
Abstract
Chimeric antigen receptor (CAR) T cell immunotherapy for the treatment of cancer is now an approved treatment for B cell malignancies. However, the use of viral vectors to provide long-term CAR expression is associated with high production costs and cumbersome quality controls, impacting the final cost of CAR T cell therapies. Nonviral integrative vectors, such as Sleeping Beauty (SB) transposons, provide an alternative to modify primary T cells. Therefore, we developed a protocol to expand SB-transfected 19BBζ CAR T cells using a lymphoblastoid cell line, and evaluated T cell phenotype as well as function along the T cell expansion. Electroporation of PBMCs with transposon plasmid decreased cell viability on day 1 but had a minor impact on the frequency of memory subpopulations when compared to mock condition. CAR+ lymphocytes showed increased proliferation compared to mock control and high cytotoxic activity towards CD19+ cells without significant differences in exhaustion markers expression. Moreover, CAR+ lymphocytes showed an increased frequency by the end of the stimulation cycle compared with day 1, suggesting that CAR expression confers a selective proliferation advantage. Immunodeficient NOD scid gamma chain knockout (NSG) mice engrafted with the human pre-B leukemic cell line RS4;11 and treated with 19BBζ CAR T cells showed improved overall survival when compared to mock T cells treated animals. The results showed that electroporation using the SB system is a simple and affordable method for inducing long-term CAR expression in T lymphocytes. Expansion of gene-modified T cells with the lymphoblastoid cell line provided up to 2 cycles of stimulations, generating effective T cells against leukemia in vitro and in vivo.
Introduction
The use of chimeric antigen receptor (CAR) T cell immunotherapy for the treatment of cancer is now approved for commercialization targeting the B cell–restricted CD19 antigen. Patients in clinical trials on different B cell malignancies experienced overall response rates of 73%, with pediatric patients bearing B cell acute lymphoblastic leukemia (B-ALL) showing the best response rates (93%). 1 Despite their successful use in clinical trials, there is no standard protocol for the generation of CAR-modified T cells, with different genetic modification vectors and expansion protocols being used. Most of the published clinical studies used viral vectors derived from the Moloney leukemia virus (MLV) or the human immunodeficiency virus (HIV), which were shown to efficiently transduce activated T cells and induce high CAR expression. 2,3 These vectors integrate the transgene sequence in the T cell genome, providing long-term expression and persistent T cell activation that is crucial for the clinical response. 4
Large-scale clinical use of viral vectors requires a dedicated production facility operating under good manufacturing practice guidelines and with a biosafety level 2. These vectors have an integration profile favoring genome sites near active genes, carrying the risk of interfering with the transcriptional regulation of neighboring regions in some contexts, with preclinical 5 and clinical 6,7 data supporting caution with their use in patients, even if T cell transformation associated to MLV or HIV transgene expression has not yet been reported despite the large numbers of patients treated, 8 Moreover, the produced vector batches must be titrated and tested for replication competent retrovirus/lentivirus, which is labor-intensive and increase the final cost of the therapy.
Transposon systems consist of plasmid-based integrative vectors that, through a cut-and-paste mechanism catalyzed by a transposase, recognize inverted terminal repeats flanking the transgene and insert it into the target cell genome. 4 Different transposon-based systems were developed, with the Sleeping Beauty (SB) being one of the most used. The SB system was shown to be very efficient in the genetic modification of T cells 9 –11 and display a nearly random integration profile when compared with other transposons, retrovirus and lentivirus, increasing its potential safety. 12,13 Moreover, the use of square wave electroporators like Lonza's Nucleofector to deliver the plasmids induces high CAR expression, rendering the generation of CAR T cells using the SB system a much more straightforward approach in terms of regulatory and biosafety aspects and with a decreased cost when compared to viral vectors. 14 These characteristics prompted the use of this system for the generation of CAR-expressing T cells in preclinical settings 10,15,16 and its use in clinical trials, with encouraging results in the treatment of patients with non-Hodgkin lymphoma and B-ALL. 17
CAR-expressing T cells can be expanded in different bioreactors, with the WAVE and G-REX expansion systems being frequently used. 18 Alternatively, gene-modified T cells can be expanded with the use of artificial antigen presenting cells (APCs) in co-culture systems, like modified K562 cell lines 14 or with allogeneic Epstein-Barr virus (EBV) transformed lymphoblastoid cell lines (LCLs). 10 In fact, the use of EBV-LCLs for CAR T cell activation induces the expansion of T cell clones that also recognize viral antigens, potentially increasing their in vivo persistence due to physiological costimulation received from endogenous APCs presenting EBV-associated peptides. 19,20 Reports using preclinical models showed that memory phenotype and differentiation stage of infused T cells can alter the outcome of the therapy, 21 –23 providing a rationale for the selection and expansion of these optimal lymphocyte subsets. 24 However, it is currently unknown how different protocols for the generation and expansion of T cells modulate these parameters and the extension of the impact of such differences on the immune response.
In the present study, we show that electroporation for the gene transfer of a SB system can be used in combination with a third party LCL-based expansion protocol in order to generate T cells expressing high and sustained levels of CARs. Both CD4 and CD8 T cells could be expanded, including memory subsets, and these cells showed high effector activity in vitro upon completion of the expansion phase. In addition, these cells showed robust antitumor activity in vivo when inoculated in tumor-bearing NOD scid gamma chain knockout (NSG) mice. We herein describe the feasibility of this robust and easy to perform protocol for the generation of CAR + T cells along with the impact of the electroporation, CAR expression, and LCL based expansion on the final cell product obtained.
Methods
Plasmids
The original 19BBζ 25 construct was kindly provided by Dr Dario Campana (St. Jude Children's Research Hospital, Memphis, TN). CAR sequences were codon-optimized by gene synthesis in pUC-57 plasmids (Genscript) and cloned into the Sleeping beauty transposon system vector pT3 using AgeI and NotI restriction enzyme sites. A Myc Tag was added to CAR 19BBζ to allow CAR detection by flow cytometry. The SB100X 26 construct was kindly provided by Dr. Sang Wang Han (Federal University of São Paulo [UNIFESP], Brazil).
Cell lines and primary cells
Nalm-6, BJAB, K562, RS4;11, and the EBV-transformed human B lymphoblastoid cell line LAZ 388 (L38827) were cultivated in RPMI (Gibco, CA) supplemented with 10% fetal calf serum, 2 mM L-Glu and 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, MO) at 37°C, 5% CO2. RS4;11 cells were transduced with a lentiviral vector encoding GFP, and positive cells were selected by fluorescence-activated cell sorting (FACS). L388 cells were irradiated (100 Gy) before co-culture with peripheral blood mononuclear cells (PBMCs). White blood cells from healthy blood donors were collected using an RS leukocyte reduction filter (Haemonetics) and washed with phosphate buffered saline (PBS). To isolate peripheral blood mononuclear cells (PBMCs), a density gradient centrifugation was performed using Ficoll-Hypaque-1077 (slow acceleration/deceleration off; centrifugation for 20 min at 890 g) followed by three washes with PBS. The use of PBMCs from healthy donors was approved by an institutional review board (Brazilian National Cancer Institute [INCA] Ethics Committee) and donors signed review board–approved informed consent.
Natural killer cell depletion
PBMCs were counted and the protocol was followed as in the manufacturer instructions. Briefly, the cells were washed and resuspended in buffer (PBS, 0.5% bovine serum albumin, 2 mM EDTA). Human CD56 microbeads (Miltenyi Biotec) were added and incubated for 15 min at 4°C. Cells were washed and loaded in LS columns (Miltenyi Biotec). The flow-through containing the unlabeled cells was recovered and used as natural killer (NK)–cell depleted cells.
T cell electroporation and expansion
PBMCs were transferred to a sterile 0.2 cm cuvette (Mirus Biotech®, Madison, WI) and electroporated as previously described.
10
Briefly, 107 mononuclear cells were resuspended in 100 μL of 1SM buffer and electroporated with 20 μg of pT3 19BBζ and 1 μg of SB100x, using the U-14 program of Lonza® Nucleofector® II electroporation system. The mock condition was electroporated with 100 μL of 1SM buffer without plasmid. After transfection, cells were gently resuspended in 1 mL of prewarmed RPMI medium supplemented with 2 mM
Flow cytometry and antibodies
Accuri C6® or FACS Canto II (BD Bioscience) were used to perform CAR expression and cell phenotype analysis. Cells were harvested, resuspended in PBS at a concentration of 106 cells/mL, and incubated with the following antibodies: anti-CD3 PercP-Cy5.5, anti-CD4-APC or -PercP, anti-CD8-PE or -APC or -APC Cy7, anti-CD20-APC, anti-CD56-FITC or -PE, anti-CD62L-FITC, anti-CD80-FITC, anti-CD86-APC, HLA-ABC-PE, anti-HLA-DR-PE, anti-PD-1-APC, anti-PD-L1-biotin Streptavidin-APC, anti-CD197-PE, anti-LAG-3-PE, anti-TIM-3-Pe-Cy7 (eBioscience), anti-CD19-FITC (BD Pharmingen), anti-Myc tag Alexa Fluor 488 (Cell Signaling Technology or Thermo-Fisher), anti-Fab purified, goat anti-mouse Alexa Fluor 488, anti-HA Tag and goat anti-rabbit PE (eBioscience). 7AAD staining (eBioscience) was performed immediately before acquisition following manufacturer instructions. Data were analyzed using the FlowJo software v10.1 (Tree Star).
Chromium release assay
Nalm-6, K562, and L388 were used as target cells at different (E:T) ratios (as specified in Fig. 1). One million target cells were labeled with 7.4 MBq of 51Cr for 1.25 h at 37°C and washed three times with RPMI medium. The target cells (5,000/well) were incubated with effector cells at the specified ratios for 4 h at 37°C in triplicates. The cells were harvested, centrifuged, and the supernatant collected. After drying the supernatant in paper filters, a Gamma Counter 550B (Beckman Coulter) was used for emission quantification. Cell lysis was calculated using the formula [(test release − spontaneous release)/(maximum release spontaneous release)] × 100.
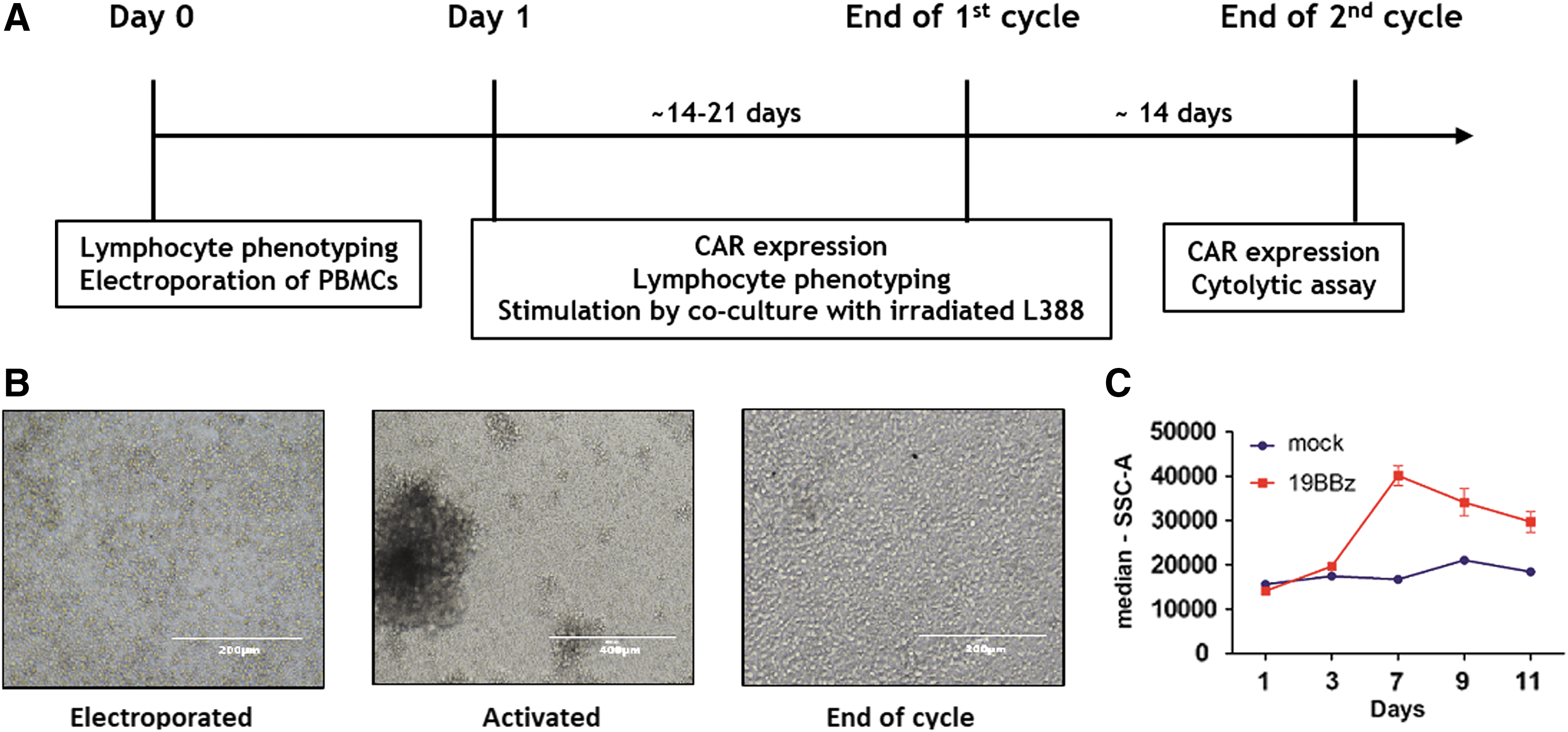
Schematic view of chimeric antigen receptor (CAR) + T cells expansion protocol.
Calcein-AM cytotoxicity assay
After 14 days (end of the first cycle) or 27 days (second cycle) of expansion of the CAR T lymphocytes, cell lytic activity was tested against RS4;11. Target cells were resuspended in complete medium with 15 μM calcein-AM (Thermo Fisher Scientific), previously diluted in dimethyl sulfoxide, for 30 min at 37°C. After two washes in complete medium, 5 × 103 target cells were placed with different ratios of effector: target cells, ranging from 50:1 to 0.75:1 in V-bottom 96-well plates in triplicates. The maximum release was done with medium plus 2% of Triton X-100, and the spontaneous release was evaluated by measuring the supernatant of target cells in medium. After 4 h of incubation at 37°C in 5% CO2, 75 μL of each supernatant was harvested and transferred into black plates and measured using SpectraMax® Gemini™ EM Microplate (excitation filter: 485 ± 9 nm; band-pass filter: 530 ± 9 nm) and the lysis was calculated using the formula [(test release − spontaneous release)/(maximum release × spontaneous release)] × 100. 28
Xenograft model
Eight- to twelve–week-old female NOD-scid IL2Rgammanull (NSG) mice obtained from Jackson laboratories were injected intravenously via tail vein (5x106 RS4;11 GFP+ tumor cells in 0.1 mL sterile PBS). After three days, mice were treated with 19BBζ CAR T cells or mock T cells (3 × 106 total cells) in 0.1 mL PBS. Animals in the vehicle control group were injected with 0.1 mL of PBS. The tumor burden was analyzed every 10 days, starting on day 20. Mice were immobilized and bled by facial vein using a 5 mm lancet (Goldenrod) and blood was collected in a tube with 0.5 M EDTA. The blood was processed with ammonium-chloride-potassium lysis buffer for 15 min, washed with PBS, and further analyzed by flow cytometry. Welfare and weight were monitored twice a week for the first three weeks and further monitored every day until death or euthanasia. The euthanasia time was determined by the loss of >15% of the weight or signs of cachexia. Weight loss, diarrhea, and skin changes were monitored as graft versus host disease (GVHD) symptoms. The CO2 chamber was used for the euthanasia procedure. After euthanasia, organs were macerated and analyzed by flow cytometry. All animal procedures were approved by the animal ethics committee of the Brazilian National Cancer Institute.
Statistical analysis
Results were expressed as means ± SEM. Statistical tests included one-way ANOVA, followed by Tukey's multiple comparison test. When applicable, paired two-tailed Student's t-tests were used. p-Values of 0.05 or less were considered to denote significance, and log-rank test was used to analyze the survival curve (*p < 0.05; **p < 0.01; ***p < 0.001; GraphPad Prism 5 or R).
Results
CAR T cells manufactured by a transposon system and expanded upon LCL-induced stimulation
We have engineered PBMCs from blood donors to express a second-generation CD19-specific CAR, called here 19BBζ. CAR transposon, and the SB100X transposase vectors were delivered by electroporation and cells were co-cultured with the irradiated L388 cell line the following day (Fig. 1). This cell line expresses receptors required for T-cell activation, such the costimulatory molecules CD80 and CD86, the B-cell lineage markers CD19 and CD20 (that can be engaged by CARs), as well as the immune checkpoint ligand PD-L1 and the major histocompatibility class 1 and 2 (Supplementary Fig. 1). We expanded CAR T cells for several days and a critical step here is determining when they return to resting state. Taking into account that T cells changes morphology and cell size upon activation (Figure 1B-C), we used these criteria to determine when to restimulate T cells with L388.
We monitored the subsets of lymphocytes before and after manufacturing to evaluate the effect of electroporation and also L388 stimulus on composition of expanded lymphocytes. Thus, three experimental conditions were included: cultured only (Neg), electroporated only (mock) and electroporated with plasmids encoding 19BBζ CAR and SB100x transposase (19BBζ). First, we have observed that electroporation alone decreased lymphocyte viability (mock condition), and the addition of 19BBζ CAR plasmid presented even greater toxicity (Fig. 2A). One day after electroporation, major lymphocyte populations were not altered amongst conditions. CD4+ and CD8+ memory subpopulations were not largely affected by 19BBζ plasmid electroporation. In CD4+ cells, a slight increase in CD4+CD62L−CCR7+ cells and a decrease in CD4+CD62L+CCR7− cells was observed when compared to other conditions. In CD8+ memory cells, electroporation of the 19BBζ plasmid led to an increase in CD8+CD62L−CCR7+ cells and a decrease in CD8+CD62L+CCR7− cells when compared with cultured condition.

Population of lymphocytes expanded after electroporation and L388-activation.
T cells were also phenotyped after the first cycle of expansion with the L388 cell line. As a human B lymphoblastoid cell line that naturally expresses CD19, this cell is able to activate 19BBζ positive CAR T cells, in line with the expansion of 20z CAR+ cells as previously described by our group. 10 Corroborating this result, CAR T cells expanded more than mock condition after LCL stimulus (Fig. 2B). By the end of the first expansion cycle, no major differences in lymphocyte subsets were observed between conditions, (n = 5). However, when comparing the first and the last day of the expansion cycle, significant changes were observed. As expected, L388 induced an increase in the frequency of CD3+ lymphocytes, and some donors showed expansion of NK cells (CD56+) in 19BBζ condition (Fig. 2E). Because CD62L and CCR7 are important molecules for T cell migration to lymph nodes, 29 we chose to analyze the expression pattern of these proteins by flow cytometry. In general, CD4+ and CD8+ subpopulations remained unaltered, with 19BBζ condition showing a similar frequency of CD62L+CCR7+ and CD62L+CCR7− cells when compared with mock condition. Altogether these results suggest that our electroporation protocol itself does not significantly affect the frequency of lymphocyte subpopulations, expanding and maintaining cells that express memory markers. Moreover, 19BBζ CAR expression minimally change the proportion of these subpopulations. As expected, L388 stimulation induces T cell enrichment, although NK cells still represented a significant proportion of the final product in a fraction of the donors.
NK cell depletion affects memory CD8 T cell populations after expansion
The optimal composition of the final therapeutic product (CD4/CD8 ratio, memory T cell subpopulations) is still a matter of debate, but as shown above, it is clear that the genetic modification and expansion protocols have an impact in these parameters. The protocol described in this paper uses an electroporation program established for T cells, which results in an extensive reduction in the number of monocytes (data not shown). Nonetheless, NK and natural killer T (NKT) cells are not excluded in this process. Given the varying degrees of NK and NKT cells expansion after L388 stimulation, which for some donors can reach up to 50% after the 1st cycle of stimulation (Fig. 2F), we sought to evaluate the impact of the presence of CD56+ cells in our expansion protocol. We therefore depleted CD56+ cells prior to L388 stimulation and evaluated the phenotype of expanded lymphocytes in our culture conditions. NK cell depletion did not affect viability on day 1 (Fig. 3A) or percentage of 19BBζ+ cells on day 1 and at the end of the first stimulation cycle (Fig. 3B). However, NK cell removal did impact the lymphocyte distribution after one cycle of L388 stimulation, with most of the cells being CD3+ (Fig. 3C); NK (CD3−CD56+) and NKT (CD3+CD56+) cells (Fig. 3D and E) were present in low levels. Importantly, CD4/CD8 ratio was not affected by NK removal. Regarding the expression of memory markers, NK cell depletion did not change significantly the frequency of these subpopulations in CD4 cells (Fig. 3G–J). However, in CD8 cells, a decrease in CD62L−CCR7− (Fig. 3K) and CD62L+CCR7− (Fig. 3M) cells was observed, while an increase was seen for CD62L−CCR7+ and CD62L+CCR7+ cells (Fig. 3L and N). These results show that NK cell depletion before L388 stimulation change the composition of CD8 subpopulations, inducing the expansion of CCR7+ cells which are associated with a central memory phenotype.

Impact of NK cell depletion on lymphocyte populations after one cycle expansion.
SB transposon system promotes stable and long-term CAR expression
At the end of each stimulation cycle, we characterized the T cell phenotype and evaluated effector function by performing cell lysis assays. As shown for a representative donor (Fig. 4A), CAR+ cells enrich in culture over time, suggesting that 19BBζ confers a selective proliferative advantage to these T lymphocytes under L388 stimulation. For this particular donor, despite reaching similar absolute T cell numbers after two stimulation cycles (Fig. 4B), 19BBζ cells showed a higher fold increase in T cell numbers compared to control cells (Fig. 4C). Importantly, 19BBζ showed high cytotoxic activity against CD19+ target cells (L388 and Nalm-6) but not control CD19− K562 myeloid cells. Mock cells showed similar cytotoxic activity towards L388 when compared with 19BBζ cells, suggesting that stimulation with third party EBV+ L388 expands virus-specific T cells. These results were confirmed in five additional donors and the results were compiled in Fig. 4G–J. The use of SB transposon/transposase system yielded up to 20% of 19BBζ+ cells at day 1 after electroporation and 60% at the end of the first cycle (Fig. 4G). Importantly, CAR T cell enrichment observed after L388 stimulation is greater than in anti-CD3/anti-CD28 beads expanded cells from the same donors (Supplementary Fig. S2). The poor viability of lymphocytes right after the electroporation protocol is largely compensated by a greater expansion capacity of 19BBζ+ cells upon stimulation with L388 (Fig. 4H–I). At the end of the second cycle of expansion, cell lysis assays were performed with four different cell lines as targets: CD19− (K562) and CD19+ (L388, BJAB and Nalm-6). As shown in Fig. 4J, lysis of the CD19− K562 cell line by 19BBζ lymphocytes was only marginal, while L388, Nalm-6, and BJAB cell lines were efficiently lysed reaching more than 80% of cell killing. The high cytotoxic activity against L388 cells in both CAR+ and CAR− cells indicates that the expansion protocol leads to the expansion of EBV-specific T cell clones (Fig. 4J).

19BBζ CAR+ cells proliferate and kill target cells in vitro.
We also evaluated the exhaustion markers PD-1, LAG-3, and TIM-3 in CAR + T 19BBζ cells 24 h after electroporation and at the end of the first expansion cycle (11–14 days of culture). The CAR T (CD4+ or CD8+) cells or mock controls acquired exhaustion receptor expression during expansion, but there was no statistical significance comparing day 1 and the end of the first cycle (Supplementary Figure 3A). No significant difference was observed between mock or 19BBζ populations at day 1, but at the end of the expansion protocol there is less of the triple negative population (CD8+LAG-3−PD-1−TIM-3−) in 19BBζ CAR T cells. Based on functional assays, our results indicate that L388 based expansion leads to fully functional effector T cells, with CAR + T cells retaining both L388 and CD19 specific lytic activities in vitro (Fig. 4).
LCL-expanded CAR T cells kill leukemia in vivo
Lastly, we sought to evaluate the in vivo antitumor activity of 19BBζ expanded by our L388−based protocol. NSG immunodeficient mice were inoculated with 5 × 106 RS4;11 GFP+ leukemia cells, and CAR + T cells were infused 3 days later. For this experiment, one group received only saline (PBS) and the two other groups received 3 × 106 cells (mock or 19BBζ CAR T cells). The 19BBζ CAR T group received the cells containing 56,5% of CD4+CAR+, and 45.1% of CD8+CAR+. The cells were expanded for 11 days and infused in mice at the end of the 1st cycle (Figure 5A). Expanded cells showed a balanced proportion of CD4 and CD8 cells, with an enrichment in 19BBζ+ cells over time as seen for other donors (Fig. 5B). Exhaustion markers were evaluated in the expanded cells before infusion, with 19BBζ cells expressing higher levels of PD-1, LAG-3, and TIM-3 compared to mock cells (Supplementary Fig. S3B). CAR T cells were able to effectively eliminate leukemia cells in vitro at the end of the first and second cycles compared to mock control (Fig. 5C and D). We also detected GFP+ leukemic cells in the blood of the PBS-treated group as soon as day 30, but not in the case of the mock or CAR T cell-treated groups (Fig. 5E). No animal showed signal of GVHD or weight loss until day 40 (Supplementary Fig. S4). Interestingly, PBS-treated mice died 45 days after tumor inoculation, while mock-treated animals started to die without detectable levels of leukemia cells in the blood (Fig. 5E and G). PBS and mock-treated animals presented leukemia cells in the brain, but not the CAR T-treated group (Fig. 5F). No tumor cells were found in spleens, bone marrow, lungs, and livers of CAR T-treated animals (data not shown). Moreover, mice treated with 19BBζ CAR T cells showed an improved overall survival when compared to mock and PBS groups, demonstrating that 19BBζ lymphocytes expanded under L388 stimulation are capable of high antitumor activity in vivo (Fig. 5G). To test the long-term persistence of CAR T cells in vivo, we re-challenged the surviving animals with 5 × 106 RS4;11 GFP+ cells at day 125 after primary tumor inoculation. Notably, in these mice previously treated with CAR T cells, no signs of tumor or circulating GFP+ cells could be observed up to day 40 post challenge (n = 4) (data not shown).
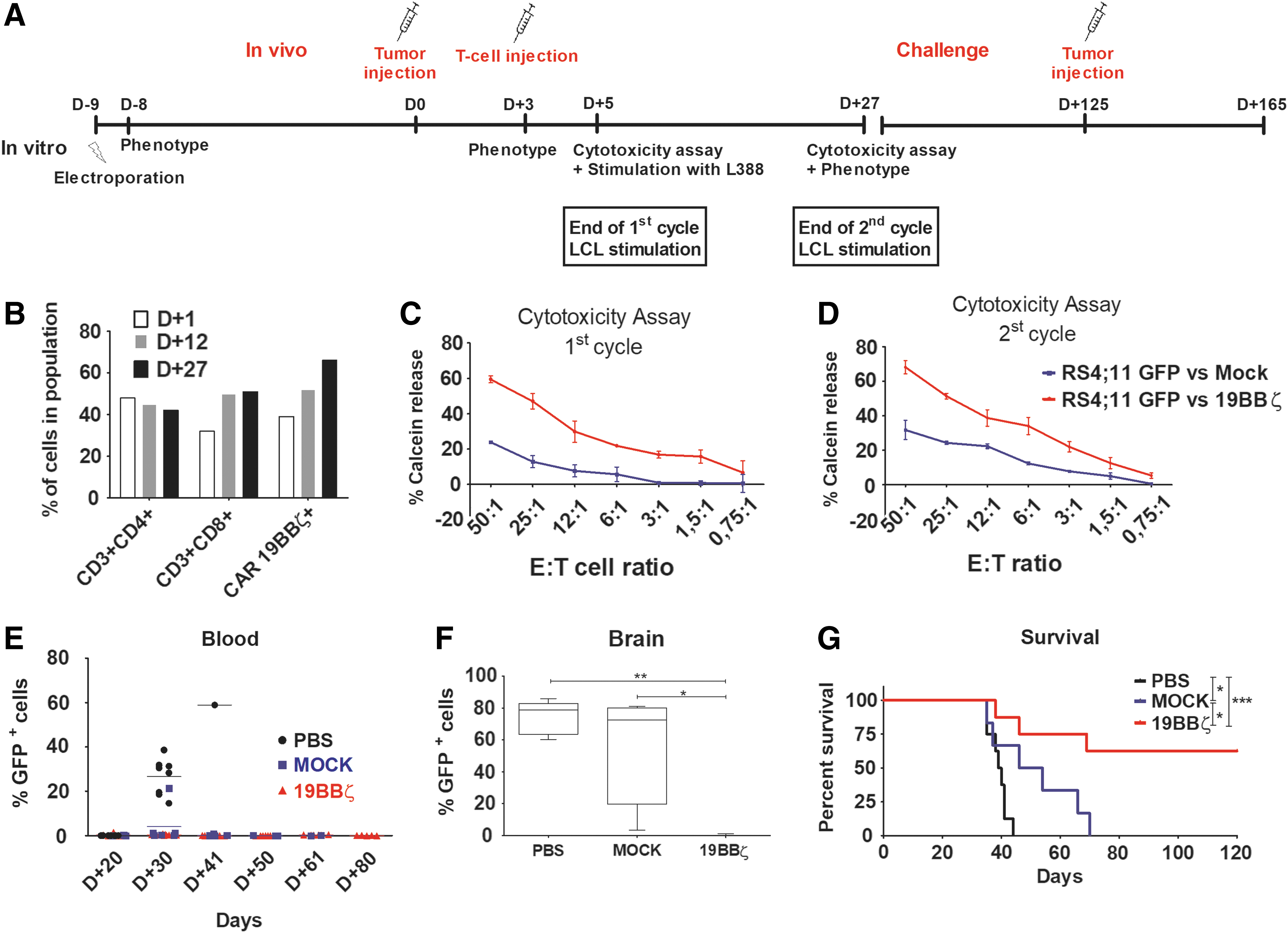
19BBζ CAR T cells protected mice from acute leukemia and confers long-term protection.
Discussion
Clinical trial data have shown that CAR T cell therapy can induce a high response rate in patients with CD19+ B cell malignancies, especially for patients with B-ALL (as reviewed elsewhere 30 –32 ). This has culminated in the recent approval by U.S. Food and Drug Administration of anti-CD19 CAR T therapy for treatment of non-Hodgkin lymphomas (axicabtegene ciloleucel, Gilead/Kite and tisangenlecleucel, Novartis) and pediatric B-ALL (only tisangenlecleucel). Projected demand for these therapies in the United States alone is on the order of 7,000 patients per year, with typical cell dose ranging from 5 × 106 to 5 × 107 CAR+ cells/kg. 33 Meeting this demand and performing all the quality controls associated with current CAR T cell production protocols involving viral vectors configure itself a challenge, particularly when failed expansions are taken into account. New approaches are being currently proposed to expand CAR-based therapy applications 34 , implicating in increased pressure for the development of alternative robust protocols based on fast, efficient, and cost-effective strategies for CAR-based T cell immunotherapies.
The results reported herein and elsewhere in the literature show that the SB transposon system is a promising alternative to retro/lentiviral vectors regarding the genetic modification of T cells. 4,14,35 SB vectors integrate into TA dinucleotides sequences with close to random pattern, promoting a safer integration profile when compared to retrovirus or lentivirus. 12,36,37 Moreover, the high costs associated with the good manufacturing practice production of viral vector batches, which are in the $300,000.00 to $500,000.00 range, together with cumbersome T cell transduction and expansion protocols, have a profound impact on the final cost of the therapy. 38 Our results show that long-term CAR expression can be achieved upon the use of the SB transposon system, which is only comprised of two plasmids and is delivered by a straightforward electroporation protocol.
The use of L388 for T cell expansion was shown to be very efficient, being able to achieve the required cell numbers for in vivo experiments and theoretically for clinical use. This might be associated with high levels of CD86, a potent costimulatory ligand, expressed by this cell line (Supplementary Fig. S1). Another important aspect is the high expression of CD19, which stimulates 19BBζ+ cells and induces the enrichment of these cells over time. Importantly, memory T cell subpopulations were present at high frequency by the end of the 1st stimulation cycle both in the CD4+ and CD8+ subsets, which show that stimulation with L388 is a suitable method for the expansion of memory cells. Furthermore, both mock electroporated and 19BBζ lymphocytes showed high cytotoxic activity towards L388, suggesting that stimulation with L388 induces the expansion of EBV-specific T cell clones. Thus, the use of CAR T cells expanded with our protocol might be useful to prevent post-transplant EBV reactivations. EBV reactivations and EBV-associated post-transplant proliferative diseases represent major risks for EBV positive patients in the post-hematopoietic cell precursor therapy setting 39 and CAR+ anti-CD19/anti-EBV dual function cells can be of great therapeutic value. The expansion of T cells with EBV transformed cell lines leads to the marked narrowing of the TCR repertoire. 40,41 This repertoire skewing leads to limited alloreactivity, reducing the risk of graft versus host disease. 42 In fact, anti-CMV or -EBV T cells are currently being used as third-party product in the non-HLA matched setting with no signs of GVHD, 43,44 reinforcing the concept that our protocol could be potentially applied to both HLA matched and unmatched individuals, although this was not formally explored in the current manuscript. It is noteworthy that we observed no signs of GVHD in NSG animals treated with the LCL expanded T cells in our experiments.
We observed that NK depletion prior to T cell stimulation impacted CD8+ T cell subpopulations. Direct or indirect crosstalk between NK and T cells early during and after T cell activation can affect their differentiation, polarization, and survival. During the initial priming phase, T cell regulation by NK cells is mainly occurring in an indirect manner via modulation of APCs (a phenomenon that we think may occur in our protocol due to antigen presentation by the L388 cell line). Furthermore, the high IFN-γ production by NK cells directs the CD4+ T cells toward a Th1 polarization. There's little evidence about the impact of NK cells in memory phenotype of T cells, but the evidence shows that NK cell did not affect the establishment of T cell memory subpopulations. Hence, establishment of T cell memory occurs both in presence and absence of NK cells during the priming phase, although higher numbers of memory CD8+ cells were observed in mice with NK cells. 45 These findings corroborate our results regarding the very little impact of NK presence in CD4 memory phenotype. On the other hand, for CAR+ CD8+ T cells, there was a dramatic change on memory cell phenotype when NK cells were depleted before T cell priming, enriching the central memory phenotype compartment.
Finally, 19BBζ lymphocytes expanded using L388 were highly efficient in vivo, with mice showing improved overall survival when compared to mock electroporated and PBS groups. Lymphocytes expressing 19BBζ were able to eliminate leukemia cells in different organs, with a marked reduction of tumor cells in the brain. The dose of T lymphocytes used for the xenograft treatment is in the range of the dosing currently applied in clinical trials 46 and in line with the transitions from preclinical to clinical data applied by several research groups.
Overall, our results show that the protocol herein described is capable of generating and expanding CAR+ cells for preclinical testing. Upon creation of a master cell bank, EBV-transformed cells (such as the L388-based protocol described here) have the potential to be adapted to fully closed electroporation and expansion systems, like Lonza's Nucleofector 4D LV and G-REX culture flasks, further improving its efficiency and reducing labor-intense cell culture protocol associated costs. The LCL based protocol could represent an alternative to strategies based on artificial APCs, 14 with the advantage of additionally generating anti-virus responses, an important feature for some patients. Adoption of automated, all-in-one closed systems, like CliniMACS Prodigy or other solutions under development, may further improve scaling up and reproducibility and will be crucial for large-scale application of CAR T cell therapy.
Footnotes
Acknowledgments
We would like to thank the Brazilian National Cancer Institute (INCA) Blood Bank and Hemotherapy Service for sample preparation and cell irradiation. Funding for this study was provided by the Brazilian Ministry of Health, Brazilian National Cancer Institute (INCA), CNPq, CAPES, INCT Synthetic Biology (465603/2014-9), Ary Frauzino Cancer Foundation (FAF), Oncobiology Program - UFRJ, FAPESP (2014/04412-4).
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.