
Abstract
Chimeric antigen receptor–modified T cells (CAR-T cells) have emerged as a promising cancer immunotherapy for solid tumors. Epithelial cell adhesion molecule (EpCAM) is overexpressed in a variety of tumors and is recognized as a biomarker for circulating tumor cells and cancer stem cells, representing an attractive target for adoptive T-cell immunotherapy. This study generated third-generation CAR-T cells with redirected specificity to EpCAM (EpCAM CAR-T) by lentiviral vector. The study demonstrated that EpCAM CAR-T cells can elicit lytic cytotoxicity to target cells in an EpCAM-dependent manner and secrete cytotoxic cytokines, including interferon gamma and tumor necrosis factor alpha. Furthermore, adoptive transfer of EpCAM CAR-T cells significantly delayed tumor growth and formation in xenograft models. In addition, the safety evaluation showed that CAR-T cells have no systemic toxicity in mice. The data confirmed the antitumor ability and safety of CAR-T cells targeting EpCAM and may provide a new target for CAR-T cell therapies in treating solid tumors.
Introduction
Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein originally discovered on colon carcinomas. 1 Subsequent research showed it to be overexpressed to varying degrees in most human carcinomas. 2 It has been shown that EpCAM abrogates E-cadherin-mediated cell–cell adhesion, thereby loosening cell–cell adhesion, promoting cell motility, proliferation, carcinogenesis, and metastasis formation. 3 Numerous studies have reported that the overexpression of EpCAM is associated with decreased overall survival of patients with many tumors. 4 –7
Recently, EpCAM was identified as a biomarker for circulating tumor cells (CTCs) and cancer stem cells (CSCs). CTCs are the potential precursors of tumor metastasis that actively invaded or have been shed from the primary tumor into the blood circulation. 8 CTC capture using EpCAM-based gating is feasible for most cancer subtypes, especially for breast cancer. 9 –12 In addition, CSCs, which maintain cancer tissues by sustaining phenotypically diverse cancer cells, are considered as pivotal target for the eradication of cancer. EpCAM is expressed on CSCs from breast, colon, pancreas, and prostate tumors. 13 –16 CSCs have a high level of resistance to chemotherapy and radiotherapy, 17 which makes EpCAM an even more interesting target for cancer immunotherapy.
EpCAM has been targeted with monoclonal antibodies (mAb) in treating various cancers. Edrecolomab was the first EpCAM-directed mAb approved in Germany for the adjuvant treatment of colorectal cancer. 18 However, the results of subsequent clinical trials for edrecolomab were invalid, and it was subsequently withdrawn from the market. 19,20 Recent advances in potentiating the antitumor effects of anti-EpCAM mAb rely on creating multifunctional antibodies, including bispecific antibodies and trifunctional antibodies capable of simultaneous binding to different targets on tumor or immune cells. Recently, a trifunctional mAb, Catumaxomab, has been approved by the European Medicines Agency (EMA) for malignant ascites in patients with EpCAM+ carcinomas. Clinical trials using anti-EpCAM mAb have met variable success in breast, colon, pancreas, prostate, and ovarian carcinoma. 20 –24 This discrepancy may be attributed to EpCAM variance in expression density and low affinity to the target antigen.
Chimeric antigen receptor–modified T cells (CAR-T cells) have recently formed a part of a broad wave of immunotherapies that are showing promise in early-phase cancer clinical trials. These CAR-T cells modified with a recombinant receptor molecule recognize cell-surface antigens directly and are independent of major histocompatibility complex (MHC) restrictions. Recent reports on the impressive efficacy of CAR-T cells against hematologic malignancies have inspired oncologists to extend their efforts for the treatment of solid tumors.
This study constructed a third-generation CAR recognizing EpCAM and transduced T cells by the lentiviral vector to redirect T cells with specificity to EpCAM (EpCAM CAR-T). Then, it evaluated whether the EpCAM CAR-T cells could inhibit the growth of solid tumors in vitro and in vivo and the safety of EpCAM CAR-T cells in xenograft mice. Finally, EpCAM CAR-T cells were shown to elicit lytic cytotoxicity to target cells in an EpCAM-dependent manner, and adoptive transfer of EpCAM CAR-T cells significantly delayed tumor growth and formation against human colon cancers, without signs of severe adverse effects. This preclinical study of EpCAM CAR-T suggests that adoptive transfer of CAR-T cells targeting EpCAM is safe and efficacious and is a promising therapeutic strategy for treating EpCAM+ solid tumors.
Methods
Cell lines
The human cervical cancer cell line Hela, human non–small cell lung cancer (NSCLC) cell line A549, human breast cancer cell line MDA-MB-231, human colon cancer cell lines HT29 and SW480, and lentivirus packaging cell line HEK 293TD were obtained from the American Type Culture Collection (Manassas, VA). All the cell lines were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum.
Human peripheral blood mononuclear cells (PBMCs) were isolated from voluntary donors with their consent. All the experiments were approved by the Ethics Committee of the State Key Laboratory of Biotherapy. After centrifugation on Ficoll-Hypaque density gradients (Sigma–Aldrich, St. Louis, MO), PBMCs were cultured in X-VIVO 15 medium (Lonza, Basel, Switzerland) supplemented with 2.5% human AB serum, 100 IU/mL recombinant human interleukin-2 (rhIL-2), and stimulated with anti-CD3 and anti-CD28 magnetic beads (Invitrogen, Carlsbad, CA).
CAR construction
The anti-EpCAM single-chain variable fragment sequence (scFv) was screened by the authors' laboratory in the early stages. The hinge region of CD8α, transmembrane domain of the CD8α, and intracellular region CD28, 4-1BB and CD3ζ, simply called 8a28BBZ, were directly synthesized by GenScript. The sequences of scFv and 8a28BBZ were spliced using overlapping polymerase chain reaction (PCR) to form the EpCAM-specific CAR. Finally, the EpCAM-specific CAR was inserted into a pWPXLD (Addgene, Cambridge, MA) lentiviral vector using BamHI and EcoRI enzymes. The whole sequence was confirmed by direct sequencing.
Lentivirus production
Lentiviruses were produced in HEK-293 TD cells using the calcium phosphate method. The pWPXLD-based plasmid and the packaging plasmids psPAX2 and PMD2.0G (Invitrogen) were used at a ratio of 2:2:1. The supernatants containing the lentivirus particles were harvested 48 and 72 h post transfection, filtered, and concentrated through ultracentrifugation at 70,000 g for 120 min. Mock lentivirus was produced using an empty pWPXLD lentiviral plasmid. The concentrated lentivirus titers were determined by Lentiviral p24 enzyme-linked immunosorbent assay (ELISA; Clontech, Mountain View, CA) or quantitative reverse transcription PCR. Finally, the concentrated lentivirus was stored at −80°C. The average ranges of the titers for the concentrated lentivirus were about 108–9 IFU/mL.
Lentivirus transduction
The isolated PBMCs described above were activated for 1 day to stimulate T cells selectively. The non-tissue culture six-well plates were coated with 1 mL RetroNectin (50 μg/mL) in phosphate-buffered saline (PBS) overnight at 4°C. The next day, the RetroNectin solution was aspirated, and the wells were blocked with 1 mL PBS solution +2% bovine serum albumin for 30 min at room temperature. The blocking solution was then aspirated, and the wells were washed twice with PBS solution. The concentrated pWPXLD-CAR-encoding lentivirus or mock lentivirus was diluted and added to each RetroNectin-coated well. The six-well plates were centrifuged at 1,000 g for 2 h at 32°C. After centrifugation, activated T cells were added to each well and incubated at 37°C with 5% CO2 for two consecutive days. Forty-eight hours later, T cells were replaced with new medium, cultured, and expanded for 1–2 weeks before analysis.
Flow cytometry
The expression levels of EpCAM on tumor cell lines were examined using flow cytometry. Briefly, 10 μL phycoerythrin (PE)-conjugated anti-human EpCAM antibody (BioLegend, San Diego, CA) was added to 1 × 106 cells suspended in 100 μL PBS. After 30 min of staining at 4°C, cells were washed three times with PBS and detected using a flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA). For analysis of CAR expression, the modified T cells were stained with PE-conjugated anti-F(ab)2 (Jackson ImmunoResearch, West Grove, PA). The phenotype of CAR-T cells was evaluated by staining with anti-CD3, anti-CD8, anti-CD4, anti-CD45RO, and anti-CD62L antibodies (BioLegend). Flow cytometry data were acquired with a FACSCalibur flow cytometer (BD Biosciences) and analyzed with FlowJo v7.6.
Cytotoxicity and co-culture assays
Cytotoxicity of CAR-T cells to target cells was first determined using a standard 51Cr release assay at a different effector cell:target cell ratio (E:T ratio = 2.5/5/10) as described previously. 25 In addition, CAR-T cells were co-cultured with a panel of EpCAM+ or EpCAM− tumor cell lines at a different E:T ratio in 96-well plates, and the supernatants were collected at 24 h. ELISA was used to detect interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) secretion (eBioscience, Inc., San Diego, CA).
Mouse xenograft model
Female NOD/SCID BALB/c mice (all 5–8 weeks of age) were obtained from the Beijing HFK Bioscience Co. Ltd. (Beijing, P.R. China). In vivo experiments were performed in accordance with the animal healthcare regulations of the State Key Laboratory of Biotherapy, Sichuan University. To examine the effect of EpCAM CAR-T cells on tumor formation, 5 × 106 HT29 and SW480 cells were respectively co-inoculated subcutaneously (hind flank) with 2 × 107 EpCAM-specific CAR-modified T lymphocytes (a mix of transduced and non-transduced cells) or mock T cells in NOD-SCID BALB/c mice. Only HT29 or SW480 tumor cells were inoculated in the NS group. Five mice per group were used. Once the tumors were palpable, the tumor growth was monitored by caliper measurement every 3 days. Tumor volume was calculated using the following formula: length × width 2 × 0.52).
Analysis of toxic effects in mice
All the NOD/SCID BALB/c mice were evaluated for evidence of acute toxic effects induced by CAR-T cell therapy. Animals were sacrificed when the control group was on the verge of death. Blood samples were collected, and serum was separated. All the values in the routine blood test and blood biochemical test were measured at the State Key Laboratory of Biotherapy (Chengdu, P.R. China). After sacrifice, the main visceral tissues in the HT29 and SW480 models were fixed by 4% polyformaldehyde solution and then processed for paraffin sections. Four-micron sections of the excised visceral tissues were stained with hematoxylin and eosin (H&E) for morphologic analysis.
Statistical analysis
All the data are expressed as the mean ± standard deviation. Statistics analysis was performed using GraphPad Prism v5.0 (GraphPad Software, Inc., San Diego, CA). Statistical analysis for IFN-γ and TNF-α secretion was performed using a paired Student's t-test. One-way analysis of variance was used to compare the tumor volume curves. p-Values of <0.05 were considered statistically significant.
Results
Construction of EpCAM-specific CAR
A third-generation EpCAM-specific CAR was successfully constructed, which was composed of a signal peptide (Sp) of hIL-2, anti-EpCAM scFv, CD8α-hinge, transmembrane of CD8α (CD8α-TM), co-stimulatory molecules CD28 and 4-1BB, and human CD3ζ domain (Fig. 1A and B). Among these, scFv is responsible for the binding of tumor antigen. CD8α-hinge mediates the dimerization of CAR. CD8α-TM anchors the CAR on the membrane of the lymphocytes. CD28 and 4-1BB are co-stimulatory molecules that can improve the cytotoxic activity and proliferation capacity of T cells, and extend the survival time of T cells. 26 –28 CD3ζ transmits the signal elicited from the binding of antigen by the scFv. The CAR with EpCAM specificity was inserted into a pWPXLD lentiviral vector using BamHI and EcoRI enzymes (Fig. 1C). The whole sequence was then confirmed by direct sequencing.
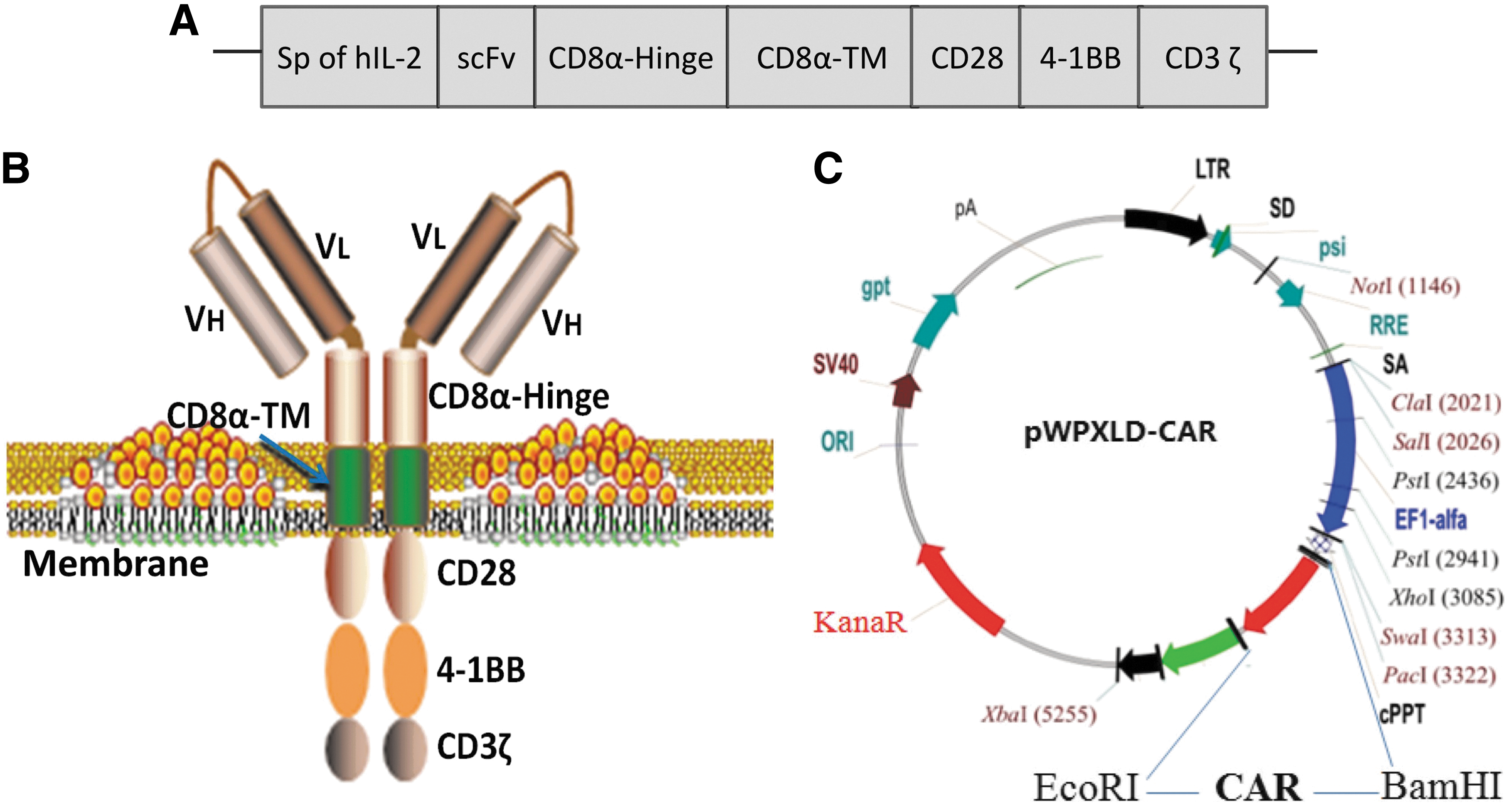
Construction of epithelial cell adhesion molecule (EpCAM)-specific chimeric antigen receptor (CAR).
Efficient generation of CAR-T cells
To achieve stable expression of CAR on T cells, first, lentivirus encoding the green fluorescent protein (GFP) was used to explore the conditions of infection. The schematic representation of pWPXLD-GFP is shown in Supplementary Fig. S1A. Human PBMCs were separated and stimulated with anti CD3/CD28 immune magnetic beads for 1 day before being transduced with lentiviruses encoding GFP. Two days later, the transduction efficiency was determined by flow cytometry and monitored under a fluorescent microscope. As shown in Supplementary Fig. 1B, 47.9% of PBMCs expressed GFP by flow cytometry (multiplicity of infection [MOI] = 5). Expression of GFP was further confirmed under a fluorescence microscope, indicating that high transfection efficiency was obtained (Supplementary Fig. 1C). As above, human PBMCs were stimulated and transduced with lentiviruses encoding the EpCAM-specific CAR. Two days later, the transduction efficiency was determined by staining with anti-F(ab)2 for flow cytometry and anti-CD3ζ antibody for Western blot analysis. As shown in Fig. 2A, 31.8% of PBMCs expressed the CAR by flow cytometry (MOI = 10). Because a CD3ζ sequence was included in the EpCAM-specific CAR (Fig. 1A and B), a protein with an expected molecular weight of 55 kD was detected by Western blot (Fig. 2B). Protein with a weight of 16 kD is endogenous CD3ζ, which could be detected in all the T lymphocytes. The result revealed that the CAR specific to EpCAM is successfully transduced and expressed on human T lymphocytes.

Expression of EpCAM-specific CAR on T lymphocytes and the phenotypes of CAR-T cells.
Ten days later, the phenotypes of all transduced T cells were examined by staining with anti-CD3, anti-CD8, and anti-CD4 antibodies. CD3 was expressed by >95% of CAR-T cells, of which 70.4% were CD8+ and 24.7% were CD4+ (Fig. 2C). The result show that the percentage of CD8+ T cells is obvious more than CD4+ T cells after amplification, which suggests that CD8+ T cells may have a stronger killing ability. Recently, CD45RO and CD62L were used to identify the type of memory T cells, which can be divided into three categories: central memory T cells (TCM, CD45RO+CD62L+), effector memory T cells (TEM, CD45RO+ CD62L−), and native T cells (TN, CD45RO−CD62L+). 29,30 In the present study, TCM accounted for the majority of the CAR-T cells (87.3%), TEM accounted for 7.08%, while TN only accounted for 4.81% (Fig. 2C).
EpCAM CAR-T cells elicit specific cytotoxicity to target cells expressing the EpCAM antigen
EpCAM expression in various tumor cell lines was independently validated by flow cytometry. EpCAM is highly expressed in the human colon cancer cell lines HT29 and SW480, and is moderately expressed in the human NSCLC cell line A549 and the human breast cancer cell line MDA-MB231, while it is not expressed in the human cervical cancer cell line Hela (Fig. 3A).

Specific interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) release of T lymphocytes transduced with EpCAM-specific CAR.
To investigate whether the EpCAM CAR-T cells were capable of specifically recognizing tumor cells, CAR-T cells were co-incubated with a panel of EpCAM+ or EpCAM− tumor cell lines (E:T ratio = 5). Twenty hours later, the amount of secreted effector cytokines including IFN-γ and TNF-α were determined by ELISA. The result showed that CAR-T cells can specifically recognize EpCAM+ tumor cell lines, including A549, MDA-MB-231, SW480, and HT29, and secrete high-dose IFN-γ (Fig. 3B) and TNF-α (Fig. 3C) compared to MOCK T cells (No-CAR). For the EpCAM− tumor cell line Hela, there was no significant difference in the level of IFN-γ and TNF-α between the EpCAM CAR-T group and No-CAR T group (Fig. 3B and C).
Next, the cytotoxicity of CAR-T cells was evaluated using a 51Cr release assay. As shown in Fig. 4A–E, T lymphocytes redirected against EpCAM were demonstrated to elicit lytic cytotoxicity to target cells in an EpCAM-dependent manner. None of the T cells killed the EpCAM− Hela cells. Thus, the result further confirmed that EpCAM-specific CAR-T cells can display specific and efficient targeting of EpCAM+ tumor cells.
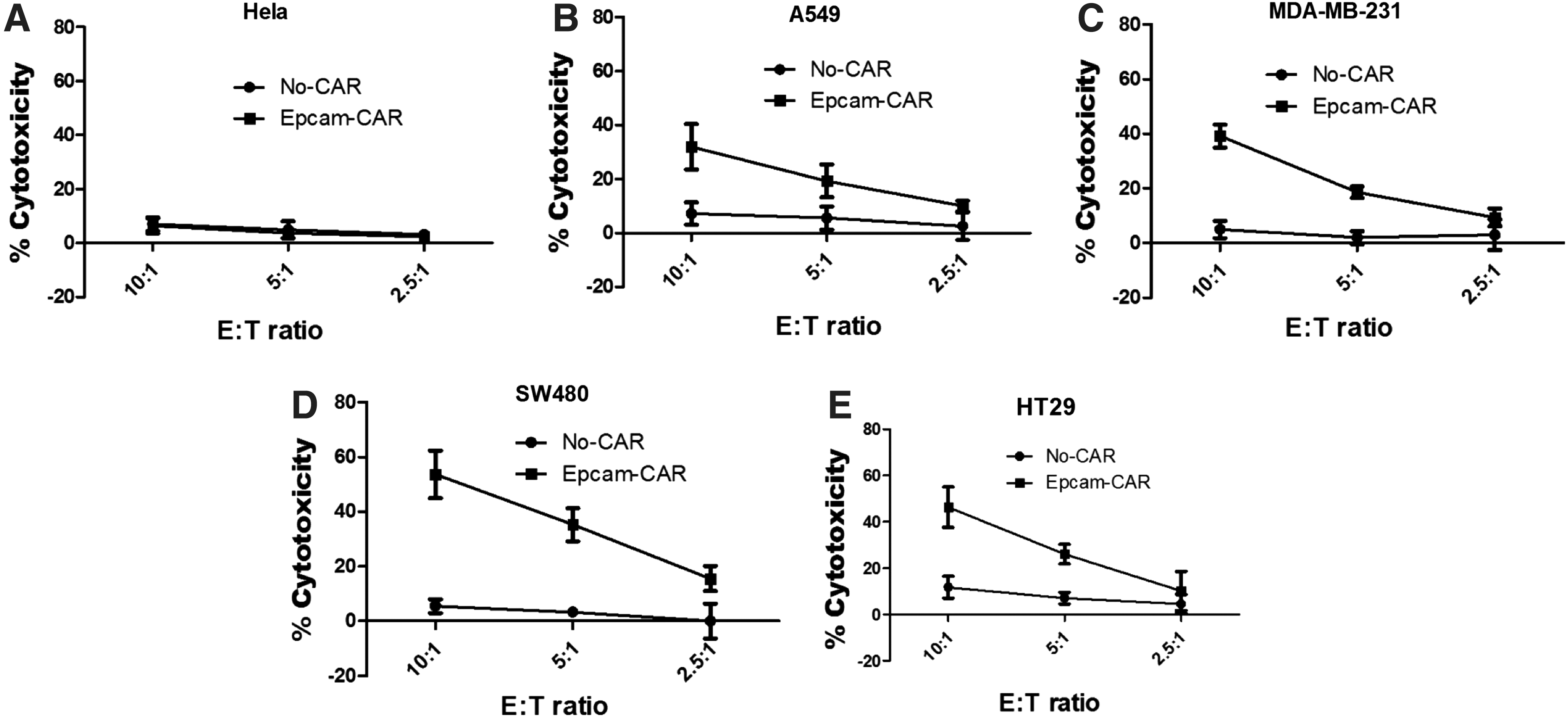
Cytotoxic function of EpCAM-specific CAR-T cells against tumor cells. A standard 51Cr release assay was performed to assess the cytotoxicity. Target cells used were EpCAM− tumor cell Hela
EpCAM-specific CAR-T cells delay the tumor growth of mice with subcutaneous EpCAM-expressing colon tumors
To investigate the ability of the EpCAM-specific CAR-T cells to control tumor growth in vivo, two colon tumor cell lines, HT29 and SW480, with high expression levels of EpCAM were selected as xenograft tumor models. EpCAM CAR-T cells or mock T cells were co-inoculated with HT29 or SW480 tumor cells in NOD/SCID BALB/c mice, and HT29 or SW480 tumor cells alone were inoculated in the NS group. Compared to control T lymphocytes, the adoptive transfer of EpCAM-specific CAR-modified T lymphocytes significantly delayed tumor formation and growth in HT29 and SW480 xenograft models (Fig. 5). By day 26, HT29 colon tumors reached an average volume of 313 ± 92 mm 3 in mice receiving EpCAM CAR-T cells compared to 725 ± 125 mm 3 in mice receiving mock T cells and 800 ± 170 mm 3 in mice in the NS group (p < 0.05; Fig. 5A and B). In the SW480 tumor model, the average tumor volume on day 32 for the CAR-T group was 159 ± 73 mm 3 compared to the average tumor volume of 407 ± 75 mm 3 observed in the mock group and 710 ± 108 mm 3 observed in NS mice (p < 0.05; Fig. 5C and D).

EpCAM-specific CAR-T cells control tumor growth in vivo. T lymphoctytes transfected with lentivirus encoding EpCAM-specific CAR (group CAR-T) or pWPXLD lentiviral vector (mock group) were co-inoculated with HT29 or SW480 tumor cells in mice at an E:T ratio of 4:1. Only tumor cells were inoculated in the NS group.
Safety evaluation of EpCAM CAR-T cells in mice
The potential toxic effects in the mice were evaluated in preclinical experiments. There were no graft-versus-host reactions such as a rash or diarrhea during the whole treatment process in any of the mice. Blood samples were collected from the mice after they were sacrificed, and all the main routine blood tests and biochemical tests were undertaken. There was no significant difference between the EpCAM CAR-T group and the control groups (Figs. 6A and B and 7A and B). Compared to mice from the NS and mock groups, no toxic pathologic changes in the heart, liver, spleen, lungs, or kidneys were detected by microscopic examination after H&E staining in the CAR-T group (Figs. 6C and 7C). Collectively, these data suggested that treatment with EpCAM CAR-T cells is safe in vivo in mice.
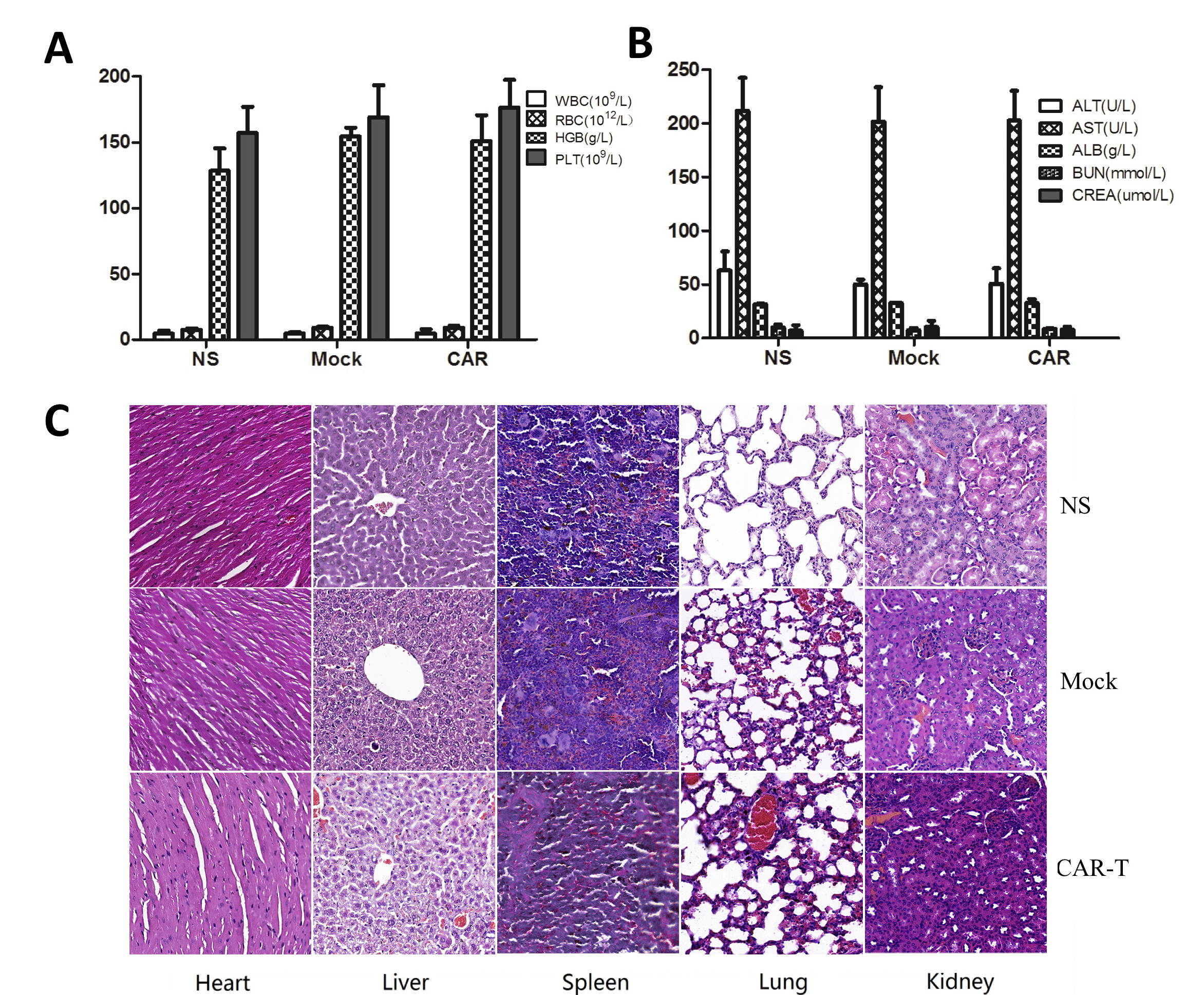
Systemic toxicity evaluation of CAR-modified T cells in the HT29 tumor model.
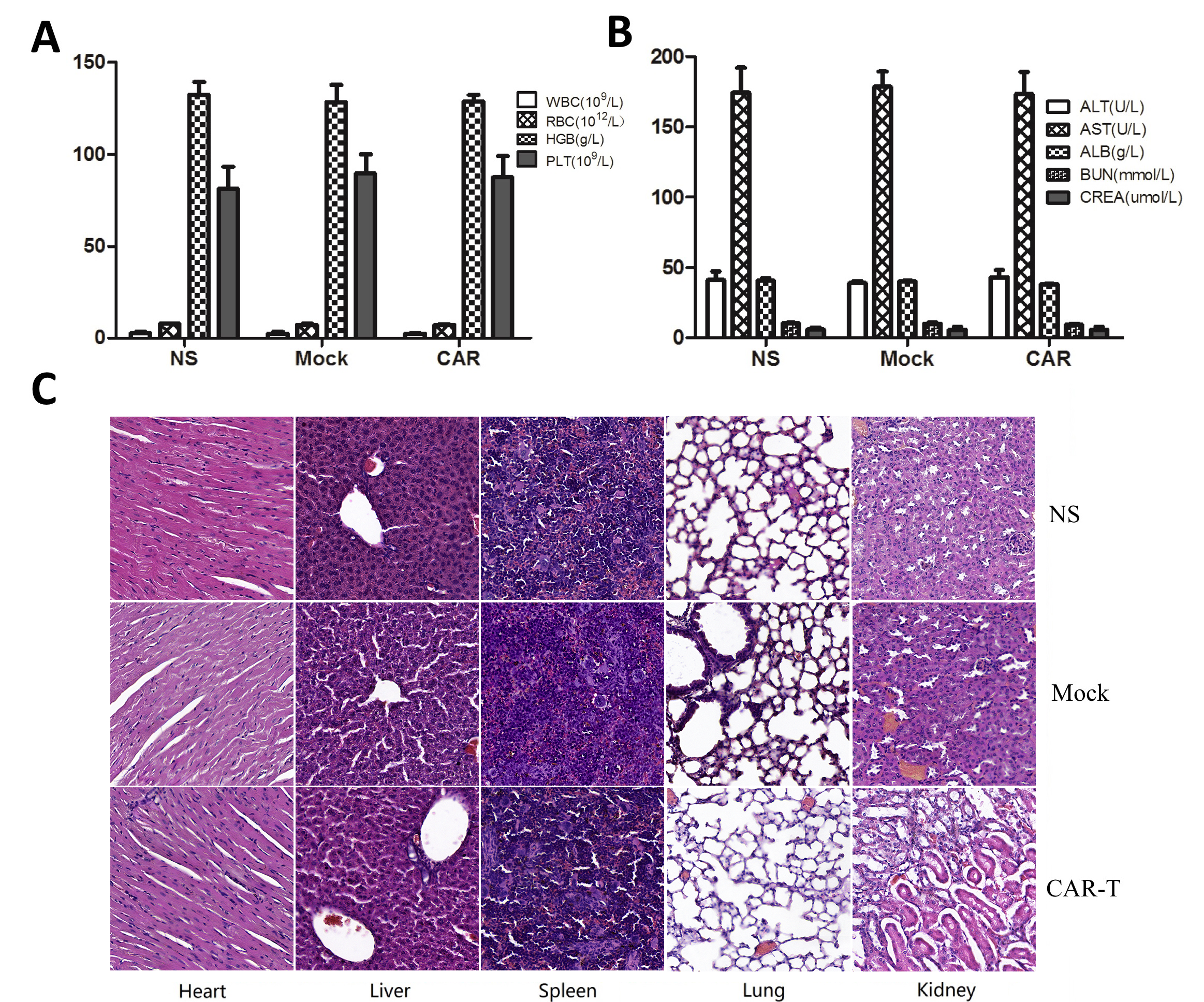
Systemic toxicity evaluation of CAR-modified T cells in the SW480 tumor model.
Discussion
The impressive therapeutic benefits of CAR-T cell therapy have been widely demonstrated in patients suffering with hematologic malignancies. 31 For solid tumors, it is critical to find an ideal tumor target antigen. EpCAM is highly expressed in various types of cancers and is involved in promoting cell proliferation, survival, and metastasis. 2,3 In addition, EpCAM has been identified as a biomarker for CTCs and CSCs. 9,14,16 Therefore, EpCAM may be an even more interesting target for cancer immunotherapy. Using flow cytometry, this study independently validated EpCAM protein expression in several solid cell lines and found that EpCAM is widely expressed in a broad panel of human tumor cells (Fig. 3A).
mAs against EpCAM are now in clinical trials for treating various cancers but do not lead to a cure, 20,21,23,24 which may be attributed to low affinity to the target antigen, as well as some other ambiguous factors. This study proposed improving the therapeutic benefits by generating a CAR that targets EpCAM. For CAR-T cell therapy, the cytotoxicity and survival of the modified T cells are very vital for antitumor efficacy. Numerous studies have reported that costimulatory factors such as CD28, 4-1BB (CD137), CD27, and OX-40 can augment the effects of the cytokine release and enhance T-cell proliferation and persistence, which consequently improved antitumor potency. 26 –28,32,33 Moreover, a large number of research studies have shown that the combination of CD28 and 4-1BB can enhance effector functions of CAR-T cells such as cytotoxicity, cytokine production, and persistence in vivo. 27,32,34 –37 To strengthen the antitumor effect of CAR-T in treating solid tumors further, this study incorporated the costimulatory factors CD28 and 4-1BB into the CAR (Fig. 1A and B).
Currently, the most popular strategy used in the transfection of T lymphocytes is the viral vector, which is an efficient tool for the genomic integration of targeted genes. This study used the lentiviral vector for transduction and achieved efficient transfection efficiency. T-cell viability could not be affected by the lentivirus, and the transfection efficiency of lymphocytes was 25–35% (Fig. 2A and B). CAR-T cells can be effectively amplified by about >100 times in 2 weeks. Although patients can be infused once with up to 108–109 lymphocytes, these CAR-T cells will gradually reduce over time. Normal memory T cells can survive for several months to many years in the body and show a strong and effective immune response to target the tumor again after being stimulated by the associated antigens. It has been reported that TCM is the strongest of the tumor-specific effector cells, playing a main role in antitumor activity, and may be the most appropriate cell type for adoptive cell therapy. 38 –40 The present data indicate that TCM constitute a major part of CAR-T cells (87.3%) after amplification (Fig. 2C).
The most critical contribution of this work is the demonstration of the applicability of EpCAM-specific CAR-T cells to target not only prostate cancer, 41 but also other solid tumors such as breast carcinoma, NSCLC, and colon cancer. First, this study demonstrated that human T cells expressing EpCAM-specific CAR lysed a panel of EpCAM+ cancer cells with high efficiency in vitro (Fig. 4). Furthermore, tumor cell killing and anti-cancer killing activity of CAR-T cells also resides in cytokine production. A significant IFN-γ and TNF-α release by EpCAM-specific CAR-T cells was found over 20 h co-cultures with EpCAM+ tumor cells (Fig. 3B and C). To evaluate EpCAM-specific CAR-T cell potential in vivo, two colon tumor models, HT29 and SW480, with high expression levels of EpCAM were adopted. EpCAM-specific CAR-T cells demonstrated powerful antitumor activity in vivo, with a dramatic inhibition of tumor growth in subcutaneous xenograft models (Fig. 5). Future studies will validate the ability of EpCAM-specific CAR-T cells to resist other solid tumors in vivo.
Unwanted toxicity is one of the major problems that limit CAR-T cell-based immunotherapy. The most common side effects of CAR-T cell therapies mainly depend on the expression levels of the tumor target in normal tissues. Currently, most of the tumor target antigens recognized by CAR-T cells are also expressed by healthy cells. Like other tumor target antigens, EpCAM is also expressed in many epithelial tissues as a cell adhesion molecule, and T lymphocytes redirected against EpCAM may lead to toxicity. However, the most toxic effects in mice were evaluated in preclinical experiments in this study, and CAR-T cell therapy did not induce any obvious systemic toxic effects. Even so, the clinical translation of this approach may thus benefit from the inclusion of a suicide gene within the CAR, which enables the CAR-modified T cells to be rapidly eliminated when undesired toxicity is observed.
In conclusion, the EpCAM-specific CAR was successfully constructed, which can be transduced into T cells and expressed steadily. CAR-T cells can proliferate quickly, mostly CD8+ and TCM. T cells expressing EpCAM-specific CAR specifically recognize EpCAM+ tumor cell lines, secrete high-dose IFN-γ and TNF-α in vitro, and also significantly inhibit the formation and growth of various tumors in vivo. This study confirmed the antitumor ability of CAR-T cells targeting EpCAM and provided a new target for CAR-T cell therapies in treating solid tumors.
Footnotes
Acknowledgments
This work is supported by the National Key Research and Development Program of China (2016YFC1303403), the National Natural and Scientific Foundation of China (81572981/H1611, 81672397/H1617, and 81703057/ H1611), the National High-tech R&D program (863 Program; 2014AA020704), and the Key Scientific and Technological Foundation in Sichuan Province (17ZDZX0037). Parts of this work were used as a poster (PO-205) at the 17th Congress of Gastroenterology China 2017 Chinese Congress of Digestive Disease.
Author Disclosure
Y-Q.W., Y-S.W., W.W., and B-L.Z. have submitted a patent concerning the methodology and application. Y-Q.W. and W.W. are scientific co-founders of Cygenpeutics and CarEne and hold the equity of the company. No conflicts of interests exist of the remaining authors.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.