
Abstract
The combination of cytotoxic treatment modalities, including oncolytic viral gene therapies and immunotherapy, usually yields a synergistic effect. In the current study, a bicistronic adenoviral vector, Ad-CD-GMCSF, carrying the cytosine deaminase (CD) and granulocyte-macrophage colony-stimulating factor (GM-CSF) transcription units driven by a cytomegalovirus promoter was constructed, and the in vitro efficacy of the vector was tested in tumor cell lines and a syngeneic mouse model of colon cancer. The tumor cells infected with Ad-CD-GMCSF vector were found to produce a substantial amount of GM-CSF in tumor cell lines. Accordingly, the vector carrying CD and GM-CSF transcription units together induced a potent antitumor immunity with a significantly increased number of tumor-specific T cells and tumor-specific T-cell cytotoxicity (p < 0.001). The tumor growth rate of Ad-CD-GMCSF-treated mice was significantly lower when compared to the control and an adenoviral vector carrying only the CD transcription unit (Ad-CD; p < 0.05). Likewise, the median overall survival of the Ad-CD-GMCSF vector group was significantly higher than that of the control and Ad-CD groups (34.0 ± 12.8 vs. 14.0 ± 0.5 and 23.0 ± 2.8 days, respectively; p < 0.001). In conclusion, along with its cytotoxic effect, the high immunostimulatory effect of the bicistronic Ad-CD-GMCSF vector has excellent potential in the treatment of cancers.
Introduction
Oncolytic viruses and viral vectors carrying suicide genes such as cytosine deaminase (CD) and thymidine kinase (TK) have been promising treatment strategies for cancer gene therapy for decades. 1 Although successful reports of in vitro studies and experimental models have been published, a limited number of clinical trials failed to yield impressive results. 2,3 The eradication of metastatic disease has long been a challenge for both oncolytic and suicide gene therapy vectors. The insufficient targeting of the therapeutic gene to the tumor cells disseminated throughout the body is a formidable problem for the success of conventional gene therapy studies. Currently, several monoclonal antibodies targeting immune checkpoints such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) or programmed death 1 (PD-1)/programmed death-ligand 1 (PD-L1) and a dendritic cell-based vaccine, sipuleucel-T, have been approved for the treatment of various tumors. 4,5 Checkpoint inhibitors have yielded long-term survival rates exceeding 25% at 5 years in metastatic melanoma patients. 6
The release of tumor-associated antigens with cytotoxic treatments has the potential to induce tumor-specific immune responses. Therefore, the combination of cytotoxic treatments aiming at tumor cell killing and immunotherapy might further increase therapeutic efficacy. Accordingly, the combination of cytotoxic treatment modalities such as chemotherapy and radiotherapy with immunotherapies seems more efficient than either strategy alone. 7,8 Likewise, an oncolytic herpes simplex virus modified to proliferate in only tumor cells and carry the granulocyte-macrophage colony-stimulating factor (GM-CSF) gene (talimogene laherparepvec [T-VEC]) has proven effective in locally advanced melanoma patients. 9 GM-CSF augments the immune response against the tumor antigens shed by the lysis of tumor cells infected with the virus. 10 The immune response induced by the virus and GM-CSF has the capability of destroying the metastatic cells. 10
The efficacy of both the replication-competent and -incompetent adenoviral vectors carrying the CD suicide gene has previously been studied in various cancer cells and experimental tumor models with success. 11 –13 The current study tested if the combination of CD/5-fluorocytosine (5-FC) gene therapy, having the capability of killing tumor cells by converting 5-FC into 5-fluorouracil (FU) in the tumor tissue, with an immunostimulatory GM-CSF gene would further increase the therapeutic efficacy and augment the magnitude of the antitumor immune response induced by the adjuvant effect of dying tumor cells. To achieve this goal, an adenoviral vector carrying both CD and GM-CSF genes driven by the cytomegalovirus (CMV) promoter was constructed. The in vivo efficacy of the new adenoviral vector design of the bicistronic transcription unit of CD-GMCSF and exogenous 5-FC was successfully tested in a syngeneic colon cancer model.
Methods
Cell lines and mice
The HEK-293 transformed human kidney cell line, the CRL-2638 mouse colonic carcinoma cell line, and the CCL-51 mouse breast cancer cell line (American Type Culture Collection, Manassas, VA) were used in the current study. Cells were maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (for HEK-293 cells), Eagle's Minimum Essential Medium (for CCL-51 cells), or RPMI-1640 (for CRL-2638) media supplemented with 10% fetal bovine serum (FBS; Invitrogen, San Diego, CA). Six- to eight-week-old female BALB/c mice (Refik Saydam Health Institute, Ankara, Turkey) were used for either the in vivo tumor model or immunological studies. All experimental procedures with the mice were conducted in accordance with the national Regulation on the Welfare and Protection of Animals Used for Experimental and Scientific Purposes, and the study protocol was approved by the Animal Experiments Local Ethics Committee of Ankara University.
Construction of recombinant adenoviruses
The recombinant adenoviral vectors were generated using the AdEasy vector system (13,14). The Ad-CD-GMCSF vector, a replication non-competent bicistronic adenoviral vector carrying the CMV promoter-driven CD and GM-CSF genes in a single continuous bicistronic transcription unit linked by an internal ribosome entry site (IRES) element, was engineered. The CD gene, amplified by using forward 5′-ACCATGAGCAATAACGCTTTACA-3′ and reverse GM-CSF 5′-GTAACCCAGTCGTTCAACGTTT-3′ primers from a pORF-codAupp plasmid (InvivoGen, San Diego, CA), was inserted next to the CMV promoter gene region of the pShuttle-CMV (Addgene, Cambridge, MA) plasmid. Then, the IRES gene, amplified by using forward 5′-ATTTTCCACCATATTGCCGT-3′ and reverse 5′-TTATCATCGTGTTTTTCA-3′ primers, was inserted. Finally, the shuttle vector of pShuttle-CMV-CD-IRES-GMCSF was completed by inserting the mouse GM-CSF gene amplified using forward 5′-ATGTGGCTGCAGAATTTACTTTTC-3′ and reverse GM-CSF 5′-TCATTTTTGGCCTGGTTTTTTG-3′ primers from pORF-mGM-CSF (InvivoGen) next to IRES (Fig. 1a). Briefly, after linearizing the constructed shuttle plasmid with PmeI, it was co-transformed into the Escherichia coli strain BJ5183 with the pAdEasy-1 (Addgene) viral DNA plasmid for homologous recombination. Likewise, Ad-CD constructed in a similar way was used as control vector (Fig. 1a).

The replication non-competent adenoviral vectors used in the study.
Expression of the CD gene and GM-CSF genes in the Ad-CD-GMCSF vector
Expression of the CD gene
The expression of the CD gene, which is not expressed in mammalian cells, in the bicistronic transcription unit of the Ad-CD-GMCSF vector was measured by extracting RNA from the tumor cells infected with the vector, and then reverse transcription polymerase chain reaction (RT-PCR) of this mRNA was used to generate cDNA for molecular weight analysis, as described previously. 13 Briefly, following the first-strand DNA by using SuperScript II Reverse Transcriptase enzyme at 25°C for 10 min, it was amplified. The predicted molecular weight of the CD fragment generated by this PCR reaction is around 1.2 Kb.
Western blotting of adenovirus CD and GM-CSF protein expression
The intracellular expression of CD and GM-CSF proteins was detected by Western blot, as described previously. 14 Briefly, 2 × 105 cells on six-well culture plates were infected with Ad-CD or Ad-CD-GMCSF vectors at a multiplicity of infection (MOI) of 10. Following 48 h of infection, the cells were collected and lysed with RIPA buffer (Mammalian Cell Lysis Kit; Sigma–Aldrich, St. Louis, MO). Equal sample amounts were loaded for separation through 12% sodium dodecyl sulfate polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). The anti-mouse GM-CSF antibody (MP1-22E9; BD Biociences, San Diego, CA), and the CD polyclonal antibody (Thermo Fisher Scientific, Waltham, MA) were used to detect GM-CSF and CD bands. Detection of immunoreactive bands was visualized by using an enhanced chemiluminescence substrate (Bio-Rad).
GM-CSF secretion from vector-infected cells
The HEK-293 and the CRL-2638 tumor cells were seeded at a density of 100,000 cells/well on six-well plates. The cells were then exposed to the Ad-CD or Ad-CD-GMCSF vectors at a MOI of 1. Following the incubation of the cells for an additional 3 days, the supernatant of the wells was collected, and GM-CSF levels were measured by enzyme-linked immunosorbent assay (ELISA; mouse GM-CSF Quantakine ELISA kit; R&D Systems, Minneapolis, MN).
Functional analysis of the CD gene in the adenoviral vectors
The tumor cells (CCL-51, and CRL-2638 cell lines) were seeded at a density of 50,000 cells/well on 96-well plates. The cells were then exposed to the Ad-CD or Ad-CD-GMCSF vectors at MOIs of 0, 1, 10, 40, and 80. After 24 h, the cultures were supplemented with 5-FC (500 μM; Sigma–Aldrich). Following the incubation of the cells for an additional 3 days, the MTT assay was performed according to the manufacturer's instructions. The percentage of surviving infected cells was calculated by taking the ratio of the surviving cells to the total cells, which were incubated without vector being added to the cultures.
Tests for the immune response
In order to test the induction of cellular immunity, the tumor-specific cytokine release was measured by enzyme-linked immunospot (ELISpot) assay and cell-mediated cytotoxicity of the splenic T cells. 15 Cell suspensions from the spleen, tumor-draining lymph nodes, and tumor tissues were prepared mechanically and from the tumor tissues by collagenase I in order to test the ratios of CD4+, CD8+, and CD4+CD25+ cells. To test the cellular immune response, three mice from each assigned group were used.
Isolation of T cells from mice
The spleens, tumor-draining lymph nodes, and tumor nodules of the mice from each test group were collected on the 14th day of either vector or phosphate-buffered saline (PBS) injection to the tumor nodules. Cell suspensions from the spleen and tumor-draining lymph nodes were prepared mechanically, while those from the tumor tissues were prepared by collagenase I following mechanical mincing. Pan T cells from the tissues were isolated using a commercial kit (Miltenyi Biotec, Auburn, CA). For cell-mediated cytotoxicity testing, the CD8+ T cells were purified by magnetic separation using a MACS CD8a+ T Cell Isolation Kit (Miltenyi Biotec). Purified T cells were maintained in RPMI-1640 medium containing gentamycin, penicillin, streptomycin, mercaptoethanol, 10% FBS, and T-cell supplement.
ELISpot assay
The CRL-2638 tumor cell–specific effector T cells were assessed by carrying out by an ELISpot assay (BD Biosciences, San Diego, CA), as previously described. 15 Briefly, 1 × 106 purified spleen T cells were added to the mitomycin C (MMC)-treated (5 μg/mL) tumor cells grown in a T-25 flask and incubated for 3 days at 37°C. Following incubation of T cells with MMC-treated tumor cells, splenic lymphocytes were used for the interferon-gamma (IFN-γ) ELISpot tests. The number of spots of IFN-γ-secreting splenic lymphocytes was determined according to the manufacturer's protocol.
Cell-mediated cytotoxicity
Cell-mediated cytotoxicity was assayed by measuring the active caspase-3 antibody apoptosis by flow cytometry (BD Pharmingen, San Diego, CA). Briefly, the splenic CD8 T cells incubated with MMC-treated tumor cells for 5 days were incubated with freshly prepared CRL-2638 cells labeled with red fluorescent dye for 4 h at 37°C with varying effector–target ratios (10:1, 5:1, and 1:1). Following the incubation, the ratio of apoptotic tumor cells was measured by flow cytometric analysis of fluorescein isothiocyanate–conjugated active caspase-3 antibody, as described previously. 14
Regulatory T cells from the tumor-draining lymph nodes and spleens of test mice
The ratio of CD4+, CD8+, and CD4+CD25+ cells of the tumor nodules and tumor-draining lymph nodes were measured quantitatively by flow cytometric analysis by labeling anti-mouse antibodies for CD4, CD8, and CD25 antigens (eBioscience, Inc., San Diego, CA). The percentage of lymphocytes positive for each antigen was determined by flow cytometry (BD Biosciences, San Jose, CA). 15
Immunosuppressive cytokine levels in the tumor microenvironment
Tumor tissues taken on the 14th day of vector or PBS injections to the tumor nodules were cut and homogenized. Briefly, approximately 50 mg fresh tumor tissue was re-suspended in 200 μL extraction buffer at 4°C. Tissues were mechanically ground using a homogenizer with an antifoam solution (antifoam Y-30 Emulsion; Sigma–Aldrich). Following the centrifugation of the homogenate at 4400 × g for 10 min at 4°C, the supernatant was recovered. Ten microliters of supernatant was used to determine the protein concentration using the Bradford protein assay. The vascular endothelial growth factor (VEGF), transforming growth factor beta 1 (TGF-β1), and interleukin (IL)-10 levels of the supernatants were measured by a colorimetric sandwich ELISA, as described by the manufacturer (R&D Systems). The supernatants of the homogenates were diluted five times for VEGF and twice for TGF-β1 measurements.
In vivo efficacy of the vector
In order to test the in vivo efficacy of the new bicistronic construct of chemotherapy sensitizing and immunostimulatory vector, a syngeneic BALB/c colon cancer model was used. CRL-2638 cells (1 × 106) were suspended in PBS and injected subcutaneously (s.c.) into 6- to 8-week-old BALB/c mice. After sufficient time had elapsed to allow the tumor nodule to develop to a size in the range 50–60 mm3, the mice were randomly divided into five groups, and 108 pfu of the replication non-competent dual-active (Ad-CD-GMCSF) vector, replication non-competent monoactive (Ad-CD) vector, or the same volume of PBS was injected intratumorally (i.t.) into seven mice for each of the treatment groups. On the same day of the vector injections, a 10-day course of intraperitoneal (i.p.) therapy (PBS or 5-FC at 500 mg/kg to the assigned groups) was started. The mice in the control and experimental groups were evaluated daily for clinical signs of toxicity, including diarrhea, loss of appetite, dehydration, infection, and body weight. Tumor sizes were measured every other day, and the tumor volume was calculated as (length × width2)/2. Mice bearing a tumor nodule with a diameter of >15 mm were sacrificed and treated as dead for the survival analysis.
Statistical analysis
Student's t-test was used to evaluate the results of the in vitro cytotoxicity tests. SPSS v10.0 (SPSS, Inc., Chicago, IL) was used for statistical analyses. One-way analysis of variance (with least significant difference post hoc comparisons) and Mann–Whitney tests were used for the comparison of tumor volumes. Tumor growth rates were evaluated by regression analysis. Survival analyses were performed using the Kaplan–Meier method.
Results
Ad-CD-GMCSF vector significantly expresses both CD and GM-CSF in mouse tumor cells
The expression of the CD gene of the Ad-CD or Ad-CD-GMCSF vector-infected tumor cells was tested by RT-PCR following 16 h of infection. The vectors carrying the CD gene as part of the mono or bicistronic transcription units (Ad-CD and Ad-CD-GMCSF) expressed the CD gene in mouse cancer cells (Fig. 1b).
The protein expression of CD and GM-CSF from adenoviral vectors was assayed by Western blotting. The intracellular protein expression was detected following 48 h of infection of Ad-CD or Ad-CD-GMCSF vectors of mouse tumor cells with a MOI of 10. While both vectors expressed CD protein in CCL-51 and CRL-2638 tumor cells, only the AD-CD-GMCSF vector caused GM-CSF production in those cells (Fig. 1c).
The GM-CSF expression of the vectors was further tested in both HEK-293 and CRL-2638 cell lines seeded at a density of 100,000 cells/well. The cells were infected at a dose of a MOI of 10 with either Ad-CD or Ad-CD-GMCSF vectors. After 72 h of incubation, the secreted GM-CSF levels were measured. While no significant GM-CSF levels were observed in the control wells and wells treated with the AD-CD vector, the Ad-CD-GMCSF vector caused prominent GM-CSF secretion in both HEK-293 and CRL-2638 cells (Fig. 1d).
Bicistronic Ad-CD-GMCSF vector has similar tumor cell killing activity as Ad-CD vector
The mouse tumor cells, including CCL-51, and CRL-2638 were seeded at 50,000 cells/well on a 96-well plate. Following 24 h of incubation, the medium was replaced with a fresh one that included various amounts of the vector at MOIs of 0, 0, 1, 10, 40, and 80. Twenty-four hours later, the medium was replaced with a fresh one, including 500 μM 5-FC to the assigned wells. After 72 h of incubation with 5-FC, the ratio of surviving cells was quantified with a MTT assay. Ad-CD-GMCSF vector did not cause significant tumor cell killing when used alone. The addition of 5-FC significantly increased the cytotoxic effect of the Ad-CD-GMCSF vector (Fig. 1d; p < 0.05). The tumor cell killing activity of both Ad-CD and the Ad-CD-GMCSF vectors was similar (Fig. 1e).
Ad-CD-GMCSF vector significantly induced a potent immune response and decreased the immunosuppressive cytokine levels in the tumor microenvironment
In order to test the tumor-specific immune response-inducing efficacy of the vectors, first tumor cell-induced IFN-γ secretion from the splenic T lymphocytes was tested. The splenocytes of the treated vector and control mice sacrificed on the 14th day of treatment were used. The lymphocytes were incubated with MMC-treated CRL-2638 cells for 3 days. Then the IFN-γ-secreting cells were determined by ELISpot assay. While there were approximately 400 spots/105 lymphocytes from the mice treated with the Ad-CD-MCSF vector bearing CRL-2638 colon cancer cells, the Ad-CD group had 200 spots, and the control mice had only 30 spots (Fig. 2a).

The Ad-CD-GMCSF vector induced a potent antitumor immune response.
Second, the cell-mediated cytotoxicity ability of CD8+ T cells was tested. The CD8+ T cells isolated from the spleen were first sensitized with MMC-treated CRL-2638 cells for 5 days and then were used to test the cytotoxic T-cell killing effect. Briefly, sensitized CD8+ cells were incubated with CRL-2638 cells at varying ratios (1:1, 1:5, and 1:10). Following a 6 h period of incubation, the ratio of apoptotic tumor cells was assayed by using a flow cytometric caspase-3 assay. The T-cell cytotoxicity rate of Ad-CD-GMCSF-treated mice was significantly higher than that of both Ad-CD-treated and PBS-treated control mice (Fig. 2b; p < 0.001).
Third, the CD8/CD4 cell ratio of tumor-draining lymph nodes of the assigned groups was determined. The Ad-CD-GMCSF vector has the highest ratio when compared to the Ad-CD vector alone and the control group (Fig. 2c).
Finally, the effects of the vector treatment on the immunosuppressive milieu in the tumor microenvironment (TME) were tested. First, the ratio of Treg cells in the tumor tissue was checked. The-Ad-CD-GMCSF vector significantly reduced the CD4+CD25+ Treg cell ratio in tumor nodules. While the majority of CD4+ T cells in the tumor tissue were CD4+CD25+ cells (83%) in the control group, the Ad-CD group had 40% and the Ad-CD-GMCSF group had 7% (Fig. 3a; p < 0.001). Likewise, the levels of immunosuppressive cytokines, including VEGF and TGF-β1, were significantly lower in the Ad-CD-GMCSF-treated group (Fig. 3b; p < 0.05). Accordingly, the levels of IL-10 in the Ad-CD-GMCSF-treated group were below the detection limit of the kit. However, the levels were 28 pg/mL for the control group and 18.5 pg/mL for the Ad-CD-treated group.

The Ad-CD-GMCSF vector decreased the immunosuppressive milieu of the tumor microenvironment.
Ad-CD-GMCSF vector significantly delays tumor growth and improves survival
BALB/c mice carrying approximately 50 mm3 of subcutaneous CRL-2638 tumor nodules were randomly divided into three groups. The groups were injected with Ad-CD (1 × 108 pfu), Ad-CD-GMCSF (1 × 108 pfu), or PBS i.t. While the mice in the control group were given 0.5 mL PBS i.p., the other groups injected with vectors were given 500 mg/kg 5-FC in 0.5 mL of volume for 10 days. There was remarkably fast tumor growth in the control group compared to the vector-injected groups (p < 0.05). The Ad-CD-GMCSF-injected group had the lowest tumor growth rate (p < 0.05; Fig. 4a). On the 25th day of 5-FC injection, three mice in the Ad-CD-GMCSF group and one in the Ad-CD group were tumor free. The tumors of those mice did not recur during the 60-day follow-up period.
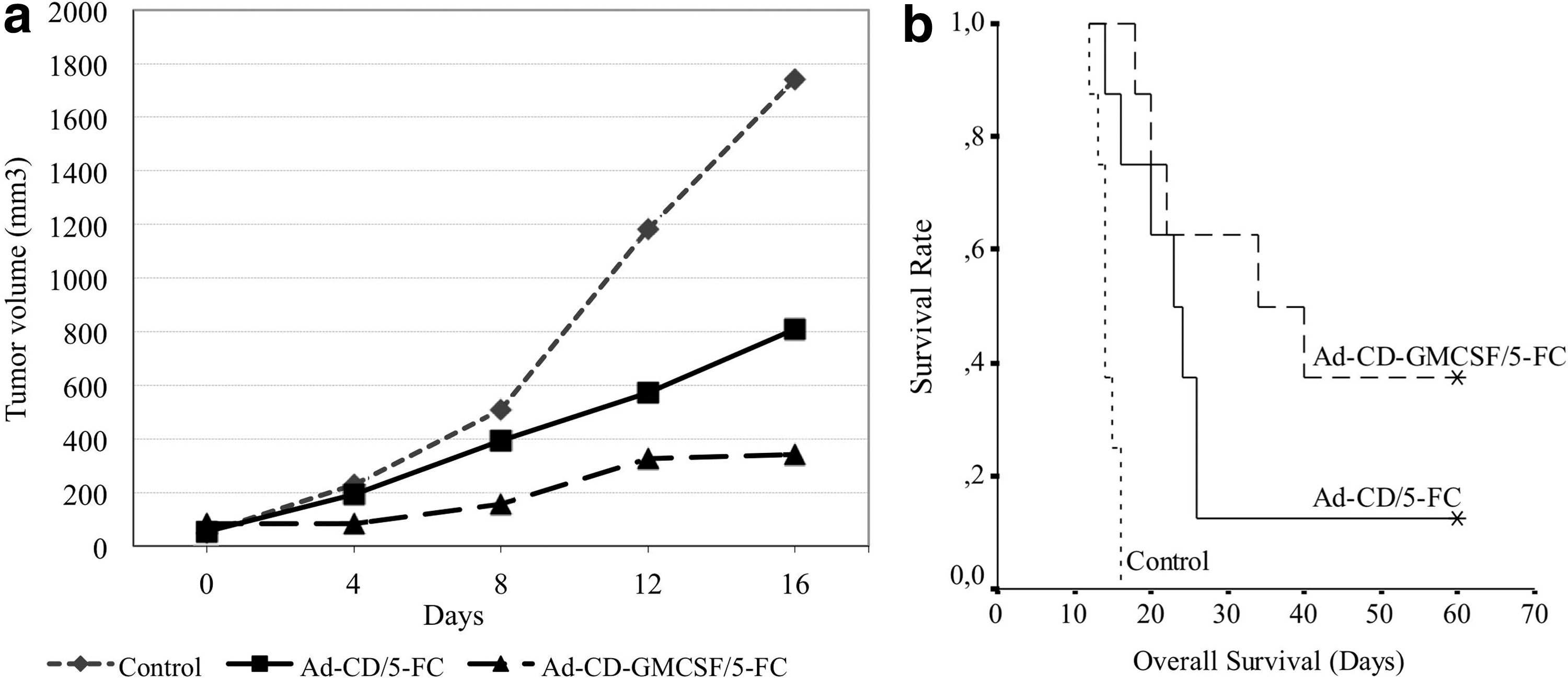
In vivo efficacy of the Ad-CD-GMCSF vector. The treatment groups: control (PBS only); Ad-CD group: intratumoral (i.t.) Ad-CD control vector followed by 10 days of intraperitoneal (i.p.) 5-FC; Ad-CD-GMCSF group: i.t. Ad-CD-GMCSF vector followed by 10 days of i.p. 5-FC.
The median overall survival times were significantly longer in the Ad-CD-GMCSF group (34.0 ± 12.8 days [confidence interval (CI) 9.0–58.9]) compared to the Ad-CD group (23.0 ± 2.8 days [CI 17.5–28.5]) and the control group (14.0 ± 0.5 days [CI 13.1–14.9]; Fig. 4b; p < 0.01).
The Ad-CD and Ad-CD-GMCSF vectors did not affect the body weights of the mice compared to the control group. The food consumption was similar in all groups. No clinical signs of toxicity, including diarrhea, dehydration, infection, or body-weight changes, attributable to vector +5-FC administration were identified.
Discussion
Previous clinical cancer gene therapy trials have yielded very little success. 2,3 Insufficient targeting of disseminated tumor cells is one of the primary reasons for the failure of previous gene therapy studies. 16,17 However, recent gene therapy trials aiming at inducing an immune response against tumor cells yielded promising results. 18
The immune system has the potential to eradicate all the tumor cells that have spread throughout the body. Though the immune cells have the advantage of targeting almost all tumor types, the inefficient expression of tumor antigens and failure to induce a strong tumor-specific immune response in the majority of tumor types are the main reasons for tumor escape. Recently, the monoclonal antibodies blocking the CTLA-4 or PD-1/PD-L1 checkpoints of cytotoxic T cells have shown substantial and durable remission in advanced cancer patients. 4 –6,19 However, those strategies work in a limited number of patients who probably have a sufficient number of cytotoxic T cells in the TME nearby, armed but suppressed by the tumor cells. 20 If one could adequately induce the immune system, then the immune cells armed against tumor cells might have the potential to eradicate the metastatic disease. Accordingly, the combination of chemotherapy with checkpoint inhibitors could provide new tumor-associated antigens and danger-associated molecules from dying tumor cells and increase the therapeutic efficacy in the clinic. 7
Oncolytic viral vectors carrying immunostimulating cytokine genes such as GM-CSF or CD40 ligand might induce both a specific antitumor immune response and objective responses. 9,21,22 In the case of T-VEC, as well as the tumor-debulking effect with the lytic effect of the vector, the local efflux of tumor antigens and danger signals also promote an immune response, which is enhanced by the viral expression of GM-CSF. 9
In conventional chemotherapeutic models, the dying tumor cells are the primary source of tumor antigens to induce an immune response. However, an insufficient number of immune cells, including dendritic cells, in the TME of dying tumor cells might result in an inefficient immune response. By giving immunostimulatory molecules such as GM-CSF, IFN-γ, IL-2, or IL-12, one of the ways to induce a tumor-specific immune response is to induce co-stimulatory molecule expression and tumor cell recognition. 23 –25 Dendritic cells armed against tumor antigens have the capability of eradicating disseminated tumor cells. In a previous study of a combination of a subcutaneous dendritic cell injection and i.t. suicide gene therapy via an adenoviral vector containing CD transcription unit, a synergistic antitumor response was shown in an experimental lung cancer model. 26 In that study, intravenous rechallenge of tumor cells did not produce lung deposits in the mice treated with s.c. dendritic cells, while the vector-only group had lung metastases. The results have enabled a strategy to be developed of combining both suicide and immunostimulatory gene therapy strategies in the same vector. A replication-incompetent adenoviral vector carrying a bicistronic transcription unit of CD and GM-CSF genes driven by a CMV promoter was designed.
While the 5-FU converted from 5-FC inside the tumor cells by the expression of CD kills the tumor cells, the expression of the gene encoding mouse GM-CSF results in local GM-CSF production to recruit and activate antigen-presenting cells, which subsequently induce tumor-specific T-cell responses. Although the transfection efficiency of adenoviral vectors in murine cells was not as high as in human cell lines, previous work with mouse cancer cell lines showed that a CD/5-FC prodrug system transduced by the adenoviral vectors could destroy those cells. 12,13 It was found that the bicistronic addition of the GM-CSF gene to the Ad-CD chemotherapy sensitization vector increased the tumor response over that induced by Ad-CD/5-FC treatment alone in a syngeneic mouse colon cancer model (Fig. 4a).
Accordingly, it was found that the mice treated with the CD-GMCSF-carrying vector have a significantly increased tumor-specific T-cell response (Fig. 2a and b). Likewise, the immunosuppressive nature of the TME was also suppressed by the AD-CD-GMCSF vector treatment. Both the ratio of the CD4+CD25+ cells and the levels of immunosuppressive cytokines such as TGF-β1, IL-10, and VEGF were significantly decreased in the TME of Ad-CD-GMCSF-treated mice (Fig. 3). The increased survival in the Ad-CD-GMCSF-treated group compared to the Ad-CD vector group suggests a potential synergistic effect of tumor cell cytotoxicity and immunostimulatory effect derived from the components of the bicistronic transcription unit of CD and GM-CSF (Fig. 4b).
In conclusion, the combination of tumor cell killing and immunostimulatory strategies in cancer treatment hold great promise in the eradication of metastatic tumors. Likewise, local injections of the current bicistronic Ad-CD-GMCSF vector into the tumor deposits directly or using guided imaging techniques could be further tested in the treatment of established tumors.
Footnotes
Acknowledgments
The authors thank Dr. Albert Deisseroth for the comments and suggestions on the design of the project and the manuscript. This study was supported by The Scientific and Technologic Research Council of Turkey (TÜBİTAK, Grant #105S093, to H.A.) and Ankara University (Grant #11B3330010 to H.A.). This work was presented at the AACR 2018 Meeting, and the abstract was published in the Proceedings of the American Association for Cancer Research 2018;59:1508 (#5911).
Author Disclosure
H.A., A.C., G.S., Y.T., and F.I. certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript. Y.T. is a shareholder of MicroVAx LLCi, Manassas, VA, but the company was not involved in the current study.