
Abstract
Machado–Joseph disease (MJD) or spinocerebellar ataxia type 3 is a neurodegenerative disorder caused by an abnormal repetition of a CAG codon in the MJD1 gene. This expansion translates into a long polyglutamine tract, leading to the misfolding of the mutant protein ataxin-3, which abnormally accumulates in the nucleus, thus leading to neurodegeneration in specific brain regions. No treatment able to modify the progression of the disease is available. However, it has previously been shown that specific silencing of mutant ataxin-3 by RNA interference with viral vectors is a promising therapeutic strategy for MJD. Nevertheless, reports of cytotoxic effects of this technology led to the safety profile of the previously tested lentiviral vectors encoding short hairpin (sh)RNAs (LV-shmutatx3) targeting mutant ataxin-3 upon brain injection being investigated. For this purpose, the vectors were injected in the mouse striata, and neuronal dysfunction, degeneration, gliosis, off-target effects, and saturation of the RNA interference machinery were evaluated. It was found that: (1) LV-shmutatx3 mediated stable and long-term expression of the shRNA in neurons of the mouse striatum; (2) neuronal dysfunction evaluated by darpp-32, NeuN, and cresyl violet staining, initially more pronounced, became indistinguishable from the phosphate-buffered saline group at 8 weeks and resolved within 20 weeks; (3) astrocytic activation was present, which resolved within 8 weeks; (4) microglial activity and proinflammatory cytokines release were present, which resolved and normalized within 20 weeks; and (5) there were no off-target effects or saturation of the endogenous RNA interference processing machinery in the mouse striatum. The data show that injection of lentiviral vectors encoding a shRNA targeting mutant ataxin-3 in the mouse brain induce transient dysfunctions, which resolve within 20 weeks. Importantly, long-term expression (up to 20 weeks post injection) of this shRNA (driven by H1 promoter) led to no toxic effect in vivo. This study thus constitutes an additional step in a future translation of gene silencing as a therapy for MJD.
Introduction
Machado–Joseph disease (MJD), or spinocerebellar ataxia type 3, is a neurodegenerative disease and the most common dominantly inherited cerebellar ataxia worldwide. 1 –4 Along with eight other conditions, they form the group of polyglutamine (polyQ) disorders, which share expanded CAG repeat mutations translating into polyQ tracts. 5,6 MJD is caused by ataxin-3 protein with an abnormal stretch of 54–84 consecutive glutamines (mutant ataxin-3) in opposition to normal ataxin-3 whose glutamine stretch ranges between 10 and 51 repetitions, and incomplete penetrance of the disease between 45 and 53 repeats. 7,8 The mutant polyQ expansion confers a toxic gain-of function to the ataxin-3 protein, leading to the formation of neuronal intranuclear inclusions (which are a hallmark of the disease), neuronal dysfunction, and degeneration. 9 Clinically, MJD symptoms comprise gait and limb ataxia, progressive postural instability, weight loss, and, in severe cases, premature death. 10 In terms of neuropathology, MJD includes severe neuronal loss in the spinal cord and in selective brain regions such as the dentate nuclei (cerebellum), pontine nuclei (brainstem), substantia nigra, and striatum. 11
Currently, there is no treatment able to change the progression of MJD (or of the other polyQ diseases), which culminates in death. However, several therapeutic strategies can be envisioned, and have already been tested in preclinical studies for MJD treatment. 12 –18 Despite promising results using these strategies, probably the most direct strategy to treat MJD would be the knockdown of the expression of the mutant gene using RNA interference (RNAi) technology. In fact, we previously demonstrated in different disease models that this therapeutic strategy could constitute an effective treatment for MJD. 19 –22 Using a lentiviral vector encoding a short hairpin (sh)RNA specifically targeting mutant ataxin-3, it was possible to reduce the MJD-related neuropathological and behavior abnormalities in different mouse models, even after the onset of the disease. 19 –21 The data showed a marked decrease in neuropathological abnormalities in the striatum and cerebellum upon mutant ataxin-3 silencing, but importantly it was also possible to rescue the ataxic phenotype in different MJD mouse models. 19 –21 The shRNA-mediated gene-silencing process was also successfully used in animal models of other neurodegenerative diseases: Huntington's disease, amyotrophic lateral sclerosis, and spinocerebellar ataxia type 1. 23 –27 Additionally, other strategies targeting RNA and using antisense oligonucleotides were also tested in several MJD models. Several studies explored the different functional mechanisms to promote exon-skipping or RNAse-H mediated degradation, which proved successful in reducing MJD-related abnormalities. 28 –31
Although RNAi technology is widely recognized to have potential as a therapeutic strategy, its efficiency has been questioned by the description of cytotoxic effects, culminating in cell dysfunction and in animal death. 32 This toxicity is associated with the RNAi effector molecules (shRNAs and siRNAs) and may be result of three essential aspects: induction of the innate immune response, 33,34 saturation of endogenous RNAi machinery, 32,35 and potential off-target effects. 36 –38 Considering these potential problems, this study investigated the safety profile of an shRNA targeting mutant ataxin-3, which had already been demonstrated to constitute an effective therapeutic strategy for MJD. Our data showed that lentiviral-mediated long-term expression of a shRNA (driven by H1 promoter) led to no toxic effect in vivo. This study constitutes an important step in the future translation of gene silencing as a therapy for MJD.
Methods
Animals
Adult males C57BL/6 wild-type mice (Charles River Laboratories), 8 weeks of age and weighing approximately 20 g, were used in this study. The animals were housed in a temperature-controlled room (22 ± 2°C) and maintained on a 12 h/12 h light/dark cycle. Food and water were available ad libitum. The experiments were carried out in accordance with the European Community Council directive (86/609/EEC) for the care and use of laboratory animals.
Lentiviral vectors
LV-shmutatx3, a lentiviral vector encoding a shRNA (Fig. 1A) targeting the human mutant ATAXIN-3 (shmutatx3), with LacZ reporter gene, 19 was produced in 293T cells with a four-plasmid system, as previously described. 39 The stocks were stored at −80°C until use.

LV-shmutatx3 mediates stable and long-term expression of the shmutatx3 in the mouse striatum.
Stereotaxic injection
Eight-week-old C57/BL6 male mice (Charles River Laboratories) were anesthetized by administration of a mixture of ketamine (100 mg/kg, Clorketam 1000; Vétaquinol) with xylazine (10 mg/kg, Rompun®; Bayer) by intraperitoneal injection. Next, each animal received a single injection of LV-shmutatx3 (1 μL, 400,000 ng p24/mL) or phosphate-buffered saline (PBS) into the striatum at the following coordinates from the bregma: caudal-rostral +0.6 mm, medial–lateral ±1.8 mm, and dorsal–ventral −3.3 mm. 12 The injections were performed with a 34G needle linked to a Hamilton syringe at a constant flow (0.25 μL/min), using an automatic injector (Stoelting Co.). The needle was left in place for an additional 5 min. Non-injected mice were used as additional controls in the study.
Tissue collection for immunohistochemical procedure/cresyl violet staining
A group of animals was sacrificed at 2, 8, and 20 weeks after the injection to obtain tissue samples for the immunohistochemical procedure/cresyl violet staining. A procedure used before was followed, in which the animals received a sodium pentobarbital overdose, 40 and afterwards were perfused through the left ventricle with 100 mL of 4% paraformaldehyde in PBS (pH 7.4) at 4°C for tissue fixation. The brains were removed, placed into the perfusion solution for 48 h at 4°C, and then placed into a cryoprotectant solution of 25% sucrose in PBS for 48 h at 4°C. After freezing at −80°C, 20 μm coronal sections of the striatum were obtained using a cryostat (Cryocut 1800; Leica Microsystems AG), collected for 48 multi-well plates with PBS containing 0.12 μmol/L sodium azide, and stored at 4°C until the immunohistochemical procedure/cresyl violet staining was performed.
Immunohistochemical procedure
Bright-field microscopy
For bright-field microscopy, 12 free-floating sections/animal, 160 μm distant from each other, were incubated in phenylhydrazine solution (1:1,000) in PBS (pH 7.4) for 30 min at 37°C to inhibit endogenous peroxidases. After three washes in PBS for 10 min each, the sections were incubated in blocking solution (PBS, 0.1% Triton X-100, 10% NGS) for 1 h at room temperature under moderate agitation, and then in blocking solution with anti-DARPP-32 primary antibody (1:1,000, rabbit polyclonal antibody, #AB10518; Millipore) or anti-NeuN primary antibody (1:1,000, rabbit polyclonal antibody, #AB78; Millipore) overnight at 4°C under moderate agitation. After three washes, the sections were incubated in blocking solution with biotinylated secondary antibody (1:200, anti-rabbit; Vector Laboratories) for 2 h at room temperature under moderate agitation. Further washes were performed, and then the sections were incubated with avidin-biotin-peroxidase solution (Vectastain ABC Elite Kit; Vector Laboratories) in PBS for 30 min at room temperature under moderate agitation. After washing, staining was performed with 3,3′-diaminobenzidine (DAB) solution (Peroxidase Substrate Kit DAB; Vector Laboratories) in Milli-Q water. Lastly, the sections were mounted on gelatin-coated slides, dehydrated with an ethanol gradient, passed through a xylene solution, and covered with Eukitt's mounting medium (Sigma–Aldrich). Images of the sections were acquired in a Zeiss Laser Microdissection PALM (Carl Zeiss MicroImaging).
Fluorescence microscopy
For fluorescence microscopy, 8 free-floating sections/animal, 160 μm distant from each other, were incubated in the same blocking solution mentioned above and then in blocking solution with anti-iba-1 primary antibody (1:1,000 rabbit polyclonal antibody, #019-19741; Wako) or anti-GFAP primary antibody (1:1,000 mouse monoclonal antibody, #3670; Cell Signaling Technology) under the same conditions. Double-staining was performed in the injected animals with anti-β-galactosidase primary antibody (1:1,000 mouse monoclonal antibody, #2372; Cell Signaling Technology) and anti-iba-1 or anti-GFAP. After washes, the sections were incubated in blocking solution with anti-rabbit secondary antibodies Alexa Fluor 488 or 594 and/or anti-mouse Alexa Fluor 488 or 594 (1:200, #A-11008, #A-11002, #A-11001, #A-11005; Life Technologies), according to the primary antibodies used under the same conditions in the dark. After further washes, the sections were incubated in DAPI solution (1:5,000; Sigma–Aldrich) in PBS for 10 min at room temperature under moderate agitation in the dark. After washing, the sections were mounted on gelatin-coated slides and covered with Mowiol mounting medium (Sigma–Aldrich). Images of the sections were acquired with a Zeiss Laser Microdissection PALM (Carl Zeiss MicroImaging).
Evaluation of DARPP-32 and NeuN depleted volume
The depleted volume of DARPP-32 and NeuN in the striatum after the injections was blindly assessed using the formula: V = d (a1 + a2 + a3 + …), where d is the distance between the serial sections (160 μm) and a1, a2, a3, (…) are DARPP-32 or NeuN depleted areas in each of the 12 sections analyzed. The DARPP-32 and NeuN depleted areas were determined using ImageJ (NIH) software. Volume was expressed in cubic millimeters. Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, Inc.), using unpaired Student's t-test and one-way analysis of variance (ANOVA).
Cresyl violet staining
Eight sections/animal were pre-mounted on gelatin-coated slides and then incubated in cresyl violet solution previously heated to 60°C for 10 min. After that, the sections were passed through Milli-Q water, dehydrated with an ethanol gradient, passed through a solution of xylene, and covered with Eukitt's mounting medium (Sigma–Aldrich). Images of the sections were acquired with a Zeiss Laser Microdissection PALM (Carl Zeiss MicroImaging).
Tissue samples for Western blot/quantitative reverse transcription polymerase chain reaction
Another group of animals was sacrificed in order to obtain tissue samples for Western blot/quantitative reverse transcription polymerase chain reaction (qRT-PCR) at 2 and 20 weeks after the injection. The brains were removed, and all of the striatal region in the right and left hemisphere was dissected by puncture with a Harris Uni-Core pen (2 mm diameter; Ted Pella, Inc.) on dry ice. Each striatum was then divided into two distinct samples: one for protein analysis (Western blot) and another for RNA analysis (qRT-PCR). Samples for Western blot were placed in microcentrifuge tubes with 200 μL of RIPA lysis solution (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing protease inhibitors (Roche) and homogenized by sonication for 5 s under ice cooling. The homogenates were centrifuged at 13,000 g for 15 min at 4°C and the supernatants frozen at −20°C. In turn, the qRT-PCR samples were frozen at −80°C in microcentrifuge tubes.
Western blot
Protein concentration was determined with the Bradford method (Bradford Protein Assay; Bio-Rad). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (4% stacking, 12% running) was performed using 40 mg of protein. Striatal samples from LV-shmutatx3-injected hemisphere, PBS-injected hemisphere, and non-injected mice were resolved on the gels. After protein electroblotting onto polyvinylidene fluoride membranes (GE Healthcare), the membranes were incubated in blocking solution (Tris-buffered saline [TBS], pH 7.6, 0.1% Tween 20, 5% milk) for 1 h at room temperature at moderate speed, and then in blocking solution with anti-GFAP primary antibody (1:1,000 mouse monoclonal antibody; Cell Signaling Technology) overnight at 4°C at moderate speed. After three washes in TBS-T (TBS, 0.1% Tween 20) for 10 min each, the membranes were incubated in blocking solution with secondary antibody conjugated to alkaline phosphatase (1:10,000, anti-mouse; Amersham Biosciences) for 2 h at room temperature at moderate speed. After further washes, membranes were detected with ECF™, and images were acquired in VersaDoc (Bio-Rad Laboratories). The membranes were subsequently washed and re-incubated with anti-β-actin primary antibody (1:5,000 mouse monoclonal antibody; Sigma–Aldrich), repeating the described process. The membranes with striatal samples from LV-shmutatx3-injected mice were further re-incubated with anti-β-galactosidase (1:1,000 mouse monoclonal antibody; Cell Signaling Technology). GFAP and β-actin (for normalization) densitometric analysis was performed with ImageJ software. Statistical analysis was carried out using GraphPad Prism version 6.0 (GraphPad Software, Inc.), using unpaired Student's t-test and one-way ANOVA.
Human neuroblastoma cells transduction
SH-SY5Y human neuroblastoma cells obtained from the American Type Culture Collection bank cultured in Dulbecco's modified Eagle's medium/F12 (Gibco) culture medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin (Gibco), and 1.2 g/L sodium bicarbonate were plated on MW12 plates (100,000 cells/well) and allowed to adhere overnight. Cells were then infected with the LV-shmutatx3 (100 ng p24/100,000 cells) for 24 h. Six to seven days later, cells were placed onto MW6 plates and were collected for qRT-PCR with 8 days of LV-shmutatx3 transduction. Cells were washed with PBS and immediately frozen at −80°C until RNA extraction. Frozen cells were slightly thawed at room temperature, and RNA extraction was performed with a NucleoSpin RNA Kit (Macherey-Nagel) using DNase digestion at the spin column, according to the manufacturer's recommendations. qRT-PCR was performed as described below.
qRT-PCR
Frozen striatum punches were slightly thawed at room temperature, and RNA extraction was performed with QIAzol® lysis reagent and an RNeasy® Mini Kit (Qiagen). Briefly, the RNA at the aqueous phase obtained with the QIAzol® lysis reagent was cleaned up with the RNeasy® Mini Kit using DNase digestion at the RNeasy® spin column, according to the manufacturer's recommendations. The RNA concentration and purity were determined with NanoDrop™ 2000 (Thermo Fisher Scientific) and stored at −80°C. cDNA synthesis was performed with iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories) from 1 μg of total RNA, according to the manufacturer's instructions. qRT-PCR was carried out with the SsoAdvanced™ SYBR® Green Supermix Kit (Bio-Rad Laboratories). The following primers were used: Ms Atxn3 (QT00161217), Ms Dicer1 (QT00114702), Ms Drosha (QT01061837), Ms Exportin-5 (QT00155309), Ms Hprt (QT00166768), Ms Il1b (QT01048355), Ms Il6 (QT00098875), Ms Taok1 (QT00292509), and Ms Tmem106b (QT00108430), all pre-designed (QuantiTect Primer Assays; Qiagen). For the human neuroblastoma cell line experiments, the primers used were: Hs HPRT (QT00059066, QuantiTect Primer Assays; Qiagen), Hs DICER 1, Hs DROSHA, Hs TMEM106B, Hs XPO5, and Hs TAOK1 (KiCqStart Pre-designed Primers; Sigma–Aldrich). Briefly, 2.5 μL of the cDNA obtained in the reverse transcription reaction diluted fourfold (for interleukin [IL]-1β and IL-6 quantification) or 10-fold (for the remaining genes) with DNase free deionized water was used. The qRT-PCR was performed as follows: one single cycle at 95°C for 30 s, followed by 45 cycles of two steps—first step of 5 s at 95°C, second step of 15 s at 60°C. The melting curve protocol started immediately after the qRT-PCR and consisted of 5 s heating at 65°C with a 0.5°C temperature increase in each step until 95°C was reached. The threshold cycle (Ct) values were generated automatically by the StepOne™ Software (Applied Biosystems). For each gene, and in each experiment, a standard curve was performed, and the qRT-PCR efficiency was determined by the software. The mRNA relative quantification with respect to control samples was determined by the Pfaffl method, taking into consideration the different amplification efficiencies of all genes in all experiments.
Bioinformatics tools to identify potential off-targeted transcripts
To investigate whether LV-shmutatx3 expression could lead to off-target effects, we analyzed endogenous transcripts with near-perfect complementarity to the shmutatx3-derived guide strand (mouse wild-type ataxin-3) and endogenous transcripts with sequences within the 3′-untranslated region that pair the microRNA-like seed region (nucleotides 2–8) of the shmutatx3-derived guide strand (Taok-1 and Tmem-106b). Briefly, the shmutatx3-derived guide strand or antisense sequence in DNA format, 5′-GATAGGTCCCG′CTGCTGCT-3′, was introduced using the siSPOTR online tool, selecting mouse for analysis. An extensive list of probable off-targeted genes, with sequences within the 3′-untranslated region that pair the microRNA-like seed region (nucleotides 2–8) of the shmutatx3-derived guide strand, rank-ordered by individual transcript probability of off-target score (TPOTS) was obtained. The first 20 genes were selected, and expression in the mouse striatum was analyzed using another bioinformatics tool—the Allen Brain Atlas. This tool provides one or more mouse striatum raw expression values for each gene, according to the number of experiments performed.
Results
LV-shmutatx3 mediates stable and long-term expression of the shRNA targeting mutant ataxin-3 in neurons of the mouse striatum
Previously, a target-specific shRNA was designed to silence human mutant ataxin-3 (shmutatx3), making use of a single nucleotide polymorphism (G987GG → C987GG) located at the 3′ end of the ataxin-3 gene immediately after the CAG repeat region 19 (Fig. 1A). It was shown that this shRNA delivered by a lentiviral vector (LV-shmutatx3) led to a marked decrease of the neuropathological abnormalities in the striatum and cerebellum and to alleviation of behavior deficits in MJD animal models. 19 –21
To examine the medium- to long-term safety of this therapeutic strategy, wild-type mice (C57BL/6) were injected bilaterally in the striatum with LV-shmutatx3 or PBS (as control) and sacrificed at 2, 8, and 20 weeks after the injection (Fig. 1B). Non-injected mice were used as additional controls. The LV-shmutatx3 contains a LacZ reporter gene, which allowed shmutatx3 expression, striatal distribution, and transduced cells to be tracked. The immunohistochemical staining for β-galactosidase revealed that LV-shmutatx3 mediated stable and long-term shmutatx3 expression in the mouse striatum (Fig. 1E, H, and K). Moreover, β-galactosidase-positive cells covered a large part of the striatum, being specific for neurons, as no co-localization with markers for astrocytes (GFAP) or microglia (iba-1) was detected (Fig. 1L–Q).
Injection of LV-shmutatx3 in the mouse brain parenchyma induces neuronal dysfunction comparable to PBS injection that resolves within 20 weeks
The expression of mutant ataxin-3 in the striatum leads to neuronal dysfunction, which can be quantified by the loss of neuronal markers, such as DARPP-32 or NeuN. 41 On the other hand, the study also showed that silencing of mutant ataxin-3 mitigates the striatal neuronal degeneration. 17
Thus, next the study investigated how LV-shmutatx3 long-term expression affects neuronal functionality and integrity. For that, immunohistochemical analyses were performed with DARPP-32 and NeuN (Fig. 2) and cresyl violet staining (Fig. 3). At 2 weeks post injection, a significantly larger region depleted of both markers was detected in the hemisphere expressing the LV-shmutatx3 compared to the PBS-injected hemisphere (Fig. 2B–C and L–M). Nevertheless, at 8 and 20 weeks after the injection, no difference in the neuronal loss was observed between the LV-shmutatx3-injected hemisphere and the control hemisphere injected with PBS (Fig. 2E and F, H and I, O and P, and R and S). In fact, the recovery at 8 weeks post injection in the neuronal integrity was far more pronounced in the hemisphere expressing the LV-shmutatx3 (91% recovery in NeuN loss at 8 weeks compared to 2 weeks post injection) compared to the PBS-injected hemisphere (79% recovery at 8 weeks compared to 2 weeks post injection), when evaluated by both DARPP-32 (Fig. 2J), and NeuN immunohistochemistry (Fig. 2T). The cresyl violet staining revealed the presence of rounded nuclei over time in both injected striatal hemispheres. At 20 weeks post injection, only a small scar of pyknotic nuclei was detected in both injected hemispheres (Fig. 3H and I), showing that there was a recovery since the time of the injection. Altogether, these data suggest that the surgical injection of LV-shmutatx3 or PBS in the mouse brain parenchyma leads to transient neuronal dysfunction characterized by a loss of DARPP-32/NeuN and the presence of pyknotic nuclei close to the LV injection site in the striatum, which resolves within 8 and 20 weeks, respectively.
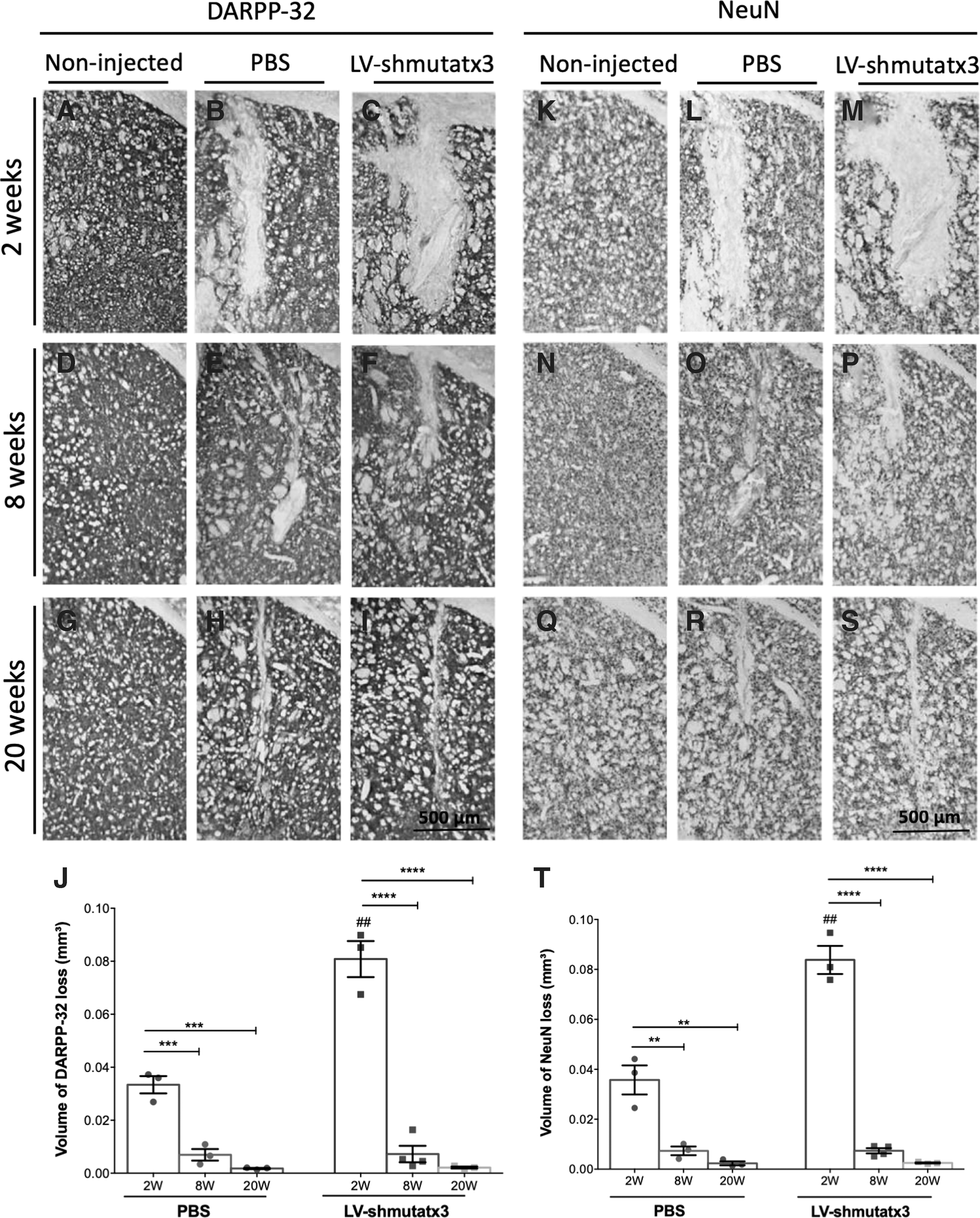
Long-term shmutatx3 expression preserves DARPP-32 and NeuN staining in the mouse striatum.
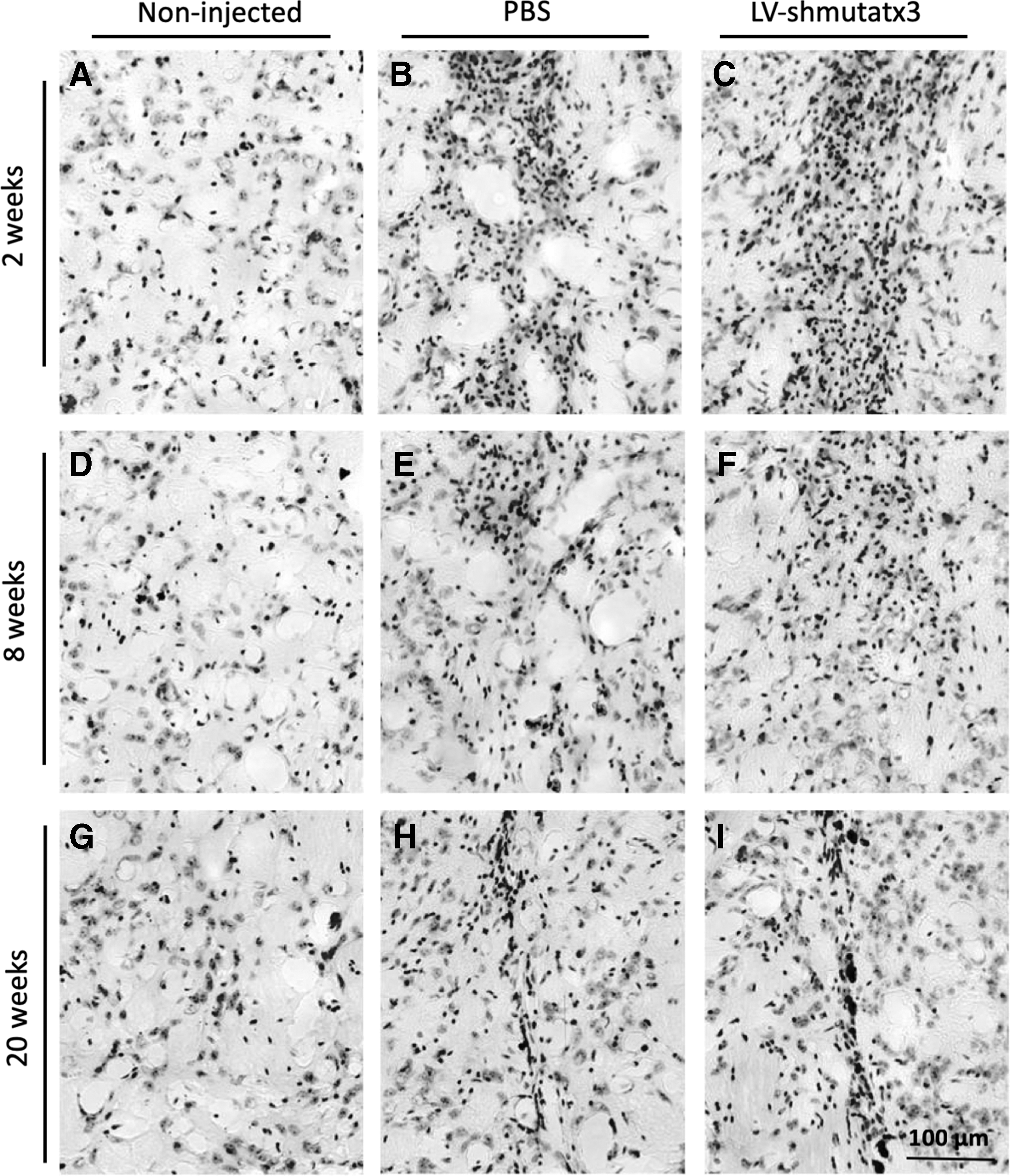
Long-term expression of the LV-shmutatx3 does not impair striatal neurons loss.
Astrocytic activation by injection of LV-shmutatx3 in the mouse striatum resolves within 8 weeks
To investigate the astrocytic response to the intracranial injection of LV-shmutatx3, immunohistochemistry and Western blot analysis of GFAP, an astrocytic marker, was performed. The GFAP labeling showed the presence of activated astrocytes over time in the striatum of both injected hemispheres. At 2 weeks post injection, local activation of astrocytes was detected in both injected hemispheres (Fig. 4B and C). At 8 weeks post injection, a minor difference in GFAP immunoreactivity was found between PBS- and LV-shmutatx3-injected hemispheres (Fig. 4E and F). At 20 weeks post injection, no astrocytic activation due to shmutatx3 expression was observed, and only a characteristic glial scar at the area corresponding to the needle track was observed in the hemisphere injected with LV-shmutatx3, as well as in the hemisphere injected with PBS (Fig. 4H and I). Consistently, Western blot analysis revealed similar GFAP levels at 8 and 20 weeks post injection between LV-shmutatx3-injected hemisphere and non-injected control mice (Fig. 4J–O). Altogether, these data indicate that LV-shmutatx3 long-term expression does not lead to an astrocytic response.
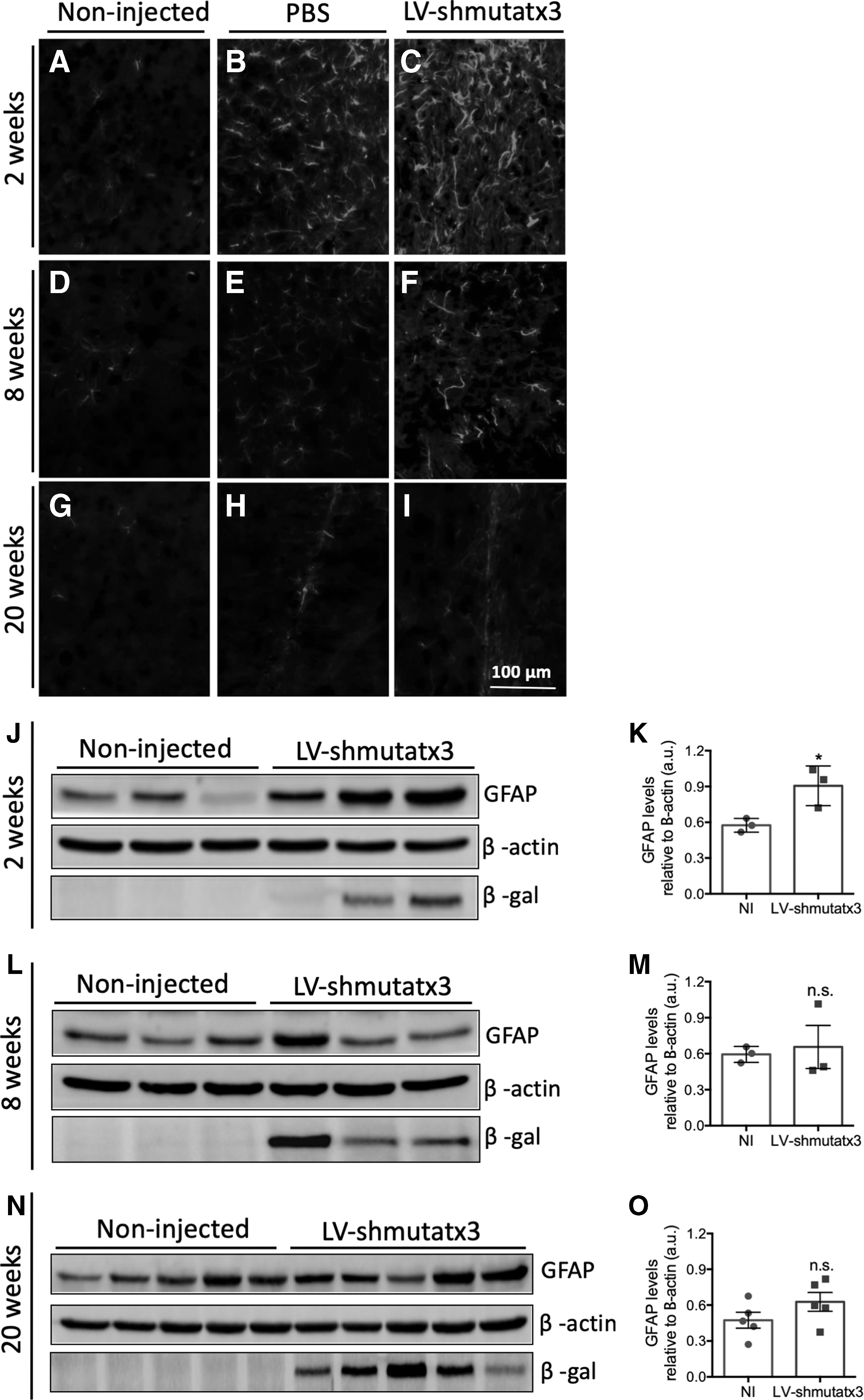
Long-term expression of the LV-shmutatx3 does not lead to sustained astrocytic activation in the mouse striatum.
Microglial activity and proinflammatory cytokines induced by injection of LV-shmutatx3 in the mouse striatum resolve and normalize within 20 weeks
Next, through immunohistochemical analysis of the microglial marker iba-1, the study evaluated whether the long-term expression of LV-shmutatx3 in the striatum would mediate activation of the resident immune cells. At 2 weeks post injection, higher iba-1 immunoreactivity was detected in the hemisphere injected with LV-shmutatx3 compared to the control injected with PBS (Fig. 5B and C). However, at 8 weeks post injection, the difference in iba-1 immunoreactivity was less pronounced between the injected hemispheres (Fig. 5E and F), while at 20 weeks post injection, no microglial activation was observed in LV-shmutatx3- or PBS-injected hemispheres (Fig. 5H and I). Next, the levels of two proinflammatory cytokines (IL-1β and IL-6) were also analyzed in the hemisphere injected with LV-shmutatx3 and compared to wild-type non-injected mice. At 2 weeks post injection, qRT-PCR analysis revealed increased IL-1β and IL-6 mRNA levels in the LV-shmutatx3-injected hemisphere compared to non-injected mice, although these levels were not statistically significant (Fig. 5J and K, left columns). Nevertheless, at 20 weeks post injection, no difference was found in IL-1β and IL-6 mRNA levels between both groups (Fig. 5J and K, right columns), indicating that the inflammation triggered by the injection procedure and the lentivirus was reverted.
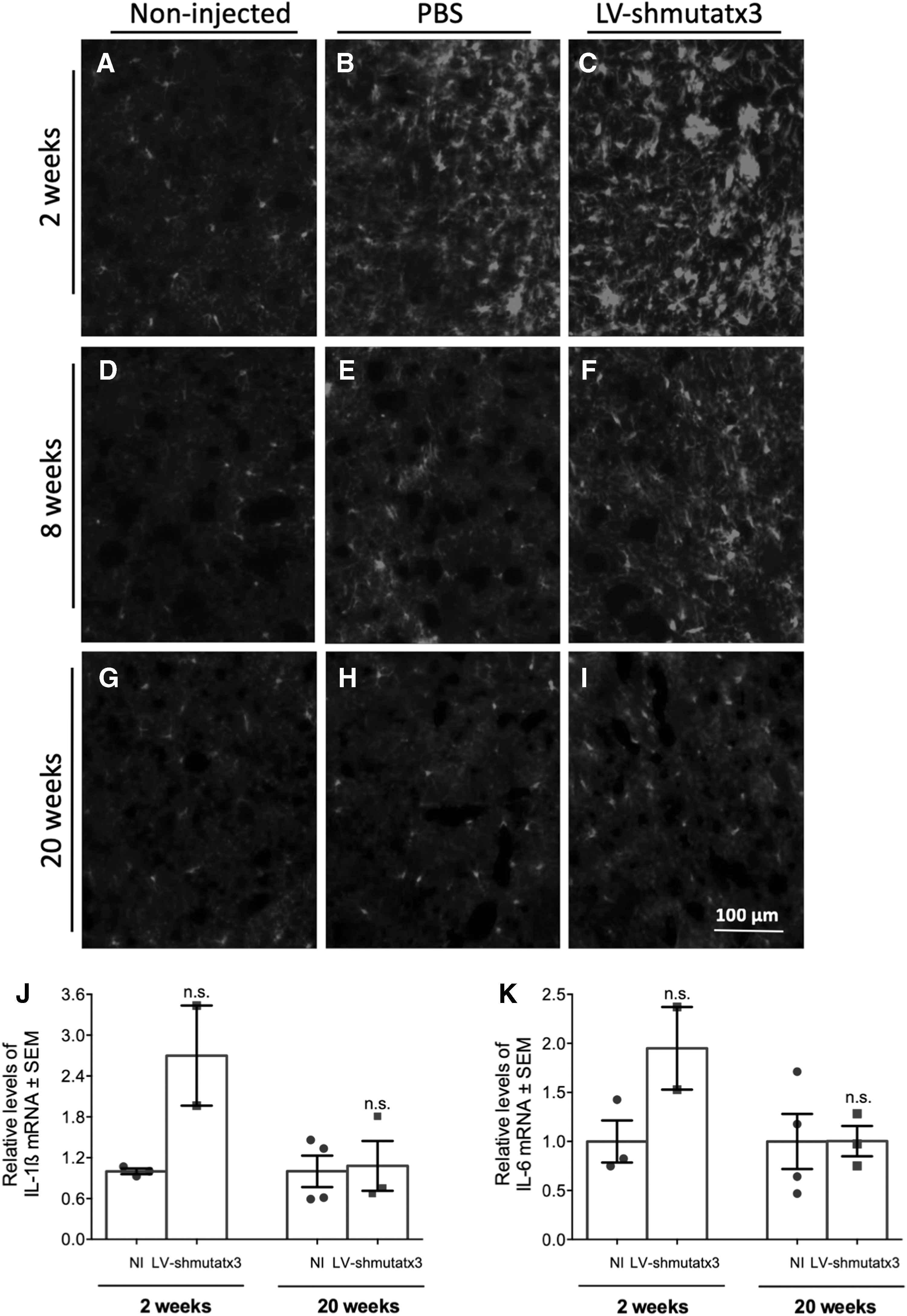
Long-term LV-shmutatx3 expression does not lead to microglial activation or sustained release of proinflammatory cytokines in the mouse striatum.
Altogether, these data indicate that LV-shmutatx3 long-term expression does not cause sustained microglial activation.
shmutatx3 expression does not cause off-target effects or saturation of the endogenous RNAi processing machinery in the mouse striatum or in human neuroblastoma cells
To investigate potential off-target effects of LV-shmutatx3 long-term expression, the study analyzed endogenous transcripts with near-perfect complementarity to the shmutatx3-derived guide strand (mouse wild-type ataxin-3) and endogenous transcripts with sequences within the 3′-untranslated region that pair the seed region (nucleotides 2–8) of the shmutatx3-derived guide strand (Taok-1 and Tmem-106b; Table 1). These later two targets were found using data from two different bioinformatics tools: siSPOTR and the Allen Brain Atlas. qRT-PCR analysis of mouse wild-type ataxin-3, Taok-1, and Tmem-106b showed no significant differences at 2 and 20 weeks post injection between hemisphere injected with LV-shmutatx3 and non-injected mice (Fig. 6A–C).

Long-term LV-shmutatx3 expression does not lead to off-target effects or saturation of the endogenous RNAi machinery.
Bioinformatics analysis to identify potential off-targeted transcripts with sequences within the 3′-untranslated region that pair the microRNA-like seed region (nuclotides 2–8) of the shmutatx3-derived guide strand
Transcripts with higher expression levels in the striatum that were later analyzed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in the samples.
To investigate whether LV-shmutatx3 expression could lead to saturation of the endogenous RNAi processing machinery, thereby disrupting biogenesis and function of miRNAs, Drosha, Dicer, and exportin-5 mRNA levels were analyzed by qRT-PCR. These levels did not significantly differ in LV-shmutatx3-injected hemisphere and non-injected mice at 2 and 20 weeks post injection (Fig. 6D–F).
Moreover, an additional evaluation was performed of the off-target effects of LV-shmutatx3 in the human neuroblastoma SH-SY5Y cell line. For that, SH-SY5 cells were transduced with LV-shmutatx3 for 2 weeks. Next, the mRNA levels of the targets evaluated before in the mouse striatum were analyzed (Fig. 7). In line with the results found in mice, no reduction was detected in wild-type ATAXIN-3, TAOK-1, and TMEM-106b levels in the cells transduced with the LV-shmutatx3 compared to the non-transduced control cells (Fig. 7A–C). Furthermore, no differences were found in RNAi pathway-related proteins between the two conditions (Fig. 7D–F). Altogether, these data show that shmutatx3 expression does not saturate RNAi machinery or lead to important off-target effects.
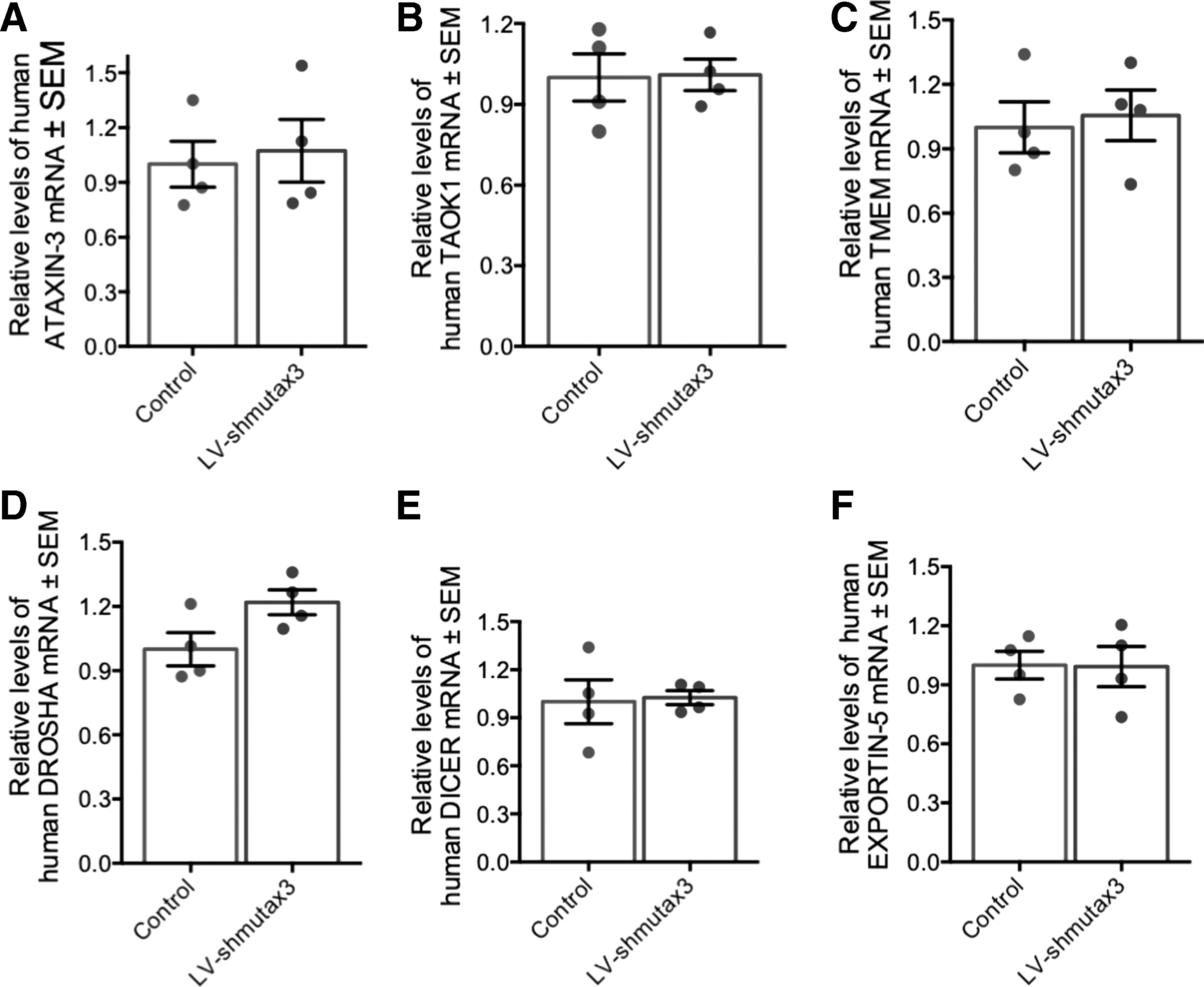
LV-shmutatx3 expression in the human neuroblastoma SH-SY5Y cell line does not lead to off-target effects or saturation of the endogenous RNAi machinery.
Discussion
This study demonstrated that shRNA long-term expression led to no sustained toxicity effects in vivo, thus constituting a safe therapeutic strategy for MJD and possibly other neurodegenerative disorders. While evidence of neuronal dysfunction and gliosis were present at initial time points, at 20 weeks post injection, no difference between the hemispheres expressing the shRNA targeting mutant ataxin-3 was detected comparing to controls injected with PBS or non-injected controls. In fact, no abnormalities were observed in neuronal integrity, immune response, and astrocytic response. Moreover, no relevant evidence of off-target effects and saturation of the endogenous RNAi processing machinery were observed.
RNAi has proven valuable as a therapeutic strategy to treat different polyglutamine diseases. 23 –27 In fact, the robust effect of shRNA-mediated silencing for MJD treatment was previously demonstrated. 19 –21 Nevertheless, previous studies have reported toxicity associated with the expression of shRNAs. 36,42 Boudreau et al. reported that local injection of shRNAs in the cerebellum (mediated by adeno-associated virus [AAV]) led to a higher toxicity when compared to artificial microRNAs. 42 In that study, the authors discussed the fact that shRNA toxicity could be related to the robust expression associated with the use of the U6 promoter, which drives higher expression compared to the H1 promoter used in this study. Moreover, in Boudreau et al.'s study, the effect of shRNA expression was only evaluated at 7 and 10 weeks after injection, whereas the present study provided data for longer-term expression, which could also contribute to higher neuronal recovery after the injection. On the other hand, Grimm et al. reported that shRNA expression in the liver (mediated by AAV) led to the death of mice due to an oversaturation of RNAi pathways. 32 When analyzing off-target effects or RNAi saturation 20 weeks post injection, no differences were detected between non-injected controls and animals expressing the shRNA in the present study. Moreover, the levels of critical components of the RNAi pathways were similar in the shRNA-injected animals at 2 and 20 weeks post injection. In line with these results, the present study also found that shmutatax3 expression did not lead to off-target effects or to the saturation of the RNAi machinery in a human neuroblastoma cell line. Moreover, the levels of human wild-type ataxin-3 were not altered, thus proving the specificity of the construction to the mutant form of the protein.
At 2 weeks post injection, some initial toxicity was observed in both injected hemispheres (shRNA and PBS), revealed by the loss of neuronal markers, astrogliosis, and microglia activation. Nevertheless, at 8 weeks post injection, the recovery was nearly complete, with almost no neuronal loss observed. Interestingly, the neuronal recovery was even more pronounced in the hemispheres injected with shRNA comparing to the hemispheres injected with PBS. The observed functional recovery of the striatal region after the lesion might be the result of neuronal sprouting, allowing the restoration of synaptic connections between neurons, 43,44 or the result of neurogenesis from the subventricular area, restoring the lesion area with neurons. 45,46 Both of these phenomena seem to be regulated by astrocytes and microglia. 47 –51 In fact, and interestingly, activation of both astrocytes and microglia was observed at 2 weeks post injection, which markedly decreased at 8 weeks post injection and was almost nonexistent at 20 weeks post injection.
At 20 weeks post injection, the expression of the shRNA was still robust. Nevertheless, no loss of neuronal markers or astrocyte and microglia activation was observed. Importantly, no difference was observed between the mRNA levels of RNAi pathway components, neither off-target effects between animals expressing the shRNA, and the non-injected mice.
Altogether, these data point to the long-term safety of this shRNA-based therapeutic strategy for MJD, which could be important for a future translational therapeutic strategy to human patients.
Footnotes
Acknowledgments
This work was supported by the European Regional Development Fund (ERDF), through the CENTRO 2020 Regional Operational Programme under project CENTRO-01-0145-FEDER-000008:BrainHealth 2020, through the COMPETE 2020—Operational Programme for Competitiveness and Internationalization and Portuguese national funds via FCT—Fundação para a Ciência e a Tecnologia, I.P., under projects POCI-01-0145-FEDER-016719 (PTDC/NEU-NMC/0084/2014), POCI-01-0145-FEDER-007440 (UID/NEU/04539/2013), and POCI-01-0145-FEDER-016390:CANCEL STEM, and through CENTRO 2020 and FCT under project CENTRO-01-0145-FEDER-022095:ViraVector; also by projects ESMI (JPCOFUND/0001/2015) and ModelPolyQ (JPCOFUND/0005/2015) under the EU Joint Program—Neurodegenerative Disease Research (JPND), the last two co-funded by the European Union H2020 program, GA No.643417 and national funds (FCT), by the French Muscular Dystrophy Association, the National Ataxia Foundation, the Richard Chin and Lily Lock Machado Joseph Disease Research Fund, and the American Portuguese Biomedical Research Fund.
Author Disclosure
The authors declare no conflicts of interest.