentiation kits (Life Technologies). 34
For immunophenotypic characterization of the engineered cells with the two vectors, cells were analyzed by flow cytometry 48 h and 7 days after transfection using a panel of mouse anti-human monoclonal antibodies (PE-conjugated) against: CD34, CD45, CD90, CD73, CD80, CD14, CD105, and human leukocyte antigen (HLA)-DR (all from Biolegend). Cells were incubated with the monoclonal antibodies for 15 min in the dark at room temperature. Then, they were washed in 2 mL of PBS and finally fixed with 1% paraformaldehyde (Sigma–Aldrich). Appropriate isotype controls (IgGγ1 and IgGγ2b) were also prepared. A minimum of 10,000 events was collected for each sample, and CellQuest (Becton Dickinson) and FlowJo® (LLC) software were used for acquisition and analysis, respectively.
Quantification of VEGF expression by the transfected cells using quantitative polymerase chain reaction and enzyme-linked immunosorbent assay
To quantify the expression of VEGF by the cells transfected with pVAX-VEGF and MC-VEGF, both real-time quantitative polymerase chain reaction PCR (qPCR) and enzyme-linked immunosorbent assay (ELISA) were performed. For qPCR, cells were harvested at each time point (days 2, 5, and 7), centrifuged, and kept at −80°C until further analysis. Total RNA was isolated using the RNeasy Mini Kit (Qiagen). RNA was quantified by ultraviolet spectrophotometry (NanoDrop), and cDNA was synthetized with the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). qPCR analysis was performed in a StepOne Real-Time PCR System (Applied Biosystems) using Fast SYBR™ Green Master Mix (Applied Biosystems), 0.5 μM of each primer, and 1 μL of cDNA in 20 μL of final reaction volume. The following primers (StabVida) were used for VEGF amplification: VEGF_fwd—GGAGGAGGGCAGAATCATCAC and VEGF_rev—GGTCTCGATTGGATGGCAGT. The 2−ΔΔCT method of relative quantification was applied to determine the fold change in mRNA expression. 36 GAPDH was used as the housekeeping gene and non-microporated MSC as a baseline.
For ELISA, culture supernatant was collected at each time point (days 2, 5, and 7), centrifuged at 500 g for 10 min and kept at −80°C until further analysis. A human VEGF-A ELISA kit (RayBiotech) was used, following the manufacturer's instructions.
Preparation of conditioned medium for functional assays
To prepare conditioned media from cultured MSC for functional assays, control and MSC engineered with pVAX-VEGF or MC-VEGF were plated at a density of 12,500 cells/cm 2 using StemPro MSC SFM XenoFree for 24 h after microporation. After 24 h, the medium was changed to Endothelial Basal Medium (EBM-2; Lonza) and maintained for 48 h. Conditioned medium was collected and normalized to cell number 72 h after transfection and kept at −80°C after centrifugation. Fresh EBM-2 and Endothelial Cell Growth Medium (EGM-2; Lonza) were used as controls.
Endothelial cell tube formation assay
A functional assay that relies in the capacity of human endothelial vein endothelial cells (HUVEC) to form tube networks when cultured in Matrigel was used to evaluate the angiogenic potential of modified cells. 37 Conditioned medium (200 μL) derived from both control cultures and gene modified MSC (pVAX-VEGF and MC-VEGF) was used to cultivate commercially available HUVEC (BD Biosciences) on 96-well plates (2 × 104 cells/well) previously coated with Matrigel Basement Membrane Matrix (Corning) for 8 h at 37°C and 5% CO2. After 8 h, tube formation was quantified, and both tube length and tube connections were measured using microscope and ImageJ (National Institutes of Health) software. 37,38
Endothelial transwell migration assay
The migration of endothelial cells in response to soluble factors, such as VEGF, is one of the crucial events during angiogenesis. 39 Herein, conditioned medium collected from MSC transfected cultures was used as a stimulus, and HUVEC's capacity to migrate through transwell inserts was quantified by using 8 μm pore transwell inserts (Millipore) previously coated with 10 μg/mL of fibronectin (Sigma–Aldrich) for 1 h at 37°C. 40,41 After a wash with PBS, 50,000 HUVEC were added to the transwells using 100 μl of EBM-2, which were then inserted and maintained on a 24-well plate with 600 μL of conditioned medium for 6 h at 37°C and 5% CO2. After 6 h, the transwells were washed with PBS, and the cells in the upper chamber were removed with cotton swabs. The cells that migrated to the lower side of the insert were stained with crystal violet 0.5% (Sigma–Aldrich) for 30 min. 42 After washing twice with PBS, each insert was observed under the microscope, and the total number of migrated HUVEC per optical field was quantified (100 × magnification).
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism v6, and significance was determined by Tukey's multiple comparison test and set at a p-value of <0.05.
Results
Minicircle production and purification
Minicircles were produced in an E. coli strain BW2P, as described. 27, 28 This strain contains a copy of the parA gene inserted in the bacterial chromosome under the control of the arabinose inducible PBAD/AraC promoter. 43 Addition of L-arabinose induces expression of ParA resolvase, the enzyme that mediates intramolecular recombination between the two MRS in the parental plasmid (PP) backbone. This recombination generates a 1,715 bp minicircle (MC) with the expression cassette (MC-VEGF) and a 2,106 bp MP with the prokaryotic backbone sequences (Fig. 1A). Recombination was confirmed by agarose gel electrophoresis analysis of purified DNA before induction (0 h) and 1 or 2 h after induction (Fig. 1B). To isolate MC-VEGF from MP and un-recombined PP, a digestion step with the nicking enzyme Nb.BbvCI was performed after cell lysis and DNA purification in order to convert supercoiled (sc) MP and PP molecules into the corresponding open circular (oc) isoforms. Since the nicking site for Nb.BbvCI is located in the prokaryotic backbone, the MC remains unaffected. Next, hydrophobic interaction chromatography was used to separate sc MC from oc DNA by exploring differences in the hydrophobicity of the molecules. 28 A phenyl-Sepharose column was used, and elution was promoted using a stepwise strategy with decreasing salt ([NH4]2SO4) concentrations (Fig. 1C). Agarose gel electrophoresis (Fig. 1D) of eluted fractions showed that un-bound MP molecules are washed out of the column at 17% buffer B (fractions 3–5 and 8–9), MC-VEGF (fractions 35–39) is eluted by increasing buffer B to 35%, and some residual RNA (fractions 52–53) is eluted during the last step by maximizing buffer B to 100%.
Characterization of human MSC engineered with VEGF-encoding vectors: proliferative capacity, cell recovery, immunophenotype, and multilineage differentiation potential
In this study, MSC from BM cultured under xenogeneic (xeno)-free conditions 34 were transfected by microporation with two non-viral vectors encoding for VEGF: a plasmid vector (pVAX-VEGF) and a minicircle (MC-VEGF). Non-transfected cells were used as a control, and cells microporated (without DNA) were also included in the study. The transfected cells were analyzed on days 2, 5, and 7 post transfection, according to a previously established transfection procedure. 30 To evaluate the impact of microporation-based gene delivery on the proliferative capability of MSC, the number of viable cells was determined (Fig. 2A). Non-transfected cells (used as a control) displayed the highest cell number at both day 2 and day 5, but on day 7, there are almost no differences between the groups. Cell recovery, which reflects the level of cell death in microporated samples (Fig. 2B), was calculated on day 2, as described elsewhere. 29 Non-transfected cells were considered to have a recovery of 100%, and lower values were observed for the conditions where cells were microporated. Cells microporated without DNA (Micro; used as a control for microporation process) or with MC-VEGF showed similar recoveries (68.8 ± 19.8% and 72.3 ± 11.2%, respectively), which were higher when compared to cells transfected with pVAX-VEGF (45.1 ± 14.0%).
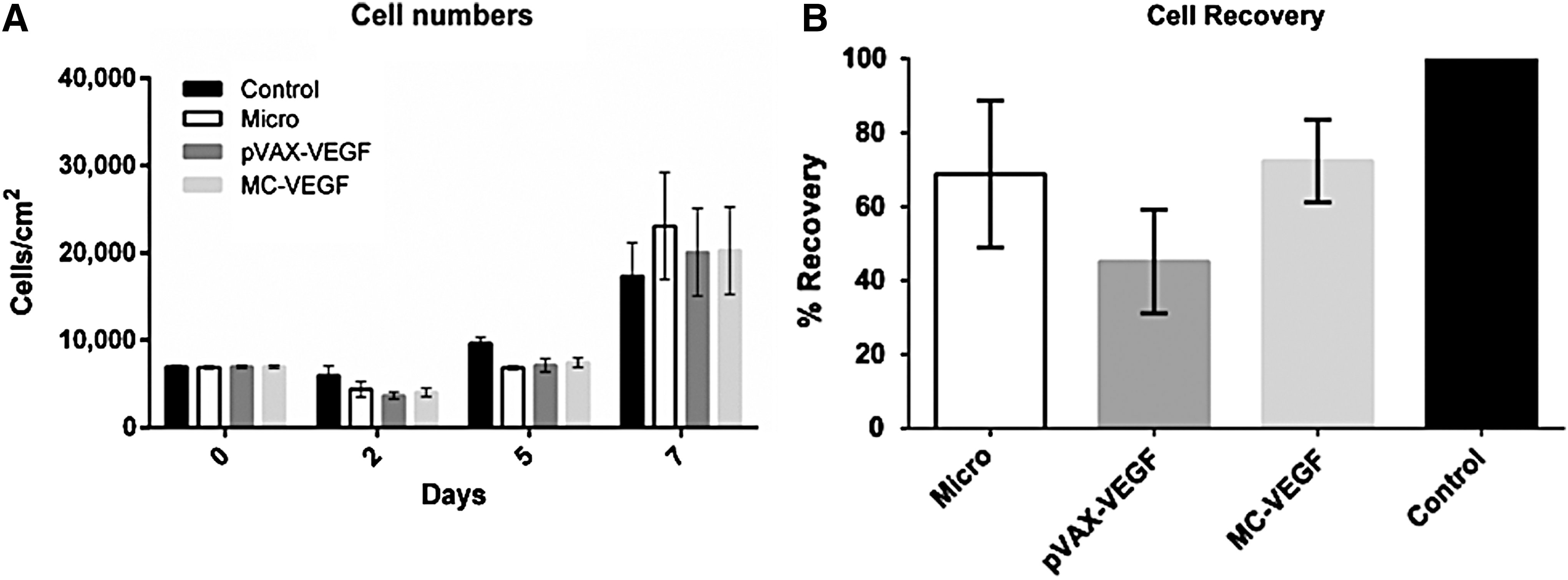
Analysis of the bone marrow (BM) mesenchymal stem/stromal cell (MSC) behavior after microporation with pVAX-vascular endothelial growth factor (VEGF) and MC-VEGF.
The differentiation potential and immunophenotype of microporated cells was assessed based on the minimal criteria proposed to define human MSC. 44 To test the effect of microporation on MSC multipotency, differentiation protocols toward osteogenic and adipogenic lineages were accomplished. By using lineage-specific stains, it was observed that cells engineered with the two vectors (pVAX-VEGF and MC-VEGF) were able to give rise to both osteocytes and adipocytes in vitro (Fig. 3A). To evaluate the immunophenotype of the modified cells, flow cytometry employing a panel of monoclonal antibodies against different surface markers was used. Regardless of the vector used, engineered MSC maintained their phenotypic profile after transfection (i.e., negative; <5%) for CD34, CD45, CD80, CD14, and HLA-DR and positive (>95%) for CD90, CD73 and CD105 (Fig. 3B).
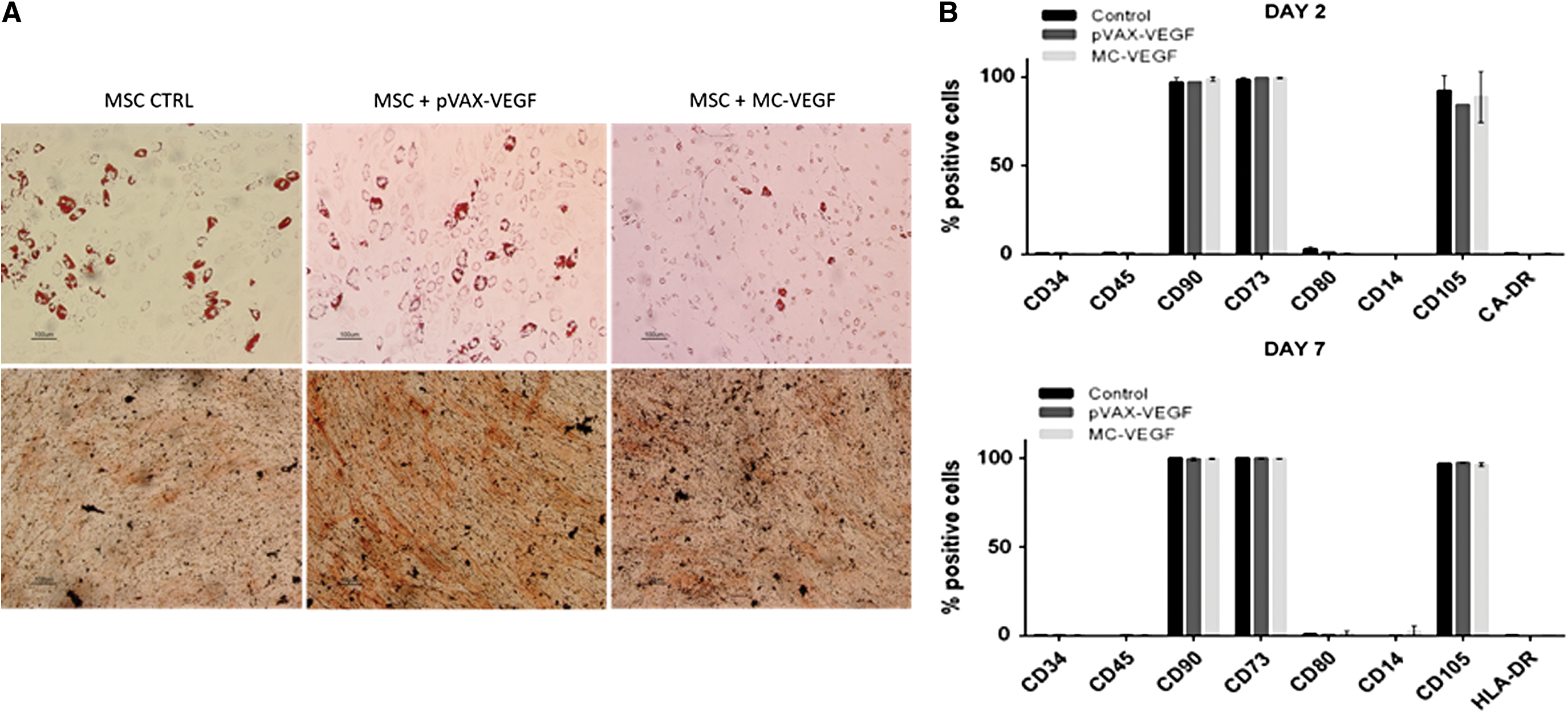
Characterization of BM MSC after transfection with pVAX-VEGF and MC-VEGF.
Quantification of VEGF expression and secretion by engineered MSC
VEGF production was evaluated on days 2, 5, and 7 by both qPCR (Fig. 4A) and ELISA (Fig. 4B). Two days after microporation, MSC transfected with MC-VEGF showed 130-fold higher mRNA copies of VEGF than non-transfected cells (control; p < 0.01), while cells transfected with pVAX-VEGF showed just a 50-fold increase on VEGF mRNA copies compared to the control (p < 0.05). The VEGF expression decreased from day 2 to day 5, remaining higher for transfected cells when compared to the control (though differences were no longer statistically significant). Finally, on day 7, the expression of VEGF was almost the same for transfected and non-transfected cells. Overall, for the three time points tested the highest VEGF expression was observed for cells transfected with MC-VEGF.
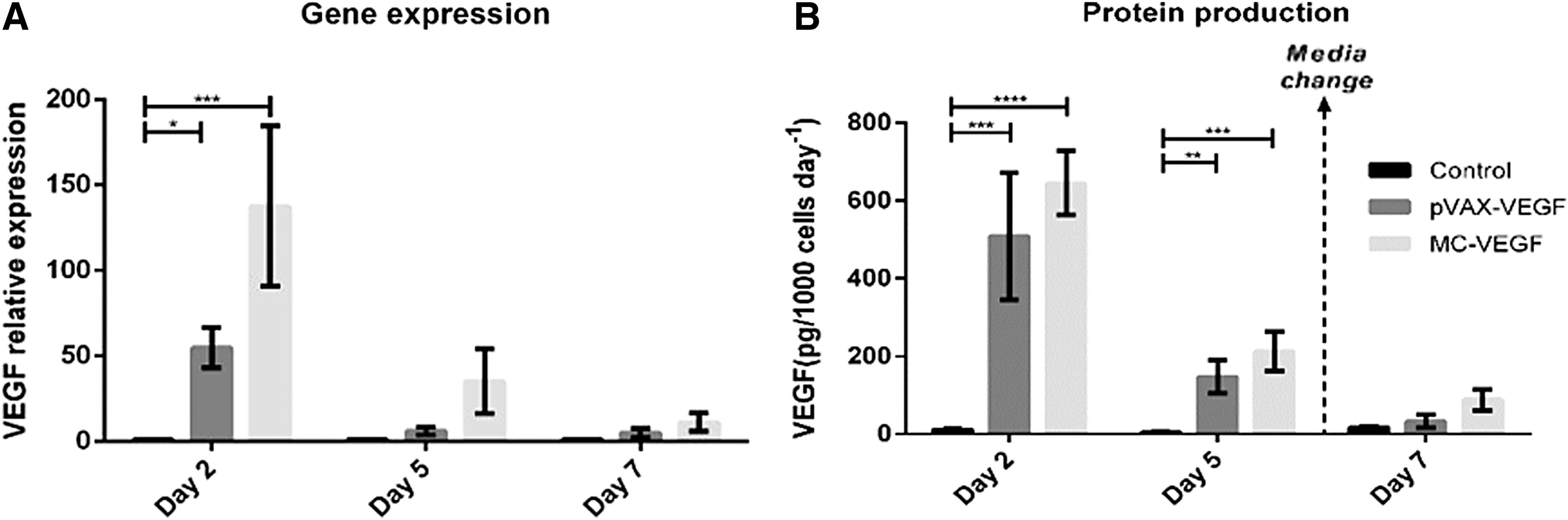
Evaluation of transgene delivery 2, 5, or 7 days after microporation with pVAX-VEGF and MC-VEGF. Analysis of BM MSC VEGF
The profiles of VEGF secretion into the culture medium were very similar. The highest VEGF secretion rates were observed on day 2 for cells transfected with MC-VEGF (644.8 ± 82.5 pg/1,000 cells/day), followed by pVAX-VEGF transfected MSC (508.3 ± 164.0 pg/1,000 cells/day). No significant VEGF secretion (11.1 ± 3.4 pg/1,000 cells/day) was observed for control MSC (non-transfected). Though the VEGF production rate decreased until day 5, the trend observed was similar to day 2, with the highest VEGF secretion for MC-VEGF (212.4 ± 50.9 pg/1,000 cells/day) supernatant and almost no VEGF on control samples (4.8 ± 1.5 pg/1,000 cells/day). On day 7, there were only smaller differences on production rates for engineered cells (87.7 ± 27.7 and 33.2 ± 16.5 pg/1,000 cells/day for MC-VEGF and pVAX-VEGF, respectively) compared to the control (17.4 ± 3.5 pg/1,000 cells/day).
Angiogenic capacity of the engineered MSC assessed by in vitro functional assays
To evaluate the angiogenic potential of modified cells, conditioned medium retrieved 72 h after microporation (or culture) from cultures of transfected and non-transfected (control) MSC, as described in the Methods, was used for the cultivation of HUVEC on Matrigel-coated surfaces (Fig. 5A). After 8 h, cell tube formation by HUVEC was observed and quantified with regard to the number of tubes and branch points (Fig. 5B and C). The conditioned media retrieved from cultures of cells modified with pVAX-VEGF and MC-VEGF induced the formation of more tubes (34.4 ± 4.0 and 36.2 ± 1.9, respectively), as well as connections (27.7 ± 1.9 and 35.4 ± 5.1, respectively), compared to the basal medium EBM-2 (negative control; p < 0.05 for pVAX-VEGF and p < 0.01 for MC-VEGF) or conditioned medium obtained from cultures of non-transfected cells (p < 0.05 for MC-VEGF). Although the differences between MC-VEGF and pVAX-VEGF were not statistically significant, conditioned medium from cells transfected with MC-VEGF showed an increased potential to induce tube formation by HUVEC, suggesting a higher angiogenic potential compared to MSC transfected with pVAX-VEGF.
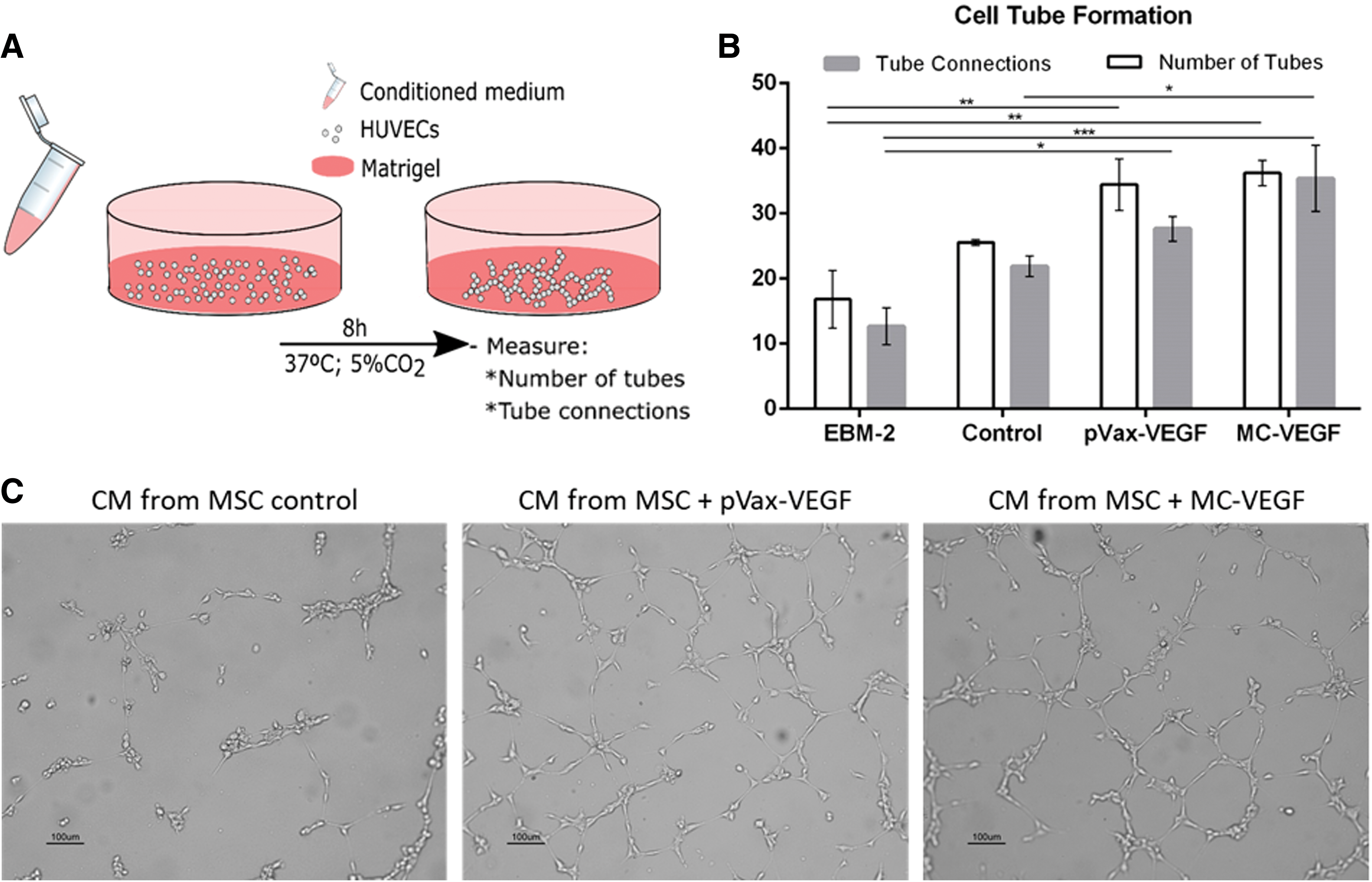
Cell tube formation assay.
The migration ability of HUVEC toward a conditioned medium obtained from cultured MSC was also evaluated, as schematized in Fig. 6A. HUVEC were cultured for 6 h in the upper part of the transwell, while conditioned medium from transfected (with pVAX-VEGF or MC-VEGF) and non-transfected cells was kept in culture plate wells where the transwells were placed. HUVEC cells that migrated through the transwell pores were counted, and the values were normalized relatively to a positive control where complete culture medium for endothelial cell growth was employed (EGM-2; Fig. 6A and B). Conditioned medium retrieved from cultures of MSC engineered with MC-VEGF cultured for 72 h induced the highest HUVEC migration (111.3 ± 4.6%). Furthermore, the difference between this and culture supernatant from control cells (non-transfected; 60.3 ± 3.6%) was statistically significant (p < 0.05). Conditioned medium obtained from cells transfected with pVAX-VEGF (also after 72 h of culture) also lead to high HUVEC migration rates (96.0 ± 2.2%), with values similar to the positive control (100%).

Transwell migration assay.
Discussion
MSC have been extensively exploited in experimental clinical studies for the treatment of a wide range of conditions (cardiovascular, neurological, or autoimmune disorders 45 –48 ) due to their unique properties, namely their trophic activity and immunomodulatory potential. In this context, strategies to improve their potential therapeutic effects have been developed, namely genetic engineering for the overexpression of specific therapeutic proteins in a setting of ex vivo gene therapy. 49,50 Although MSC are able to overexpress genes transferred by viral vectors efficiently, this strategy has the inherent potential risks of oncogene activation or tumor-suppressor gene inactivation, as well as immunogenic responses. 51,52 Although safer than viral vectors, plasmid DNA vectors can still produce immunological responses and silence transgene expression due to the presence of bacterial regions in the backbone (e.g., bacterial origin of replication, antibiotic resistance marker). 53 –55 Minicircles, on the other hand, can be less immunogenic (and thus safer) due to the reduced content of bacterial sequences rich in unmethylated CpG residues that may activate the immune system, and are more efficiently delivered into stem cells by microporation due to their lower size compared to plasmid vectors. 18,56
The angiogenic factor VEGF has been extensively investigated in the context of ischemia treatment in PAD in both preclinical studies 2,57 –59 and clinical trials. 60 –62 In fact, there is already an approved gene therapy product for PAD treatment (Neovasculgen®), which is based on a plasmid DNA encoding the VEGF165 gene under the control of the CMV promoter. 62
Efforts have also been made to explore the angiogenic potential of MSC to treat PAD. 63 Nevertheless, these non-stimulated cells secrete limited levels of VEGF (<20 pg/1,000 cells/day), as shown by the authors and others. 6 This work sought to exploit the inherent regenerative properties of MSC and the angiogenic role of VEGF by transiently engineering these cells to overexpress this factor. VEGF-containing minicircles (MC-VEGF) produced and purified by a recently established protocol, 27,28 as well as a conventional plasmid vector (pVAX-VEGF), were efficiently used to transfect BM MSC by microporation, an electroporation-based method with reduced electrode surface area to diminish cell mortality. 64 The therapeutic features of the modified cells were verified by VEGF quantification, but also by in vitro functional assays that were developed to confirm the angiogenic potential of these cells and predict their effect in vivo. 65 All the work was developed under xeno(geneic)-free conditions already established for MSC culture in order to achieve a reproducible, safe, and reliable MSC cell therapy product. 34
In most studies where minicircles have been tested for the genetic modification of human MSC, commercially available molecules 66 or minicircles purified with a different method were used. 67 –70 The purification approach used in the study by Kay et al. is based on an inducible-SceI nuclease integrated in the bacterial chromosome that degrades MP species in vivo. 67 The authors' methodology for vector manufacturing has at least two advantages: (1) it takes place in vitro, so is easier to control and does not impose an extra metabolic burden on producer cells; and (2) the use of HIC allows separation of supercoiled from other MC isoforms. 28 The method was effective for the purification of MC-VEGF, and isolated fractions of supercoiled MC-VEGF were obtained that were ready for the transfection of MSC.
The parameter used to evaluate cell integrity after transfection in most published works is cell viability, 71,72 but high viabilities can be observed even when few cells are recovered from the electroporation process. Herein, cell recovery was evaluated (Fig. 2B), which was observed to be higher when cells were microporated with MC-VEGF (72.3 ± 11.2%) compared to pVAX-VEGF (45.1 ±14.0%). The cell recoveries observed for MC-VEGF were greater than those reported by Aluigi et al. (around 45%), who used the same cell type but another electroporation method—nucleofection—and a different DNA molecule. 73 Another report using a similar protocol showed comparable cell recoveries for the microporation of BM MSC. 29 The high recoveries observed herein for MC-VEGF may represent an advantage over common plasmids from the clinical standpoint, as an increased number of cells can be recovered from the same initial cell number.
Transfection with either MC-VEGF or pVAX-VEGF did not change MSC features, namely in vitro differentiation capacity and immunophenotype, proposed as minimal criteria for MSC identity, 44 in agreement with other studies using plasmid vectors. 29,71,72 Of note, although the microporation process seems to slow down the proliferation rate of the cells in culture, even without DNA, microporated MSC have their growth potential completely recovered after 7 days, reaching the same numbers as non-transfected control cells. Furthermore, for MSC microporated with MC-VEGF, the recoveries were higher (72.3 ± 11.2%) and similar to microporation control (68.8 ± 19.8%) when compared to values obtained for pVAX-VEGF (45.1 ± 14.0%), showing, as expected, that small MC-VEGF molecules causes less cell damage than conventional plasmids. 74
The efficiency of the protocol of MSC microporation with MC-VEGF or pVAX-VEGF was first evaluated by quantifying VEGF levels in terms of gene expression (qPCR) and protein production (ELISA). Two days after transfection, cells transfected with minicircles showed higher VEGF production (644.8 ± 82.5 pg/1,000 cells/day) than cells modified with pVAX (508.3 ± 164.0 pg/1,000 cells/day), reflected not only by protein amount in culture supernatant but also by VEGF mRNA copies. Analysis of VEGF copies revealed an increase of around 130-fold and around 50-fold for BM MSC transfected with MC-VEGF and pVAX-VEGF, respectively, when compared to non-modified cells. The rise observed in protein production rate compared to the control was around 59-fold for MC-VEGF and around 46-fold for pVAX-VEGF. In both analyses, the same tendency was verified, with higher values observed for MC-VEGF samples. The smaller change observed between the two tested vectors in what concerns protein amount, when compared to mRNA copies, can be due to the short half-life of the VEGF protein, which is approximately 50 min. 75
Even though viral vectors have been proposed as more efficient for cell engineering, the transfection protocol showed an increased VEGF expression compared to some reports where virus were used. Beegle et al. reported that using lentivirus to overexpress VEGF in BM MSC for hind-limb ischemia led to a 10-fold increase in VEGF protein production compared to non-transduced cells measured by ELISA 72 h after viral transduction. 14 In another study, a combination of adenovirus transduction with microencapsulation was developed to induce VEGF overexpression and promote vascularization of tissue-engineered dermis. Here, maximum VEGF production rates measured by ELISA were observed 8 days after transduction, with a 20-fold increase in VEGF production rates relative to non-transduced cells. 76 Although in the mentioned work VEGF levels remained elevated for a longer period, being still detectable after 14 days, the strategy detailed herein led to a higher fold increase in VEGF production (59-fold) regarding basal levels observed in non-modified cells.
In a recent study reporting the use of minicircles to transfect BM MSC using microporation, the transfection efficiency observed was around 40%. 69 Although the current system cannot be directly compared to this, since a reporter gene (luciferase) was used to evaluate transfection and a different therapeutic gene (CXCR4) was tested, similar and promising results were shown in the present study using a therapeutic pro-angiogenic protein (without the use of a reporter that may affect VEGF expression) in a completely xeno-free setting. The analysis was not focused only on transgene expression, but also on the function of the obtained cell therapy product. So, the modified cells were tested with in vitro functional studies that mimic in vivo angiogenesis processes.
VEGF is known to recruit and promote migration of endothelial cells, two important steps in blood vessel formation and remodeling. In order to evaluate the biological activity of the expressed VEGF in this regard, functional assays were performed to predict and evaluate the angiogenic potential of BM MSC engineered with pVAX-VEGF and MC-VEGF by microporation. 77 Experiments were thus set up where conditioned media from cultures of gene modified and control cells were used as a stimulus. In particular, endothelial cell tube formation assays were used to evaluate the capacity of HUVEC to form tubular structures, and transwell migration assays were used to assess the ability of endothelial cells to migrate toward a stimulus, in this case the high VEGF levels, since this is one of the key mechanisms underlying angiogenesis. 39,40
The results for cell tube formation were in accordance with the results for VEGF expression, since conditioned medium from cells modified with MC-VEGF leads to the formation of the highest number of tubes (36.2 ± 1.9) and branch points (35.4 ± 5.1) by HUVEC. Supernatants retrieved from cultures transfected with pVAX-VEGF also contributed to an increased amount of tubes (34.4 ± 3.9) and tube connections (27.7 ± 1.9) compared to non-modified cells (25.6 ± 0.4 tubes and 21.9 ± 1.6 connections). It was expected that high VEGF levels would maximize HUVEC tube formation capacity, since, as an angiogenic factor, VEGF increases the potential of endothelial cells to reorganize and form these tube-like structures that resemble blood vessels. 38 Similar results were observed for cell migration studies performed with HUVEC using conditioned media retrieved from MSC cultures, where induction with complete endothelial growth medium (EGM-2) was set as 100% migration. For the conditioned medium obtained from cultures of MC-VEGF-transfected cells, the highest migration (111.3 ± 4.6%) of HUVEC throughout the transwell pores was observed, followed by the supernatant from cells transfected with pVAX-VEGF (96.0 ± 2.1%) that led to a migration similar to the set control (100%). Once again, these results are related with VEGF amount in culture medium that is also recognized as recruiter of endothelial cells, thus promoting HUVEC migration. 38 These results suggest that modification of MSC using VEGF-containing vectors may contribute to an improvement of the angiogenic potential of these cells, verified by an increase in endothelial cell migration and organization into vessel-like structures.
The ex vivo gene therapy strategy developed herein primes an increased VEGF secretion by MSC within a short time frame (around 2–5 days), which can be advantageous for some critical situations where a fast intervention is required. In this scenario, upon the administration of the modified MSC, the peak of VEGF production will start by promoting endothelial cell migration and proliferation, thus accelerating angiogenesis at the injury site. Then, the decrease in VEGF secretion will avoid an uncontrolled angiogenic process and therefore an abnormal blood vessel growth, which in most severe cases can potentially lead to hemangiomas or even cancer. 78,79
Overall, transfection of MC-VEGF into BM MSC not only induced higher VEGF levels compared to pVAX-VEGF, but also resulted in an increased angiogenic potential of these cells as assessed in in vitro functional studies. Although the differences may not be statistically significant, the use of these smaller vectors presents other advantages, safety being one of the most important. Minicircles have lower size and have reduced numbers of unmethylated CpG sequences, which are commonly found in bacterial DNA and have been documented to trigger immune response through TLR9 activation. 74,80,81 Further studies are required in this field, such as evaluation of TLR9 expression after transfection, since an increase in TLR9 was recently observed, even when small DNA molecules as minicircles are used. 74
Even though further studies are required, namely assessment of therapeutic potential in vivo on a limb ischemia model to evaluate and predict the efficacy of this approach in PAD, this study showed encouraging results on the efficient non-viral gene modification of BM MSC toward the establishment of an angiogenic ex vivo gene therapy strategy to improve the intrinsic therapeutic features of MSC further.
Footnotes
Acknowledgments
Funding received by iBB—Institute for Bioengineering and Biosciences from the FCT-Portuguese Foundation for Science and Technology (UID/BIO/04565/2013) and from Programa Operacional Regional de Lisboa 2020 (project no. 007317) is acknowledged. We also acknowledge the funding received from Programa Operacional Regional de Lisboa 2020 through the project PRECISE—Accelerating progress toward the new era of precision medicine (project no. 16394). J.S. and C.A. acknowledge FCT for the PhD fellowships PD/BD/52477/2014 and PD/BD/116842/ 2016, respectively.
Author Disclosure
No competing financial interests exist.