
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR) editing is being considered as a potential gene repair therapy to treat Duchenne muscular dystrophy, a dystrophin-deficient lethal muscle disease affecting all muscles in the body. A recent preliminary study from the Olson laboratory (Amoasii et al. Science 2018;362:89–91) showed robust dystrophin restoration in a canine Duchenne muscular dystrophy model following intramuscular or intravenous delivery of the CRISPR editing machinery by adeno-associated virus serotype 9. Despite the limitation of the small sample size, short study duration, and the lack of muscle function data, the Olson lab findings have provided important proof of principle for scaling up CRISPR therapy from rodents to large mammals. Future large-scale, long-term, and comprehensive studies are warranted to establish the safety and efficacy of CRISPR editing therapy in large mammals.
Clustered regularly interspaced short palindromic repeats (CRISPR) is a novel gene editing technology that became widely used about 5 years ago. 1 –3 Despite its recent debut, it is rapidly advancing from a research tool to a potential therapeutic modality. CRISPR technology is relatively easy to understand and administer. A small sequence-specific guide RNA (gRNA) is used to direct the CRISPR-associated (Cas) nuclease to an intended genomic location for site-specific modification. For therapeutic application, a vector, such as adeno-associated virus (AAV), is used to deliver the gRNA and Cas nuclease to the diseased cells. The disease-causing mutation can be either corrected or removed by CRISPR/Cas-mediated editing in vivo, holding the promise of providing one-time curative therapeutic outcomes for patients. Though highly promising, significant challenges remain in applying CRISPR technology in human gene therapy. Creative strategies are needed to deliver the CRISPR/Cas system to the target cells efficiently and selectively, to achieve a sufficient level of editing for disease amelioration, to avoid and/or attenuate immune responses induced by the bacterial Cas protein and/or delivery vectors, and to prevent and/or minimize untoward off-target genomic alterations. 4
Duchenne muscular dystrophy (DMD) is a relatively common X-linked inherited muscle disease caused by reading frame-aborting mutations in the DMD gene. The DMD gene is one of the largest genes in the body, and it encodes the 427 kD dystrophin protein. Dystrophin is a sub-sarcolemmal cytoskeletal protein, and it protects muscle from contraction-induced injuries. Muscle is essential for body movement, posture maintenance, respiration, and blood pumping. The absence of dystrophin leads to muscle degeneration, necrosis, inflammation, and fibrosis. As a consequence, DMD patients become wheelchair-bound in their early teens and die prematurely in their 20s or 30s from respiratory and/or heart failure. The human body has >600 muscles. These muscles consist of ∼45% body mass and are distributed throughout the body. Given the large size of the gene, the presence of muscle throughout the body, and the highly inflamed nature of the dystrophic muscle, DMD is considered one of the most challenging monogenic diseases amenable to gene therapy. Over the last 40 years, numerous viral and non-viral vector-delivered gene therapy strategies have been tested in mouse models of DMD. 5 –7 Many of these strategies have resulted in remarkable levels of dystrophin restoration and function improvement in mouse muscles. Yet, none of these “mouse therapies” has directly led to equivalent results in human patients. Preclinical study in the symptomatic canine model may help bridge the gap between mice and humans. 8,9
CRISPR technology was first introduced for editing DMD gene mutations in 2014 to correct the germline DNA in mdx mice, a mouse DMD model (Table 1). 10 Subsequently, a series of studies showed restoration of dystrophin expression in patient cells in vitro by co-transfection of the gRNA and Streptococcus pyogenes Cas9 (SpCas9) or Lachnospiraceae Cpf1 (a homolog of Cas9; Table 1). 11 –21 In vivo editing in postnatal animals was first achieved in 2016 in mdx mice by co-delivering the gRNA and Staphylococcus aureus Cas9 (SaCas9) or SpCas9 using adenovirus or AAV (Table 1). 22 –25 These studies revealed a moderate level of DNA editing (≤10%). However, robust dystrophin expression and muscle function improvement were observed in newborn and young adult mdx mice following local or systemic AAV injection. These findings were subsequently validated by other laboratories in different mouse models using SaCas9, SpCas9, Campylobacter jejuni Cas9, and adenine-based editors (Table 1). 20,26 –32 The next logical step was to see if this success can be reproduced in the canine model. 33
CRISPR editing of DMD gene mutations from 2014 to 2018
AAV, adeno-associated virus; ABE, adenine base editors (engineered adenine deaminase and the Streptococcus pyogenes Cas9); Cas9, CRISPR associated protein 9; Cpf1, CRISPR from Prevotella and Francisella 1; CjCas9, Campylobacter jejuni Cas9; DMD, Duchenne muscular dystrophy; EDL, extensor digitorum longus; EHM, engineered heart muscle; gRNA, guide RNA; HDR, homologous recombination; i.m., intramuscular injection; i.m.e., intramuscular electroporation; iPSCs, induced pluripotent stem cells; i.p., intraperitonial injection; i.v., intravenous injection; LbCpf1, Lachnospiraceae bacterium Cpf1; ND, not done; NHEJ, non-homologous end joining; P1–3, postnatal days 1–3; P1–18, postnatal days 1–18; P2, postnatal day 2; P3, postnatal day 3; P4–12, postnatal days 4–12; r.o., retro-orbital injection; SaCas9, Streptococcus aureus Cas9; SpCas9, Streptococcus pyogenes Cas9; SphcCas9/hSpCas9, human codon-optimized SpCas9; TA, tibialis anterior.
This milestone has now been accomplished according to a recent study by Amoasii et al. (Fig. 1). 34 The authors packaged the gRNA and SpCas9 in two AAV serotype 9 (AAV9) vectors and tested them in the ΔEx50 canine DMD model (Fig. 1). In this model, a point mutation at the beginning of intron 50 of the DMD gene results in exon 50 skipping in the RNA transcript, reading-frame shift, and loss of dystrophin expression. 35 Dystrophin expression can be recovered by reframing or removing exon 51. In the Cas9 vector, the authors used the muscle specific CKe promoter. In the gRNA vector, three copies of the same gRNA were expressed using three different pol III promoters. This gRNA targets the SpCas9 to the splicing acceptor site of exon 51. This genomic modification results in the skipping of exon 51 or reframing of the disrupted reading frame. The authors co-injected these two vectors into 1-month-old ΔEx50 puppies either intramuscularly in the absence of immune suppression (n = 2; 1.2 × 1013 vg/vector into the cranial tibialis muscle) or intravenously under immune suppression (n = 2; 2 × 1013 vg/kg/vector in one puppy and 1 × 1014 vg/kg/vector in another puppy; Fig. 1). 34 The outcome of CRISPR editing was studied 6–8 weeks after injection. Specifically, the authors examined (1) gene editing on genomic DNA and cDNA by tracking indels by decomposition analysis and deep sequencing of the polymerase chain reaction product amplified from the target site; (2) dystrophin expression by immunostaining and Western blot; (3) dystrophin-associated glycoprotein complex restoration by β-dystroglycan immunostaining; (4) disease amelioration by qualitative hematoxylin and eosin (H&E) staining of muscle sections, developmental myosin heavy chain (a regeneration marker) expression, and serum creatine kinase levels; (5) safety by hematology, blood biochemistry, target site indel formation, and Cas9 expression in the testes, and deep sequencing of the predicted top off-target sites in muscle; (6) immune response by CD4+ and CD8+ T-cell immunostaining in the locally injected muscle.
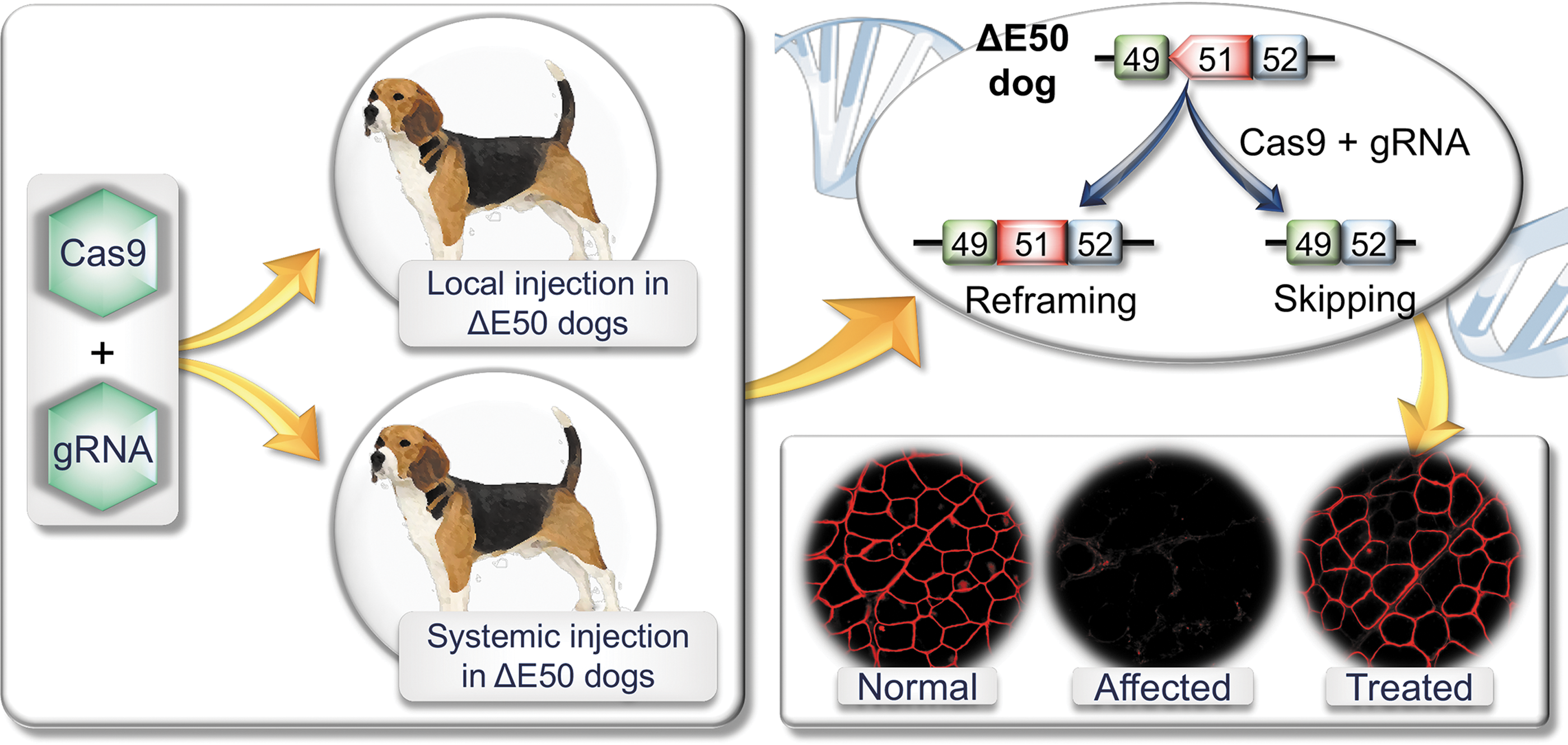
Schematic illustration of clustered regularly interspaced short palindromic repeats (CRISPR) editing in the ΔE50 canine Duchenne muscular dystrophy model. An adeno-associated virus (AAV) CRISPR-associated protein 9 vector and an AAV guide RNA vector were co-delivered to ΔE50 dogs by intramuscular or intravenous injection. In ΔE50 dogs, exon 50 is skipped in the RNA transcript by a naturally existing point mutation. This leads to frame-shift and dystrophin deficiency. CRISPR therapy restores dystrophin expression by reframing or skipping exon 51. Please note: immunostaining images are for illustration purposes. They are not from the Amoasii et al. publication.
DNA-level editing was similar to that seen in mouse studies (1–14%). 22 –24 Immunostaining revealed widespread dystrophin expression. Western blots showed dystrophin restoration to 3–92% of wild-type levels. Remarkably robust dystrophin expression was detected in the heart and diaphragm (∼90% and 50% of normal, respectively, on Western blot) in the puppy that was systemically injected at the dose of 1 × 1014 vg/kg/vector. In the puppy that received the lower dose (2 × 1013 vg/kg/vector), dystrophin expression was consistently lower and more sparse. The authors also found some evidence suggesting a reduction of muscle damage in treated puppies. Importantly, in this short-term study, the authors did not detect T-cell response and germ cell editing. No safety concern was raised either.
The Amoasii et al. study is significant to the development of therapeutic CRISPR editing for DMD. First, this study demonstrated feasibility of systemic CRISPR editing in a diseased large mammal. Second, the level of dystrophin restoration, especially systemic delivery at the dose of 1 × 1014 vg/kg/vector, has reached the range considered to benefit patients according to mouse studies and clinical case reports. 36 This dose is well tolerated in newborns with spinal muscular atrophy type 1 and is in line with the dose used in ongoing systemic AAV micro-dystrophin trials in DMD patients. 37,38 Third, the strategy described by the authors may be directly applicable to ∼13% of DMD patients. 39
As a proof of principle, the findings reported by Amoasii et al. are encouraging. However, the implication of the study is considerably limited due to the sample size, age of injection, duration of the treatment, and methods used to characterize the treatment outcome. A sample size of two for local injection and one for each dose of systemic injection is too small for statistical analysis. Additional studies in more dogs are needed in order to validate the findings.
In the Amoasii et al. study, the affected dogs were treated at 1 month of age. This is analogous to ∼1.5 years in humans. DMD patients are usually diagnosed between 3 and 5 years of age. Hence, the strategy described by Amoasii et al. might be suitable for treating neonatal patients identified through newborn screening. However, to meet the need of existing patients, the protocol has to be optimized in older affected dogs.
DMD is a chronic disease. DMD treatment requires continuous dystrophin expression. 40 It was recently shown that persistent (up to 18 months) dystrophin restoration and muscle and cardiac function improvement can be achieved in young adult mdx mice from a single systemic AAV CRISPR injection (Table 1). 29 Amoasii et al. terminated the study before treated puppies were weaned (2.5–3 months of age). At this age, affected puppies usually show mild clinical symptoms and signs. 41 The short study duration makes it difficult to monitor clinical improvement, durability of gene editing, and potential untoward adverse events that arise later.
As discussed above, Amoasii et al. performed a panel of assays to study editing effect, dystrophin restoration, and disease amelioration. There is no doubt that all these studies are important. However, a more thorough analysis should be included in future studies to assess the consequences of AAV CRISPR delivery fully in dystrophic large mammals. A significant safety concern of in vivo CRISPR editing is off-target editing. The authors only quantified indel formation in the predicted off-target sites in muscle. Unbiased approaches such as the recently reported verification of in vivo off-targets (VIVO) method should provide a more comprehensive view on genome-wide off-target editing. 42 Besides indels, other important genetic changes such as AAV vector genome integration and large chromosomal alterations (e.g., large deletion, inversion, translocation, and duplications) should also be investigated. It is worth noting that a significant portion of systemically injected AAV vectors will end up in the liver. Hence, in addition to muscle, the liver should also be carefully examined for potential off-target genomic modification. To determine the safety, the authors also studied hematology and blood biochemistry. This can be further expanded to include the growth curve (body weight) and histology examination of major internal organs such as the liver, kidney, lung, and brain. Germline editing is an ethical/safety concern. 43,44 Amoasii et al. did not find Cas9 expression and target-site indel formation in the testes. Similar analysis in germ cell forming tissues (the testes and ovaries) should be included in future large-scale, long-term studies.
Immune responses have been at the center stage of gene therapy for decades. 45 These include innate, cellular, and humoral responses to the viral capsid, vector genome, and transgene product. The cellular immune response has been a major hurdle when intramuscular AAV delivery was initially tested in dog muscle. Robust cytotoxic immune rejection was evoked in normal adult dogs by AAV2, AAV6, and AAV9 that express the transgene from a ubiquitous promoter. 46 –48 In one study, cellular immunity was even detected in neonatal normal dogs. 46 Interestingly, nominal CD4+ and CD8+ T-cell infiltration was found in locally injected dogs by Amoasii et al. This suggests that the use of the tissue-specific promoter is a powerful approach to prevent untoward cellular immune responses. While this finding is encouraging, a generalized conclusion that the CRISPR/Cas system does not induce severe innate and/or cellular immune response will require more thorough examination. For instance, (1) some muscle-specific promoters (such as the desmin and SPc5-12 promoter) have been shown to drive expression in antigen-presenting cells (APCs), 49,50 (2) some AAV capsids may transduce APCs more effectively, and (3) age and breed influence the activity and profile of the dog immune system. 51 There is a high likelihood that the T-cell response may still be a major hurdle for future AAV CRISPR local injection studies in the canine DMD model if a different promoter, different AAV capsid, different age, or different breed is used.
All three types of immune responses have been reported following high-dose systemic AAV delivery in neonatal large animals (reviewed by Duan 36 ). Of note are two recent reports showing a fatal innate immune response in nonhuman primates and piglets. 52,53 In the Amoasii et al. study, both systemically injected puppies survived to the scheduled experiment termination date, suggesting acute immunological death might not be a major concern if CRISPR AAV vectors are delivered to affected puppies using an experimental design and study protocol identical to that described by Amoasii et al. 34 Several recent reports suggest that immune responses to the bacterial-originated Cas protein cannot be underestimated. 54 –56 Evaluation on anti-Cas9 T cells (by CD4, CD8, and Treg immunostaining, tetramer staining, INF-γ enzyme-linked immunosorbent spot, T-cell lineage, and functionality study and intracellular cytokine staining), anti-Cas9 antibodies (by in vitro neutralizing assay for the neutralizing antibody and by enzyme-linked immunosorbent assay [ELISA] for the binding antibody), and serum cytokines and complement (by bead-based multiplex cytokine analysis and complement pathway ELISA assay) should be included in future studies. 57,58
The ultimate goal of CRISPR therapy is to mitigate dystrophic muscle disease. In this regard, the treatment effect should be quantified by morphometric analysis of muscle histology and physiological measurement of muscle function. Histopathology evaluation should include, but not be limited to, quantification of the percentage of centrally nucleated myofibers, analysis of myofiber size distribution, evaluation of macrophage and neutrophil infiltration, and qualitative (ideally quantitative) study of muscle fibrosis by Masson trichrome staining or picrosirius red staining (or by biochemical quantification of the collagen content in muscle). Similar assays have been routinely used to characterize morphological changes in dystrophic canine muscles in the literature. 59 –61
The mouse and dog DMD models were established at about the same time.
62,63
While a quite comprehensive list of methods has been available for studying muscle function in mice, methods to study dog muscle function remain underdeveloped until recently. Over the past few years, a number of new assays have been developed, including in situ evaluation of a single dog muscle contractility,
64
hind-limb muscle force measurement,
65
method to study sympatholysis and functional ischemia in canine muscle,
66
electrical impedance myography,
67
noninvasive gait analysis,
68
–70
and noninvasive whole-body activity assay.
69,71
The standard operating procedures have been published for many of these protocols and are freely accessible on the Treat-NMD Neuromuscular Network Web site (
Likely due to the limited sample size and the termination of the study at the pre-symptomatic age, the above-mentioned histological and physiological evaluations were not performed in the Amoasii et al. study. Inclusion of these assays in future studies will better inform the potential outcome when systemic CRISPR therapy moves to human patients.
There are hundreds of muscles in the body (∼700 in a dog). Muscle disease in DMD patients is shaped by the level and extent of dystrophic changes in all body muscles, in particular the heart, diaphragm, and major pelvic limb muscles (e.g., quadriceps) because dystrophy in these muscles leads directly to death or ambulation loss. Following systemic injection, Amoasii et al. performed H&E staining on three skeletal muscles, dystrophin immunostaining on the heart and six skeletal muscles, dystrophin Western blot on the heart and five skeletal muscles, and Cas9 Western blot on the heart (left ventricle, right ventricle, and septum) and 10 skeletal muscles. The authors also quantified level of developmental myosin heavy chain in two skeletal muscles and on-target indel formation on the heart (left ventricle, right ventricle, and septum) and four skeletal muscles. Although Amoasii et al. included the heart and diaphragm in all these assays, the total number of skeletal muscles examined remains low compared to how many muscles a dog has. It will be worthwhile to include more muscles in future studies to appreciate bodywide dystrophin restoration and disease amelioration better.
In summary, the findings reported by Amoasii et al. are encouraging because they provide the critical proof of principle for continuing to explore systemic CRISPR editing as a potential therapy for DMD. 33 The results of Amoasii et al. also suggest that the immune response to AAV9-mediated Cas9 expression may, under certain circumstances (as in the Amoasii et al. study), not be a severe concern as has been speculated. Validating these observations with additional studies, as suggested in this article, should prove to be extremely advantageous in the development of CRISPR technology as a therapeutic modality. The Amoasii et al. study has provided a good starting point for experimentally testing AAV-mediated CRISPR therapy in the canine DMD model (or other large mammalian models of human diseases). Large-scale, comprehensive, long-term studies are needed to determine the risk–benefit ratio of systemic AAV CRISPR therapy for DMD before considering testing this promising therapy in patients.
Footnotes
Acknowledgments
The authors thank the support from the National Institutes of Health (NIH; AR-69085 to D.D., GM-063732 and GM-117059 to S.C.), Department of Defense (MD150133 to D.D.), Hope for Javier (to D.D.), Jackson Freel DMD Research Fund (to D.D.), and Intramural Research Program of the NIH, NCATS (to N.N.Y. and C.H.H.).
Author Disclosure
D.D. is a member of the scientific advisory board for Solid Biosciences and an equity holder of Solid Biosciences. The Duan lab has received research supports unrelated to CRISPR editing from Solid Biosciences. C.H.H., D.D., N.B.W. have filed a patent disclosure on systemic CRISPR therapy.