
Abstract
Glucocorticoids have been commonly used in clinic for their anti-inflammatory and immunosuppressive effects, and it has been proposed that they be used to prevent liver toxicity when systemic administration of adeno-associated virus (AAV) vectors is needed in patients with central nervous system diseases and muscular disorders. Glucocorticoids also enable modulation of vascular permeability. First, this study investigated the impact of dexamethasone on AAV vascular permeability after systemic injection. When a low dose of AAV9 was injected into mice treated with dexamethasone, global transduction and vector biodistribution were not significantly different in most tissues, other than the liver and the heart, when compared to control mice. When AAV9 vectors were used at a high dose, both the transgene expression and the AAV vector genome copy number were significantly decreased in the majority of murine tissues. However, no effect on global transduction was observed when dexamethasone was administered 2 h after AAV vector injection. The study on the kinetics of AAV virus clearance demonstrated that dexamethasone slowed down the clearance of AAV9 in the blood after systemic application. The mechanism study showed that dexamethasone inhibited the enhancement of AAV9 vascular permeability mediated by serum proteins. The findings indicate that dexamethasone is able to inhibit the vascular permeability of AAV and compromise the therapeutic effect after systemic administration of AAV vector. In conclusion, this study provides valuable information for the design of future clinical studies when glucocorticoids are needed to be compatible with the systemic administration of AAV vectors in patients with central nervous system and muscular diseases.
Introduction
Adeno-associated virus (AAV), a nonpathogenic parvovirus that requires a helper virus for efficient replication, has been utilized as a viral vector for gene therapy because of its safety and simplicity. To date, 13 different AAV serotypes and >100 variants have been isolated and used for gene delivery vehicles. 1,2 Several AAV serotypes have been used in clinical trials. 2 –5 As the first characterized capsid, AAV2 has been the most widely studied as an effective delivery vehicle for different therapeutic transgenes, such as RPE65 for Leber congenital amaurosis, factor IX (FIX) for hemophilia B, nerve growth factors for Alzheimer's disease, and glutamic acid decarboxylase for Parkinson's disease. 6 –10 AAV5 and AAV8 have also been applied in several clinical trials for patients with hemophilia B. 3,11 –13 AAV4, AAV9, and rh10 have been used to target the eye and brain, 2,14 and AAV1 and its derived mutant AAV2.5 have been used for muscular transduction in patients with alpha-1 antitrypsin deficiency and muscular dystrophy. 5,15
For central nervous system (CNS) disorders, an intraparenchymal injection is commonly used for AAV vector administration in preclinical and clinical studies. 16,17 This direct delivery of AAV vectors into the brain parenchyma circumvents the blood–brain barrier (BBB), but it has a poor vector spread and limits the transgene expression at the site of injection. 17,18 Moreover, other studies have demonstrated that an intramuscular injection of AAV is able to transduce the motor neurons in animal models of motor neuron diseases because of the retrograde transport properties of AAV vectors. However, transgene expression in the spinal cord motor neurons was at a relatively low level. 19 Additionally, many CNS disorder diseases, such as amyotrophic lateral sclerosis, Huntington's disease, leukodystrophies, and lysosomal storage diseases, occur in multiple brain regions. Therefore, using the local injection of AAV vectors, it is difficult to achieve therapeutic intervention for patients with these neurological disorders. When gene therapy with AAV vectors is used for muscular diseases, especially muscular dystrophies, it achieves a greater therapeutic benefit if every muscle fiber, including the heart, is targeted. For this reason, phenotypic correction should focus on all muscles in the body. Therefore, systemic administration of AAV vectors has been proposed for these CNS and muscle diseases.
In order to achieve effective CNS and whole-body muscle transduction after systemic administration, an AAV vector should be able to cross the vascular barrier. Among the serotypes, AAV8 and AAV9 have been reported to cross the endothelial barrier and transduce various tissues effectively. 20 –22 In particular, AAV9 has gained much attention for its global transduction after systemic administration. 17 AAV9 vectors have the ability to cross either the capillary endothelial cell barriers or the BBB via transcytosis. 23 –26 Transcytosis is a widespread activity found in many endothelial and epithelia cells. At present, AAV4, AAV5, and AAV8 have also been found to be able to cross the endothelial layer via transcytosis. 27
It is well known that glucocorticoids, such as prednisone and dexamethasone, have a potent anti-inflammatory and immunosuppressive function. In clinical trials with AAV vectors, glucocorticoids have been used to rescue transgene expression and ameliorate liver damage by blocking the immune response in patients with hemophilia after systemic administration. 11,28 –30 It is also well known that glucocorticoids function to maintain vascular permeability during infection or anaphylaxis. In recent clinical trials targeting the CNS or muscle tissues with systemic administration of AAV vectors, glucocorticoids have been used to prevent liver toxicities related to high doses of AAV vector. 31 Therefore, it is important to elucidate whether glucocorticoids have an effect on whole-body transduction of AAV after systemic administration.
This study investigated the effect of glucocorticoids on the vascular permeability of AAV vectors in vitro and in vivo. When mice were treated with dexamethasone before application of AAV9 vectors at a high dose, global transduction was decreased. However, no effect on AAV9 global transduction was found if dexamethasone was used 2 h after AAV administration. Mechanism studies showed that pretreatment with dexamethasone inhibited AAV9's capacity to cross the endothelial cell layer, which resulted in decreased global transduction. This study provides valuable information for the rational application of dexamethasone with AAV vectors in future clinical trials.
Methods
Cell lines
HEK293 and Caco-2 cells were maintained at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 1% penicillin-streptomycin. To obtain the cell monolayer from Caco-2 cells on a permeable membrane, cells were seeded on 0.4 μm pore size polycarbonate filters in 6.5 mm Transwell Chambers (Costar, MA), and then cultured for 14 days prior to AAV infection. The medium was replaced at intervals of 2–3 days.
Recombinant AAV production
Recombinant AAV was produced by a triple-plasmid transfection system, as previously described. 32 A 15 cm dish of HEK293 cells was transfected with 9 μg of the AAV transgene plasmid pTR/CBA-Luc, 12 μg of the AAV helper plasmid containing AAV Rep and Cap genes, and 15 μg of the Ad helper plasmid pXX6-80. HEK293 cells were then collected and lysed 60 h post transfection. The supernatant was subjected to a CsCl gradient ultracentrifugation. Virus titer was determined by quantitative polymerase chain reaction (qPCR).
Animal study
Animal experiments performed in this study were conducted with C57BL/6 mice. The mice were maintained in accordance to National Institutes of Health guidelines, as approved by the UNC Institutional Animal Care and Use Committee (IACUC). Dexamethasone (0.2 mg/mouse) or phosphate-buffered saline (PBS) was injected into mice via intraperitoneal (i.p.) injection 2 h before the mice received the AAV9 vectors (low dose: 2 × 1010 vector genomes [vg]/mouse; high dose: 2 × 1011 vg/mouse) via retro-orbital injection. The same doses of dexamethasone or PBS were further injected each day for the next 6 days (Fig. 1A). To test whether the doses of dexamethasone affected AAV9 global transduction, dexamethasone at doses of 0.2, 0.067, or 0.023 mg was injected into the mice before AAV9 administration (2 × 1010 vg/mouse) and then each day for the next 6 days. To test administration timing of dexamethasone, 0.2 mg of dexamethasone was injected at −2, 2, and 24 h after AAV9 administration at a dose of 2 × 1010 vg. The same dose of dexamethasone was repeated each day for the next 6 days (Fig. 3A). To optimize administration timing without impact on global transduction with the high dose of AAV9 vector (2 × 1011 vg/mouse), 0.2 mg of dexamethasone was injected into the mice at 0, 2, 8, and 24 h after AAV9 administration and repeated daily for the next 6 days (Fig. 6A).

Effect of dexamethasone on the global transduction of adeno-associated virus serotype 9 (AAV9) at a low dose.
To study the kinetics of AAV9 vector clearance in the blood, blood samples were obtained 5 min and 2, 24, 48, and 72 h after AAV9 was injected. Blood samples were centrifuged at 1,000 g, and the plasma was frozen at −70°C within 30 min after the blood sample was taken. The vector genome of AAV9 from the plasma samples was tested by qPCR.
Luciferase expression was imaged using a Xenogen IVIS Lumina (Caliper Lifesciences, Waltham, MA) following the i.p. injection of D-luciferin substrate (NanoLight, Pinetop, AZ). Bioluminescent images were analyzed using Living Image (PerkinElmer, Waltham, MA). Acquisition and quantification were performed using Living Image software version 2.20. Quantification was analyzed by defining regions of interest. Global transduction compared in this study was the difference between whole-body transduction and liver-specific transduction.
AAV genome copy number analysis
On days 1, 7, or 10 after AAV9 administration, mice were sacrificed, and tissues were harvested. The minced tissue samples were treated by proteinase K. The total genomic DNA was isolated with a DNeasy Blood & Tissue kit (Qiagen, Valencia, CA). The luciferase gene was detected by qPCR assay. The mouse lamin or GAPDH gene served as an internal control.
Permeability assay in vitro
The medium of the upper and lower chambers was replaced by serum-free medium before dexamethasone treatment. The Caco-2 cell monolayer was pretreated with dexamethasone or PBS for 1 h, and then AAV9 vectors (2 × 109 vg/well) were added to the medium of the upper chamber of the Transwell. At the 0.5, 2, 6, and 24 h post-infection time points, the medium in the lower (basolateral) chamber was collected and tested for the presence of transcytosed AAV9 vector genome DNA by qPCR.
To study whether the application of dexamethasone impacted the effect of serum proteins on AAV9 vascular permeability, the Caco-2 cell monolayer was pretreated with dexamethasone or PBS for 1 h. Human serum and AAV9 (2 × 109 vg/well) were incubated at 4°C for 2 h. The mixture was added to the medium of the upper chamber of the Transwell. At 6 h post infection, the medium in the lower (basolateral) chamber was collected and tested for the presence of AAV9 vector genome DNA by qPCR.
Statistical analysis
The data are presented as the mean ± standard deviation. Student's t-test was used to compare the differences between two groups, while Tukey's multiple comparison test was used between three or more groups. p-Values of <0.05 were considered statistically significant.
Results
Dexamethasone has no significant effect on the global transduction of AAV9 vector at a low dose of systemic administration
Dexamethasone is typically used at multiple doses in clinics for AAV systemic administration. 11,12 This study investigated whether the long-term application of dexamethasone had an effect on AAV global transduction. The first dose of dexamethasone was injected 2 h before AAV9 (2 × 1010 vg) administration, and then once a day for the next six consecutive days (Fig. 1A). Imaging was carried out on days 1, 3, and 7 after AAV administration. As shown in Fig. 1, transduction efficiency of the whole body (minus the liver) was not significantly different between the mice treated with seven doses of dexamethasone or PBS, while higher liver transduction was observed in mice receiving dexamethasone (Fig. 1B and C). On day 7 after the last dose of dexamethasone treatment, mice were euthanized, and different tissues were collected for AAV genome copy number analysis. Significantly fewer viral genomes were observed in the hearts of mice receiving seven doses of dexamethasone, while the viral genomes were much higher in the livers of the mice with dexamethasone treatment than those of the PBS-treated mice (Fig. 1D). To some extent, the results suggest that dexamethasone injected at multiple doses might affect the transduction of some major tissues after low-dose AAV9 vectors were systemically administered, although no effect on whole-body transduction minus the liver was found.
Administration doses and timing for the first dose of dexamethasone do not affect AAV9 global transduction
To study the effect of different doses of dexamethasone on whole-body transduction of AAV9, dexamethasone at three different dosages (0.20, 0.067, and 0.023 mg/mouse) was used daily for 7 days. On day 7 after the last dose of dexamethasone, the mice were imaged (Fig. 2A). There was no significant difference in whole-body transduction of AAV9 among the mice, regardless of the doses of dexamethasone administered (Fig. 2B), although enhanced transduction in the liver was dose dependent (Fig. 2C). The highest liver transduction was achieved in mice receiving 0.2 mg of dexamethasone (Fig. 2A and C).
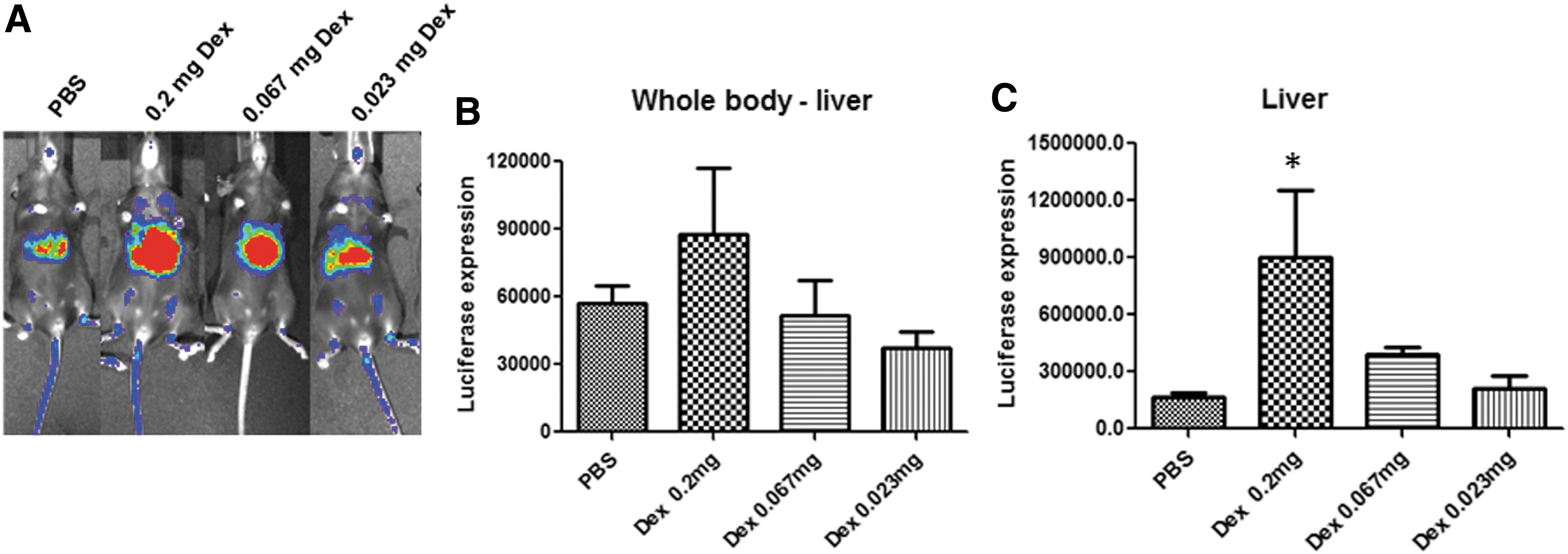
Effect of dexamethasone at different doses on the global transduction of AAV9 at a low dose. Dexamethasone (0.2, 0.067, or 0.023 mg) was administered daily for 7 days following the scheme described in Fig. 1A.
Next, the study investigated whether the administration timing for the first dose of dexamethasone affected AAV9 global transduction. Dexamethasone at a dose of 0.2 mg was administered 2 h before or 2 and 24 h after AAV9 injection followed by daily application for the next 6 days (Fig. 3A). Consistently, there was no difference in global transduction observed in mice, regardless of the application timing for the first dose of dexamethasone (Fig. 3B and C). However, it was interesting to note that enhanced liver transduction was only seen in mice administered dexamethasone before AAV9 injection (Fig. 3D).

Effect of dexamethasone at different administration timings on the global transduction of AAV9 at a low dose.
Dexamethasone inhibits transduction in the major tissues of mice at a high dose of AAV9 vector administration
As stated above, dexamethasone only had an effect on transduction in the heart and the liver but not in other tissues when AAV9 vector was systemically administered at a low dose of 2 × 1010 vg/mouse. In clinical trials, a much higher dose of AAV vector has been proposed for brain or whole-body muscular transduction with systemic administration of AAV vector at a minimum of 1 × 1013 vg of AAV/kg. The study therefore investigated whether dexamethasone had an effect on global transduction when a high dose of AAV vector was used. Mice were treated with 0.2 mg of dexamethasone 2 h prior to injection of 2 × 1011 vg/mouse of AAV9 (1 × 1013/kg) via the retro-orbital vein and were then injected once every 24 h for the next six consecutive days. Imaging was carried out on days 1 and 7 after the last dose of dexamethasone administration (Fig. 4A). Different from the results with the low dose of AAV vectors, a much lower global (minus the liver) transduction was observed in mice treated with dexamethasone after systemic administration of a high dose of AAV vector (Fig. 4A). Similarly, transduction efficiency in the liver was much higher in mice treated with dexamethasone than in control mice treated with PBS. Mouse tissues were harvested on days 1 and 9 after AAV9 injection for vector biodistribution analysis. Genomic DNA was isolated, and the AAV genome number was determined by qPCR assay. The results showed that much less AAV genomic copy number was detected in the hearts, kidneys, and brains from mice treated with dexamethasone than from control mice (Fig. 4B). Consistent with transgene expression, the AAV genomic copy number was increased in the livers of mice receiving dexamethasone treatment (Fig. 4B). The results further indicate that the administration of dexamethasone has the potential to inhibit AAV9 vector vascular permeability and could result in lower transduction in other tissues, except for the liver.

Dexamethasone inhibits AAV9 distribution after systemic infection of AAV9 vectors at a high dose. Dexamethasone at a dose of 0.2 mg was administered 2 h before 2 × 1011 vg of AAV9 injection following the scheme described in Fig. 1A. Imaging was carried out on days 1 and 7 after the AAV vectors were administered
Delayed injection of dexamethasone has no effect on the global transduction of AAV9 vectors
As stated above, when the first dose was administered 2 h before high-dose AAV9 injection, dexamethasone decreased global transduction (Fig. 4). To investigate whether the first dose of dexamethasone administered at different times had an effect on global transgene expression efficiency of AAV vector, dexamethasone was applied at 0, 2, 8, and 24 h after AAV9 injection at a dose of 2 × 1011 vg and was followed by daily treatment for the next 6 days (Fig. 5A). Imaging was carried out 7 days post AAV injection. Mice treated with dexamethasone showed enhanced liver transduction, irrespective of the time when the first dose of dexamethasone was administered (Fig. 5B). On day 10 post AAV9 injection, mice were euthanized for analysis of luciferase expression and AAV genome copy number in the heart, liver, lung, kidney, muscle, and brain (Fig. 5C and D). The results showed that there was no change in AAV9 global transduction in the mice when the first dose of dexamethasone was used at 2 h or later after AAV9 administration. However, decreased transduction was observed in the heart when dexamethasone was injected at the same time as AAV9 administration (Fig. 5C and D). In addition, transgene expression showed no difference in the lung, but decreased gene copy numbers were detected when dexamethasone was injected at 0 and 2 h after injection of AAV9 vectors (Fig. 5C and D). The results suggest that dexamethasone could be administered 2 h after systemic administration of AAV vectors, which would not impact global transduction.
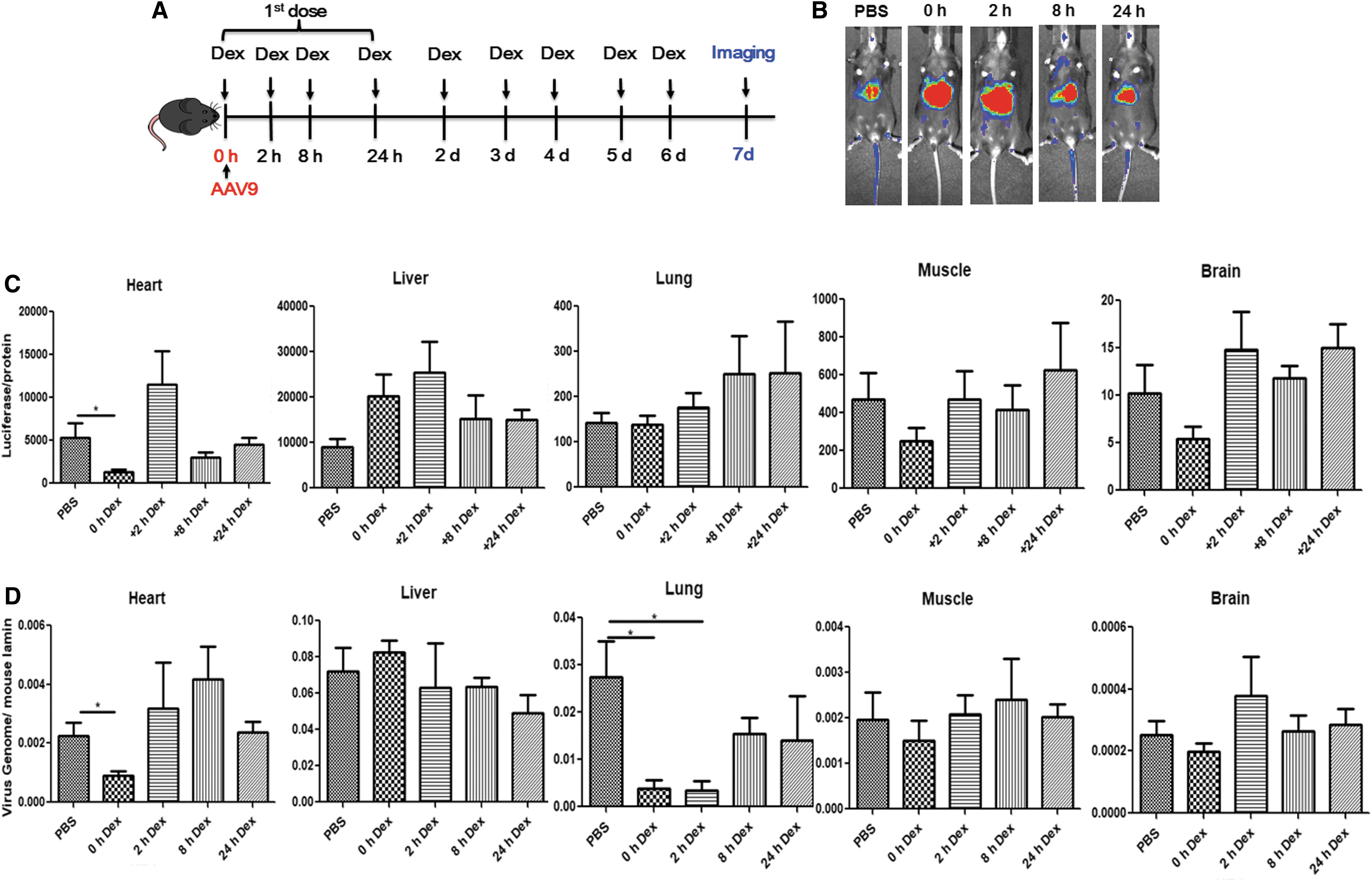
Dexamethasone did not impact global transduction in mice with late administration.
Dexamethasone decreases the clearance of AAV9 vectors in the blood
The above studies demonstrated that dexamethasone inhibited the permeability of AAV9 and then decreased the global transduction after systemic administration. It has been suggested that there is a relationship between whole-body transduction and the kinetics of vector clearance in the blood after systemic administration of AAV vectors. 25,33 Next, the study examined whether dexamethasone had an effect on the clearance of AAV9 vectors in the blood of mice. To do so, the amount of AAV9 vectors in the plasma was monitored by qPCR assay at different time points after systemic administration of AAV9. Higher copy numbers of AAV9 vectors in the blood were detected in mice treated with dexamethasone than in control mice at the early time points (5 min and 2 h) post AAV9 injection (Fig. 6). Regardless of treatment with dexamethasone, there was no significant difference in the number of AAV9 viruses in the blood of the mice at 24, 48, and 72 h post AAV9 administration (Fig. 6). This result further supports the blocking effect of dexamethasone on AAV vascular permeability after systemic injection.

Dexamethasone slows down the clearance of AAV9 in the blood. Dexamethasone at a dose of 0.2 mg was injected 2 h before AAV9 was administered. Blood was collected 5 min and 2, 24, 48, and 72 h after AAV9 injection. AAV genome in the plasma was detected by qPCR. ***p < 0.05.
Dexamethasone inhibits the permeability of AAV9 in vitro
Some AAV vectors have been reported to be able to cross the epithelial barriers via transcytosis. 20,27 To determine whether dexamethasone could change the permeability of AAV9 in endothelial cells during infection, Caco-2 cells were seeded on the inserted wells of a Transwell plate to establish monolayers. Dexamethasone (cohort: Dex-AAV9) or PBS (cohort: PBS-AAV9) was added to the cells 2 h prior to AAV9 vector infection (2 × 109 vg/well). In the third cohort, dexamethasone and AAV9 were added at the same time (cohort: Dex + AAV9). The medium was collected from the lower chambers at the indicated time points (0.5, 2, 6, and 24 h) post infection, and tested by qPCR for the AAV9 vector genome copy number. The AAV genome copy number in the cohort Dex-AAV9 was much less than that in the cohort PBS-AAV9 at 6 h (Fig. 7). However, the copy number of the AAV genome showed no significant difference at 0.5, 2, and 24 h, regardless of the dexamethasone treatment (Fig. 7). In addition, when dexamethasone and AAV9 were added simultaneously (cohort Dex + AAV9), dexamethasone had no effect on AAV9 permeability (Fig. 7). This result suggests that pretreated dexamethasone decreases AAV global transduction by inhibiting AAV vascular permeability.
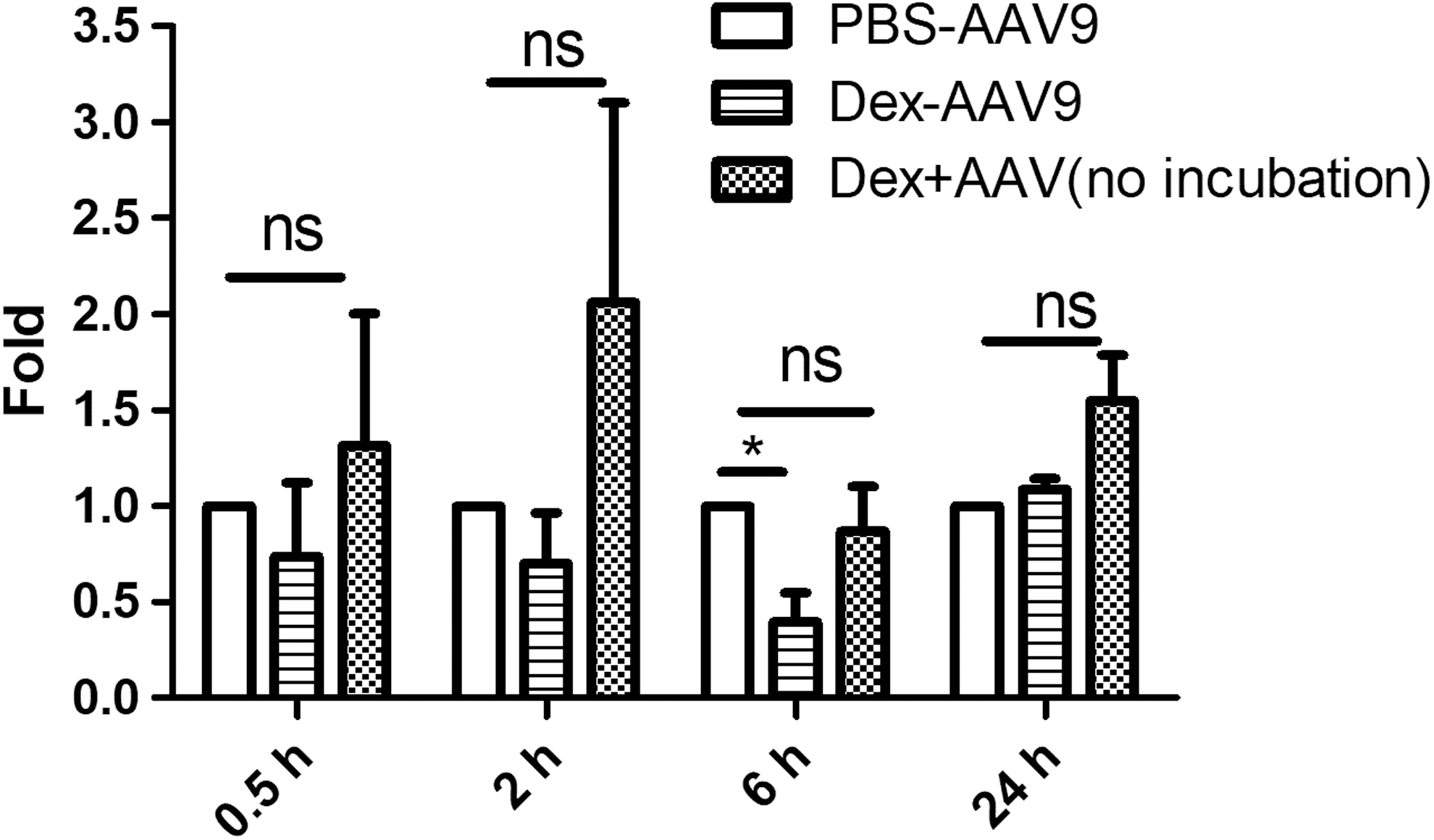
Dexamethasone inhibits the permeability of AAV9 in vitro. Caco-2 cells were pretreated with dexamethasone or phosphate-buffered saline (PBS) before AAV9 incubation for 1 h. At 0.5, 2, 6, and 24 h after AAV infection, the medium was collected from the lower chamber and was tested by qPCR for the AAV9 virus genome. The experiment was performed independently three times. Each experiment contained three replicated Transwells. The y-axis represents the fold change between the wells that contained dexamethasone or PBS. *p < 0.05.
A previous study demonstrated that human serum proteins are able to enhance AAV9 global transduction in mice by increasing AAV9 vascular permeability. 33 Next, this study investigated whether dexamethasone had an effect on enhancing AAV9 vascular permeability mediated by serum proteins. AAV9 vector (2 × 109 vg/well) was incubated with human serum or PBS for 2 h, and then the mixture was added to the Caco-2 cells pretreated with dexamethasone (cohorts: Dex-Serum or Dex) or PBS (cohorts: PBS-Serum or PBS) for 2 h. At 6 h post AAV addition, the medium from the lower chamber was harvested for AAV genome copy number analysis. Consistent with previous results, 33 the AAV genome copy number increased in the media with AAV9 vector incubated with human serum (PBS-Serum vs. PBS; Fig. 8). However, when the cells were pretreated with dexamethasone, the AAV genome copy number in the lower chamber was similar between Dex and Dex-Serum cohorts, but much lower than in the PBS cohort (Fig. 8). These results indicate that dexamethasone is able to block completely the serum enhancement effect on the vascular permeability of AAV9 after systemic administration.
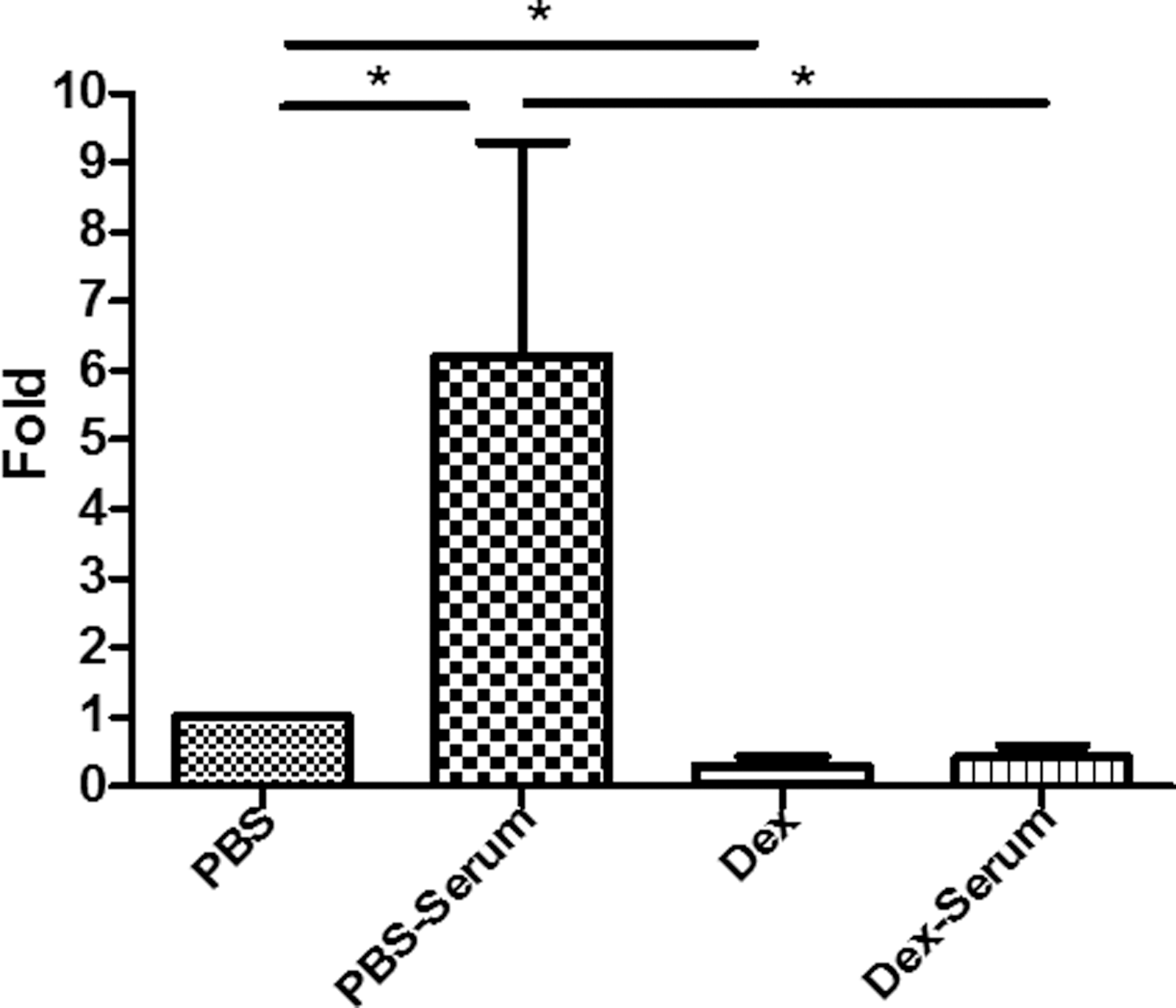
Dexamethasone blocks the enhanced effect of serum proteins on the permeability of AAV9. Serum and AAV9 vectors were incubated for 2 h, and were then added to Caco-2 cells pretreated with Dex or PBS. At 6 h post AAV9 infection, the media was collected from the lower chamber and was tested by qPCR for the AAV9 virus genome. The experiment was performed independently three times. Each experiment contained three replicated Transwells. The y-axis represents the fold change between the wells that contained dexamethasone or PBS. *p < 0.05.
Discussion
Glucocorticoids have been proposed to be included in regimens for the systemic administration of AAV vectors in patients with CNS and muscular disorders. 34 In addition to its modulation of the immune system, glucocorticoids are also widely used in clinics to retain vascular permeability in patients with inflammation or allergies. It is necessary to elucidate the effect of glucocorticoids on the epithelial and endothelial permeability and on the global transduction of AAV after systemic administration. In this study, when dexamethasone was applied before or at the same time as AAV9 systemic administration at a high dose, a significant decrease in transduction was observed in the majority of mouse tissues, except for the liver. However, dexamethasone did not show any adverse effects on AAV9 global transduction if it was used 2 h after injection of AAV vector. The administration of dexamethasone slowed down the blood clearance of AAV9 vectors after peripheral vein injection. The mechanism study demonstrated that dexamethasone inhibited vascular permeability of AAV9 vectors in vitro. Most importantly, dexamethasone completely blocked the enhanced effect of serum proteins on AAV9 vascular permeability.
Microvascular endothelial cells are able to regulate paracellular and transcellular pathways dynamically for the transportation of plasma proteins, solutes, and liquids. Transcellular pathways are used to transport large molecules, while smaller molecules are transported via paracellular pathways, that is, by the regulation of tight junction permeability between adjacent cells. 35,36 Several studies have demonstrated that AAV vectors are able to cross the endothelial layer via transcytosis, especial AAV8 and AAV9. 20,22,37 Plasma mainly contains various proteins, and extravasation of plasma proteins serves various functions, such as maintaining balanced blood and interstitial pressures and helping other molecules cross the vessel wall. 38 A recent study reported that serum proteins, such as fibrinogen, enhance the permeability of AAV9 vector in vitro and augment global transduction in mice. 33
Glucocorticoids, which are potent suppressors of inflammation and allergic pathologies, are broadly used in clinics. Glucocorticoids are able to interfere with the function of microcirculation, which is associated with an inflammatory response by inhibiting the vasodilation in arteriolar and capillary vessels, therefore maintaining the blood flow. 39 Glucocorticoids also retain vascular permeability in the capillary and post-capillary venules when inflammation occurs, and then reduce exudation. 39 Corticosteroids exert their function by binding to the cytoplasmic receptors and inhibiting the transcription of a large number of genes whose products are involved in inflammation, vascular leakage, and angiogenesis. 40 –42 It has been reported that dexamethasone also changes the expression of tight junction proteins, thereby reducing the permeability in multiplex cell species. 43 –46
This study demonstrated that dexamethasone was able to inhibit AAV9 vascular permeability. In particular, it was able to block the enhancing effect of serum proteins on AAV9 vascular permeability completely. This finding on the vascular permeability assay was also echoed by the effect of dexamethasone on viral clearance in the blood after the systemic administration of AAV9 vectors, in which dexamethasone slowed down viral clearance in the blood. Based on these observations, a possible mechanism for the inhibiting effect of dexamethasone on AAV9 global transduction is that dexamethasone is able to restrict the transport and diffusion of AAV9 vectors through tight junction permeability, but it may not affect AAV9 transcytosis.
Due to their anti-inflammatory immune-suppressing function, it has been proposed that glucocorticoids be used for the prevention of liver toxicities with the systemic administration of high-dose AAV vectors in patients with CNS or muscular disorders. In clinical trials for patients with hemophilia, liver damage has manifested in some patients after the systemic administration of AAV vector for liver targeting. It has been suggested that the capsid-specific cytotoxic T lymphocytes (CTLs) mediating elimination of AAV transduced hepatocytes can be attributed to these liver toxicities. Treatment with prednisolone resolved this transient liver damage. 4,12 Nevertheless, what has been neglected is that glucocorticoids have an effect on maintaining vascular integrity, which potentially impacts the ability of AAV to cross the blood vessel for whole-body transduction. Indeed, in this study, dexamethasone significantly decreased whole-body transduction, except for the liver, especially when a high dose of the AAV9 vector was systemically administered. This inhibition was not obvious when a low dose of the AAV9 vector was used. The reason why dexamethasone only impacts the global transduction of AAV9 with a high dose but not a low dose is unclear. Perhaps AAV9 may use several mechanisms to cross the vascular barrier. When a low dose of AAV9 is used, AAV9 may use the preferred pathway (e.g., transcytosis) to cross the endothelial layer of blood vessels. When a high dose is applied, the preferred pathway is saturated, and the remaining AAV vectors may employ other secondary pathways to cross the vascular barrier (e.g., intercellular tight junction or gap), and these non-preferred pathways could be impacted by glucocorticoids. It was noted that mice with low-dose vector showed decreased transduction in the heart, which also suggests the dexamethasone potentially inhibited the vascular permeability and induced higher liver transduction. Consistently, increased liver transduction was observed in mice with high-dose AAV9 vector administration, while the global transduction was decreased. These results suggest that decreased global transduction may correspond with increased liver transduction.
High liver transduction from the dexamethasone treatment may raise concerns relating to the AAV capsid-specific CTL response, as described above. 28,47,48 The capsid antigen presentation in AAV target cells is dose dependent. 49,50 In recent clinical trials of patients with hemophilia B, at weeks 6–10 after the systemic administration of higher doses of AAV vectors, the clotting factor IX was decreased. 11 Further studies suggest that therapeutic failure might result from the capsid-specific CTL response mediated elimination of AAV transduced hepatocytes. 11 After administration of dexamethasone, significantly more AAV virions are detected in the liver, which will induce more antigen presentation on the surface of the hepatocytes for recognition by capsid-specific CTLs and lead to more destruction of the AAV-transduced hepatocytes and liver dysfunction. Therefore, the study attempted to optimize the administration of dexamethasone in order to decrease high liver transduction and maintain the global transduction efficiency of AAV9. It was found that the delayed administration of dexamethasone avoided the adverse effect on global transduction. This result provides valuable information for the optimal administration of dexamethasone in clinics. Alternatively, the application of liver-detargeting AAV vectors with the ability to cross the vascular barrier and with tropism in other tissues may address this concern. 51
Since glucocorticoids are able to affect immune cell function and execute immunosuppressive actions, a previous publication reported that treatment with prednisolone plus rapamycin resulted in a significant decrease in immunoglobulin G and AAV-specific antibody producing cells, which may be a potential approach for AAV gene delivery in patients with pre-existing antibodies or AAV re-administration. 52 However, prednisolone treatment alone did not have any effect on antibody depletion. Moreover, the immune-suppression regimens needed to be applied for a relatively long period time (up to 4 and 8 weeks in their study). 52 In this study, dexamethasone was used for a short time period (1 week) and may not be able to reduce the production of anti-vector antibodies. Therefore, the regimen of dexamethasone in this study would not have any benefit for AAV re-administration.
In summary, dexamethasone decreased the permeability of AAV9, which led to less whole-body transduction after AAV9 systemic administration, perhaps because dexamethasone inhibited the enhancement of serum proteins on AAV9 global transduction. Delayed administration of dexamethasone may avoid the adverse effect of AAV vector on global transduction. This study highlights the concern about the effects of dexamethasone on whole-body transduction of AAV9 vectors, and provides useful information to optimize glucocorticoid applications when systemic administration of AAV vectors is required in future clinical trials.
Footnotes
Acknowledgments
We thank Violeta Zaric for her technical assistance. The authors acknowledge the UNC Biomedical Research Imaging Center and the Small Animal Imaging facility for their assistance in mouse imaging. This work was supported by National Institutes of Health grants R01AI117408 and R01HL125749 (to C.L.).
Author Disclosure
R.J.S. is the founder and a shareholder at Asklepios BioPharmaceutical and Bamboo Therapeutics, Inc. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking. No competing financial interests exist for the remaining authors.