
Abstract
The introduction of chimeric antigen receptors (CARs) to augment the anticancer activity of immune cells represents one of the major clinical advances in recent years. This work demonstrates that sorted CAR natural killer (NK) cells have improved antileukemia activity compared to control NK cells that lack a functional CAR. However, in terms of viability, effectiveness, risk of side effects, and clinical practicality and applicability, an important question is whether gene-modified NK cell lines represent better CAR effector cells than primary human donor CAR-NK (CAR-dNK) cells. Comparison of the functional activities of sorted CAR-NK cells generated using the NK-92 cell line with those generated from primary human dNK cells demonstrated that CAR-NK-92 cells had stronger cytotoxic activity against leukemia cells compared to CAR-dNK cells. CAR-NK-92 and CAR-dNK cells had similar CD107a surface expression upon co-incubation with leukemia cells. However, CAR-NK-92 cells secreted higher granzyme A and interleukin-17A levels, while CAR-dNK cells secreted more tumor necrosis factor alpha, interferon gamma, and granulysin. In addition, CAR-NK-92 cells revealed a significantly higher potential for adverse side effects against nonmalignant cells. In short, this work shows the feasibility for further development of CAR-NK strategies to treat leukemia.
Introduction
Acute myeloid leukemia (AML) is a clonal disease characterized by impaired hematopoiesis due to malignant accumulation of immature myeloid cells. Adult AML remains a difficult-to-treat disease, with 5-year overall survival rates of <30%. Thus, even with the advent of precision medicine, there remains a clear unmet clinical need to develop improved treatment strategies for AML patients. 1 Cancer cells can employ various immune escape mechanisms to evade the immunosurveillance of host immune cells. For example, AML cells were shown to avoid natural killer (NK) cell recognition by downregulating expression of ligands for the activating receptor NKG2D (e.g., major histocompatibility complex class I–related molecules MICA and MICB or ULPB4). 2 Thus, many resources are currently devoted to develop adoptive immunotherapies to overcome this, including application of NK cells, which are cytotoxic innate lymphoid cells that recognize and eliminate virally infected cells and cancer cells without the need for prior sensitization. However, manufacturing a standardized, functionally active NK cell product derived from third parties and cryopreserved as an “off-the-shelf” NK product is an essential prerequisite to allow continuous access to cell therapeutics for clinical application to several cancer patients. In the past, it was difficult to realize this goal with primary donor NK cells by long-term cultivation and cryopreservation processes, but this important requirement was achieved by the generation of the highly active, resistant, and interleukin (IL)-2-dependent NK cell line NK-92 (NantKwest, Culver City, CA). 3 NK-92 was derived from the peripheral blood of a 50-year-old male patient with non-Hodgkin's lymphoma. 4,5 Currently, this effector cell line is available for various clinical applications from a current Good Manufacturing Practice–adequate master cell bank. For example, improved survival of AML patients who were transplanted from NK-alloreactive donors demonstrated the antileukemia activity of donor NK cells. 6,7 Infusion of irradiated (10 Gy dose) NK-92 cells was shown to be safe, with evidence of efficiency in patients with refractory hematologic malignancies who relapsed after autologous hematopoietic stem-cell transplantation. 8
Another promising strategy for anticancer therapy is to redirect natural killer (NK) cells and exploit their innate cytotoxic capacities via genetic modification with chimeric antigen receptors (CARs) designed to target tumor-specific antigens and enhance NK cell-mediated anticancer activity. CARs are synthetic receptors that consist of a single-chain variable fragment (scFv) for antigen recognition, a hinge region for flexibility, a transmembrane sequence, and an endodomain to promote activation of cytotoxic signaling cascades. CAR-NK cells are currently tested in several different cancer settings. The efficacy of CAR-NK-92 cells to target carcinoembryonic antigen (CEA)-expressing colorectal cancer cells was dependent upon CEA expression, with greater CAR-NK-induced cytotoxicity against the high CEA-expressing tumor cells. 9 Combination of the multikinase inhibitor regorafenib and CAR-NK-92 cells designed to target epithelial cell adhesion molecule led to improved therapeutic efficacy in human colorectal cancer xenografts. 10 Dual-specific CAR-NK-92 cells that target both the epidermal growth factor receptor (EGFR) and the mutant EGFRvIII had better activity against glioblastoma xenografts. 11 Treatment of a breast cancer brain metastases model with EGFR-CAR-NK-92 cells resulted in greatly reduced tumor burden and significantly increased survival time of tumor-bearing mice when applied in combination with oncolytic herpes simplex virus. 12 Anti-CD133-CAR-NK-92 cells exhibited enhanced activity against CD133+ ovarian cancer stem cells and remained cytotoxic when used in combination with cisplatin. 13
The antileukemia activity of CD123-CAR-T cells has previously been shown. 14 CD123, the IL-3 receptor alpha chain, was demonstrated to be highly expressed on the surface of many primary AML cells. 15 Interestingly, in recent decades, NK manufacturing and ex vivo “up-scaling” expansion processes of primary human donor NK cells has been improved and optimized to provide more stable and highly active primary NK cells in sufficiently high cell numbers for future clinical application. 16,17 In parallel early transduction experiments, those expanded and activated NK cells were gene modified at different cultivation periods to express anti-CD123-CARs against AML cells, which resulted in an effector cell composition containing non-modified and CAR-engineered NK cells. Those non-sorted NK compositions (effector cell mixture of non-modified and transduced NK cells) showed improved killing activity, degranulation, and cytokine release against the CD123+ AML cells.
To test the hypotheses that NK cells engineered to express a CAR specific for antigens associated with AML show enhanced antileukemia activity, and, furthermore, whether primary NK cells could be a powerful alternative NK cell source compared to the established CAR-NK-92 cell line, both NK cell types were sorted after CAR transduction, and relevant parameters of effector-relevant functional cytotoxicity were assessed. Therefore, an alpharetroviral vector system was used to generate CD123-CAR-NK cells. Stable expression of the CD123-CAR was demonstrated in NK-92 cells and primary human donor NK (dNK) cells. After cell sorting, CAR-NK-92 and CAR-dNK cells both had improved activity against the CD123+ AML cell line KG-1a and against primary human leukemia cells. However, comparison of CAR-NK cell function also revealed some differences that are likely due to the NK cell source used to generate CAR-NK cells.
Methods
Cell lines
Human embryonic kidney 293T and human HT1080 fibroblasts were cultured in Dulbecco's modified Eagle's medium supplemented with stable glutamine (Biochrom, Berlin, Germany), 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 1 mM sodium pyruvate (NaPyr; PAA, Pasching, Austria). NK-92 cells were cultured in RPMI-1640 medium supplemented with stable glutamine, 10% FBS, 1% P/S, 1 mM NaPyr, and 400 IU/mL IL-2 (Proleukin S; Novartis Pharma GmbH, Nuremburg, Germany). KG-1a cells were cultured in Iscove's modified Dulbecco's medium (IMDM) supplemented with stable glutamine (Biochrom), 15% FBS, 1% P/S, and 1 mM NaPyr.
Primary dNK cells
Following informed consent and in accordance with the Hannover Medical School ethics commission, primary human dNK cells were purified from different human buffy coats by hands-on immunomagnetic separation (MACSxpress® Whole Blood NK Cell Isolation Kit; Miltenyi Biotec, Bergisch-Gladbach, Germany), and cultures with an initial cell concentration of approximately 2–4 × 106 dNK cells/mL were expanded for up to 14 days in NK MACS (NK MACS basal medium; Miltenyi Biotec) medium supplemented with 5% human serum type AB (Biochrom), 2% NK MACS Supplement® (Miltenyi Biotec), and IL-2 (1,000 IU/mL, Proleukin S; Novartis Pharma GmbH). Cell cultivation was initiated in 25 cm 2 flasks and expanded to 75 cm 2 flasks on expansion day 8. Growth medium was changed every 2–3 days.
Primary human AML cells
The functional killing properties of non-modified and CAR-engineered sorted dNK and NK-92 cells were carried out with native AML blasts isolated from the peripheral blood of three different patients who were classified as described before by Klöß et al. 17 Briefly, samples were from an 80-year-old female with de novo AML (FAB subtype M0) containing FLT3-ITD, a 78-year-old male with secondary AML (FAB subtype M5) containing mutated NPM1, and a 53-year-old male with relapsed AML (FAB subtype M5). These primary AML samples were thawed, washed in phosphate-buffered saline (PBS) containing 10% FBS, and treated with DNase I (0.1 mg/mL) for 15 min. Informed consent was obtained from all patients according to the principles of the Declaration of Helsinki and the Hannover Medical School ethics committee (approved protocol #2613-2015). Cells were cultivated in IMDM supplemented with 10% FBS, 1% P/S, L-glutamine, IL-3 (20 ng/mL), IL-6 (20 ng/mL), stem-cell factor (20 ng/mL), granulocyte colony stimulating factor (CSF; 50 ng/mL), and granulocyte macrophage CSF (50 ng/mL) at 37°C and 5% CO2. Prior to use in cytotoxicity assays, AML blasts were washed with PBS and re-suspended at 2.5 × 105 cells/mL in TexMACS medium supplemented with 5% human serum albumin (HSA). CD123 surface expression on patient samples was determined by no-wash, single-platform flow cytometry (FCM) using an anti-CD123-APC antibody (BD Biosciences, San Jose, CA) and 7-aminoactinomycin D (7-AAD; Beckman Coulter, Brea, CA) to estimate leukemia cell viability.
Alpharetroviral constructs
The CD123-specific third-generation CAR construct has been described previously. 18 Briefly, self-inactivating (SIN) alpharetroviral vectors containing a myeloproliferative sarcoma virus promoter and a woodchuck hepatitis virus post-transcriptional regulatory element were used to deliver and express third-generation anti-CD123 CAR constructs (Fig. 1A). For some constructs, a hemagglutinin (HA) tag was introduced between the signal peptide and the codon-optimized scFv for direct detection of CAR expression. All CAR constructs also express the enhanced green fluorescent protein (EGFP) via an internal ribosomal entry site (IRES).
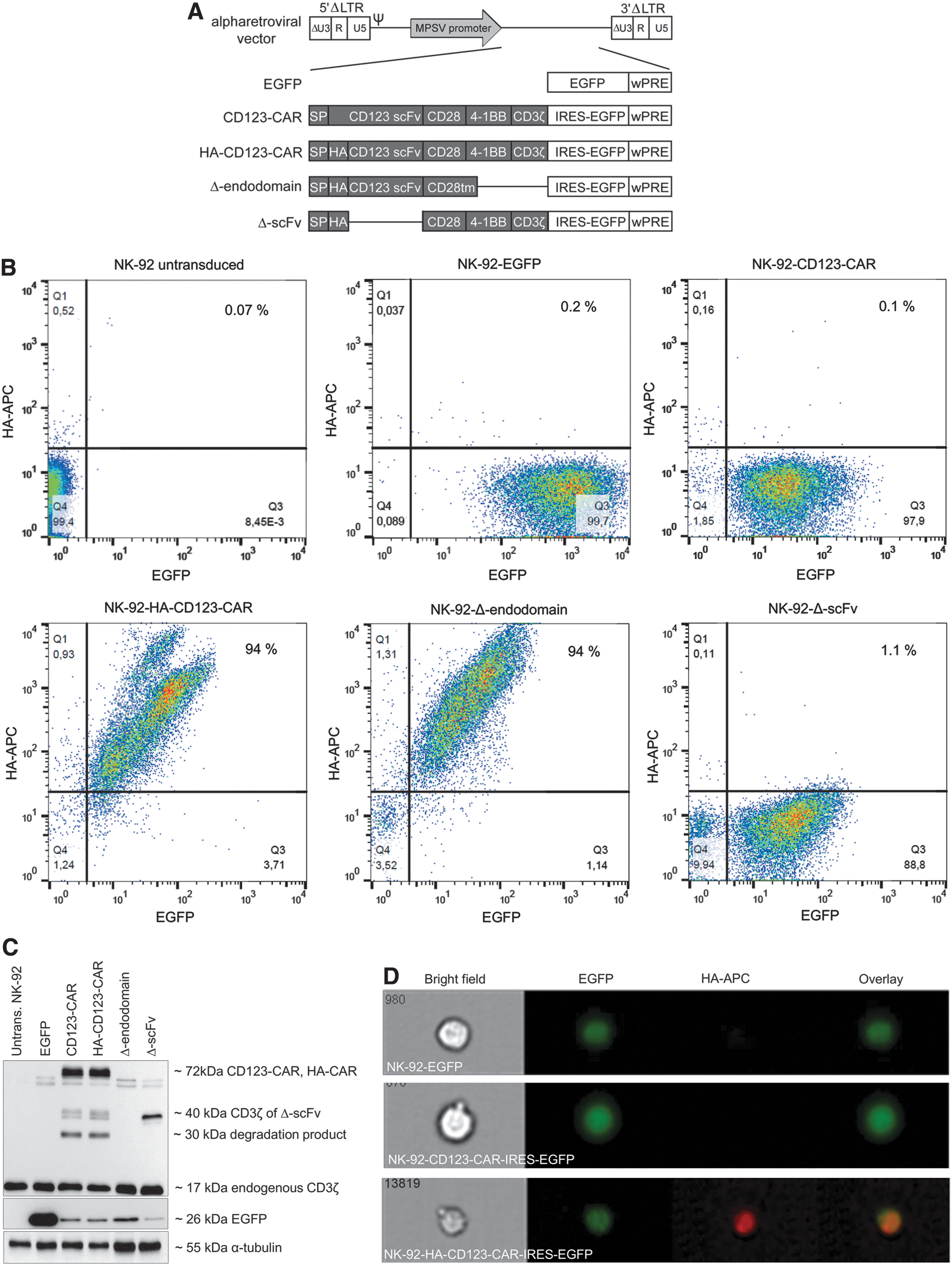
Alpharetroviral vector supernatant production and NK cell transduction
Alpharetroviral vector supernatants were produced by calcium phosphate transfection of 293T cells with 5 μg of the vector, 2.5 μg of the codon-optimized alpharetroviral gag/pol, and 2 μg of the RD114/TR envelope (expression plasmid kindly provided by Dr. F.L. Cosset, Lyon, France) plasmids per 10 cm cell culture dish (TPP, Trasadingen, Switzerland). Viral supernatants were collected 36 and 48 h after transfection, filtered through Millex-GP 0.22 μm filters (Millipore, Schwalbach, Germany), concentrated by ultracentrifugation, and stored at −80°C. Transduction of HT1080 fibroblasts by spinoculation (1 h, 400 g, ∼35°C) in the presence of 4 μg/mL protamine sulfate and different volumes of viral vector supernatants was accomplished to determine viral vector titers. NK cells were transduced with known multiplicities of infection (MOI) as described. 19 Transduced NK cells were enriched by flow cytometric sorting, and sorted cells with similar CAR expression were used for all experiments.
Immunoblot
Cell extracts were prepared, and immunoblotting was performed via standard techniques. Briefly, cell pellets were re-suspended in lysis buffer (50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl, 100 mM NaF, 1% Triton X-100, 1 mM Na3VO4, 1 mM dithiothreitol, and protease inhibitors [cOmplete Mini; Roche Diagnostics, Mannheim, Germany]) and incubated for 20 min on ice. Lysates were clarified by centrifugation (15 min, 4°C, 13,000 g), and supernatants were collected. Cellular protein concentrations were determined using the Coomassie dye-binding assay according to Bradford 20 (Bio-Rad Laboratories, Hercules, CA). Total cellular protein amounts were adjusted for equal loading in all experiments, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (GE Healthcare Life Science, Solingen, Germany). Membranes were blocked in Tris-buffered saline containing 5% (w/v) milk powder, probed with specific antibodies (anti-HA; 1:2,000; Cell Signaling Technology, Danvers, MA), anti-EGFP (1:2,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-CD3ζ-horseradish peroxidase (HRP; 1:1,000; Santa Cruz Biotechnology, Inc.), anti-tubulin (1:10,000; Sigma–Aldrich, St. Louis, MO). After incubation with the appropriate HRP-conjugated secondary antibodies (except for the anti-CD3ζ antibody, which is already coupled to HRP), proteins were detected by chemiluminescence (Amersham Biosciences, Little Chalfont, United Kingdom), and images were captured with the Fusion imaging system (Molecular Devices, Biberach, Germany).
FCM analysis
NK-92 cells were harvested, washed with PBS, and re-suspended in PBS at a cell density of 1 × 107 cells/mL. Cells were then stained (30 min at 4°C) with an anti-HA-APC antibody (mouse clone GG8-1F3.3.1; Miltenyi Biotec), washed with PBS, and analyzed on FlowSight (Merck, Darmstadt, Germany) to detect the HA-tagged anti-CD123-CAR localized to the cell membrane.
Target and effector cells were stained with monoclonal antibodies (mABs) for specific surface markers and subsequently analyzed on a Navios FCM (Beckman Coulter) in a no-wash, single-platform procedure using Flow-Count® fluorospheres (Beckman Coulter), as described by Kloess et al. 16 Dead cells were excluded by 7-AAD staining. The cell phenotypes were assessed with the following mABs: NKG2D (NK group 2 member D; CD314)-PE (phycoerythrin [PE]), NKp30 (CD337)-PE, NKp44 (CD336)-PE, NKp46 (CD335)-PC7 (phycoerythrin-cyanin-7), CD69-ECD (phycoerythrin-Texas Red®-X), CD137-APC (allophycocyanin), CD178 (Fas ligand; FasL)-APC, CD253 (tumor necrosis factor [TNF]-related apoptosis inducing ligand [TRAIL])-APC, CD3-PB (Pacific Blue), CD19-FITC (fluorescein isothiocyanate), CD14-ECD, CD56-PC7, CD16-APC or -PB, CD107a-APC, CD3-PB, and CD45-KO (Krome Orange). All antibodies were purchased from Beckman Coulter, except CD137-APC, CD178-APC, and CD253-APC (BD Biosciences).
CD107a degranulation assay
Primary NK and NK-92 cells were co-incubated with K562 (effector/target [E/T] ratios of 0.5:1, 1:1, and 5:1 as an internal control), or KG-1a and primary AML blasts (E/T of 0.5:1 and 1:1) in the presence of CD107a (PE) antibody (Beckman Coulter) for 1 h at 37°C and 5% CO2. Monensin and GolgiPlug (both 1:1,000; BD Biosciences) were then added. After a total incubation time of 5 h, cells were washed in PBS, stained, and CD107a expression was quantified by FCM.
Cytotoxicity assay
The cytotoxicity of dNK or NK-92 cells was assessed against K562 cells for 4 h as an internal control for effector-based killing activity at E/T ratios of 0.5:1, 1:1, and 5:1 (data not shown). Specific killing effects of sorted, non-modified EGFP, and anti-CD123 CAR dNK or NK-92 cells against KG-1a or native leukemic blasts for up to 24 h were assayed at an E/T ratio of 5:1. Co-cultivations of effector cells and target cells were performed in NK MACS medium containing 1,000 IU/mL IL-2 and supplemented with 5% HSA. Detection of viable effector and target cells after cytotoxic interaction was based on a no-wash, single-platform FCM (Navios; Beckman Coulter), including Flow-Count® fluorospheres, and depended on surface expression of specific antigens such as CD16-APC or -PB, CD56-PC7 or -APC, and CD45-KO for NK cells, CD15-FITC for K562, and CD34-PC7 and CD123-APC for KG-1a and native blasts. Dead cells were excluded by 7-AAD staining. All mABs were obtained from Beckman Coulter. Lytic activity and viability during the cytotoxic reactions of the different effector cells was calculated as described by Klöß et al. 17
Side-effect experiments in response to sorted, non-modified CD123-CAR- or EGFP-expressing dNK or NK-92 cells, respectively, were assessed by co-incubation (E/T ratio: 1:1 under the same culture conditions; see above) with peripheral blood mononuclear cells (PBMCs), human lung microvascular endothelial cells (HLMVECs; PELO Biotech, Planegg, Germany) or human primary lung fibroblasts (HPLFs; Lonza, Basel, Switzerland). NK-induced effects against these normal cell types were characterized by 10-color FCM analysis and compared between modified, sorted CAR-expressing dNK and NK-92 cells.
For blocking experiments, sorted CD123-CAR-dNK and -NK-92 cells were preincubated with antigen CD123-polyHIS (1 μg/mL final concentration; 30 min at 4°C) before co-cultivation with KG-1a or primary blasts at an E/T ratio of 5:1. Target cells were preincubated for 30 min at room temperature with anti-CD123 monoclonal antibody (Becton Dickinson, Franklin Lakes, NJ). Experiments in which effector and target cells were both blocked are referred to as “double blocked.” FCM analysis started after incubation of 0–24 h using 7-AAD, CD34-PC7, CD56-APC, CD16-PB, and CD45-KO (all monoclonal antibodies purchased from Beckman Coulter).
Cytokine analysis
Cytokine production of NK cells was assessed in supernatants with the 13-plex flow assay kit LEGENDPLEX® Human CD8 Panel (BioLegend®). FCM-mediated measurements of soluble pro- and anti-inflammatory cytokines and pro-apoptotic markers such as TNF-α, interferon gamma (IFN-γ), IL-4, IL-6, IL-10, IL-17A, sFas, sFasL, perforin, granzyme A (GraA), granzyme B (GraB), and granulysin were done according to the manufacturer's instructions.
Time-lapse microscopy
Redirected and nonspecific E/T cell–cell contacts and cell cluster formations between target and effector cells of the AML cell line KG-1a and transduced primary human NK and NK-92 cells were monitored by fluorescence tracking microscopy using an IX81 microscope (Olympus, Tokyo, Japan). All transduced primary NK and NK-92 cells containing co-expressed EGFP (green emission) were additionally stained with monoclonal antibody CD56-PE (Miltenyi Biotec). KG-1a cells were labeled intracellularly with the cell proliferation dye eFluor® 450 (Affymetrix®; eBioscience, San Diego, CA). Transduced NK cells were co-incubated with KG-1a cells (E/T ratio of 5:1) on chamber slides at 37°C and 5% CO2 for up to 8 h after starting time-lapse recording. Time-lapse imaging experiments were accomplished to document redirected cell migrations, specific E/T interactions, and brief, insignificant effector-mediated scanning of target cells using newly designed tracking protocols, which also allowed evaluation of all recorded images (every 30 s) containing specific E/T and E/E cell contacts and cluster formations by quantitative analyses using the Olympus scanR automated image and data analysis.
Statistical analyses
GraphPad Prism v6.05 (GraphPad Software, Inc.) was used to analyze and compare effector-mediated properties between primary dNK cells and the NK-92 cell line. In detail, significant differences for effector cell–mediated killing activity, CD107a degranulation, and cytokine concentrations and secretions (fg/cell) were statistically evaluated using an unpaired t-test with Welch's correction for unequal variances. In this regard, differences in results of four independent experiments each accomplished in duplicate are indicated as the mean ± standard deviation (SD) and were considered significant when p ≤ 0.05, while p-values ≥0.05 were considered nonsignificant (n.s.). Moreover, results of effector cell viability, transduction frequencies, effector cell–target cell interactions/cluster formation, and surface expression of specified receptors on effector and target cells were statistically evaluated and are indicated as percentile medians with ranges.
Results
CAR vectors and expression in NK-92 and primary donor NK cells
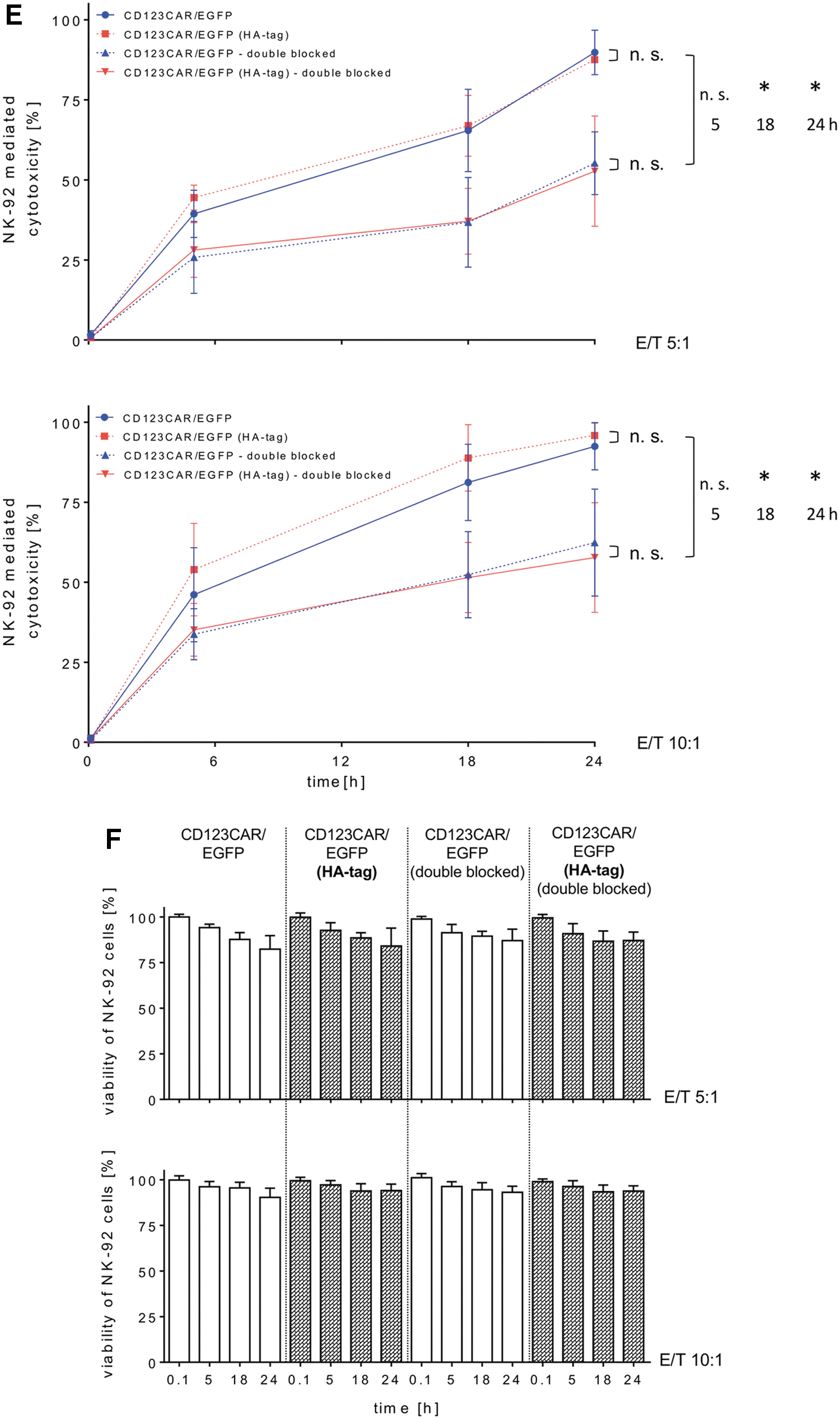
The alpharetroviral vectors shown in Fig. 1A were used to generate RD114/TR-pseudotyped viral vector particles, which were titered on HT1080 cells and then used to modify the NK-92 cell line. After transduction, vector modified NK-92 cells were enriched by flow cytometric sorting for EGFP expression (96–99% purities; Fig. 1B). In addition to EGFP expression, the HA-tagged CD123-CAR expression was also stable over several months, as demonstrated by FCM after staining with an antibody to detect the HA tag (Fig. 1B, and data not shown). The Δ-endodomain-NK-92 cells, which lack the signaling domain that drives CAR-induced cytotoxicity, were similarly detected by FCM for EGFP and HA-tag expression, while the Δ-scFv-NK-92 cells, which lack the scFv, were detected by FCM for EGFP but not HA-tag expression. This may be due to sequestration of the HA-tag by the cell membrane, as immunoblotting experiments using an antibody to detect the CD3ζ domain that is expressed in each of the CAR constructs, except for the Δ-endodomain construct, demonstrated expression as expected. A signal specific for the full-length CD123-CAR was detected at an apparent mass of 72 kDa in whole-cell lysates from both HA-CAR- and non-tagged CAR-expressing NK-92 cells, and the Δ-scFv construct was detected at approximately 40 kDa (Fig. 1C). Unspecific double bands just below the CAR-specific signal were also detected in all samples, including unmodified NK-92 cells as well as HT-1080 cells (Fig. 1C, and data not shown). The signals at approximately 30 kDa that were observed in lysates from the two full-length CAR constructs and the Δ-scFv construct are presumably degradation products. As expected, the anti-CD3ζ antibody also detected a 17 kDa signal that corresponds to endogenous CD3ζ in all NK-92 lysates, including untransduced controls (Fig. 1C). The EGFP marker protein was detected in all transduced but not untransduced control NK-92 cells, and an anti-α-Tubulin antibody demonstrated protein loading in each lane (Fig. 1C). FlowSight analyses using an anti-HA antibody demonstrated localization of the HA-CD123-CAR on the cell surface of HA-CD123-CAR-NK-92 cells but not on EGFP-NK-92 cells or CD123-CAR-NK-92 cells, indicating specific detection of HA-CD123-CAR-NK-92 cells and proper spatial orientation of the scFv (Fig. 1D). NK-92 cells modified with the CD123-CAR or HA-CD123-CAR exhibited similar cytotoxic activity against the CD123+ AML cell line KG-1a, indicating that the HA tag did not influence CAR-NK-92 mediated killing, which was tested at two different effector to target ratios (5:1 and 10:1) and measured at several time points over 24 h (Fig. 1E). Double-blocking CAR-NK-92 cell cytotoxicity was accomplished by blocking the scFv of CAR-NK-92 cells with a histidine-tagged IL3RA protein and blocking KG-1a cells with an anti-CD123 antibody. Selectivity of the CAR was demonstrated, as double blocking reduced the anti-leukemia activity of CAR-NK-92 cells as quantified by reduced elimination of KG-1a cells (Fig. 1E). The viability of CAR-NK-92 cells remained stable throughout the experiments at both E/T ratios tested (Fig. 1F). The CD123-CAR-induced specific cell killing was further supported by cytotoxicity experiments in which NK-92 cells were transduced with CAR constructs lacking either the ectodomain (antigen recognition) or the endodomain (cytotoxic signaling; Fig. 1A). Deletion of either of these domains eliminated the enhanced CD123-CAR-NK-92 cell cytotoxicity against AML cells (Supplementary Fig. S1).
In order to allow comparison of the antileukemia activity of CAR-NK-92 cells with primary CAR-NK cells, primary dNK cells were transduced with RD114-TR pseudotyped alpharetroviral vector constructs to express EGFP alone or the CD123-CAR, which also expresses EGFP via an IRES element (Fig. 1A). Flow cytometric sorting for EGFP+ cells resulted in enrichment of transduced dNK cell populations to >90% (median 94.2%, range 90.1–97.5%) for the EGFP control vector and >61% (median 61.7%, range 50.7–85.9%) for dNK transduced with the EGFP/CD123-CAR construct (Fig. 2). Direct staining for the scFv of the CD123-CAR showed even greater enrichment (median 92.7%, range 78.8–96.8%) of surface CD123-CAR transduced dNK, indicating that measurement of EGFP expression may lead to under estimation of dNK transduction levels when EGFP is expressed via an IRES element. The expression level of a second gene via an IRES element, like the EGFP in the CD123-CAR vectors, is often greatly reduced compared to the gene expressed directly following the promoter. 21
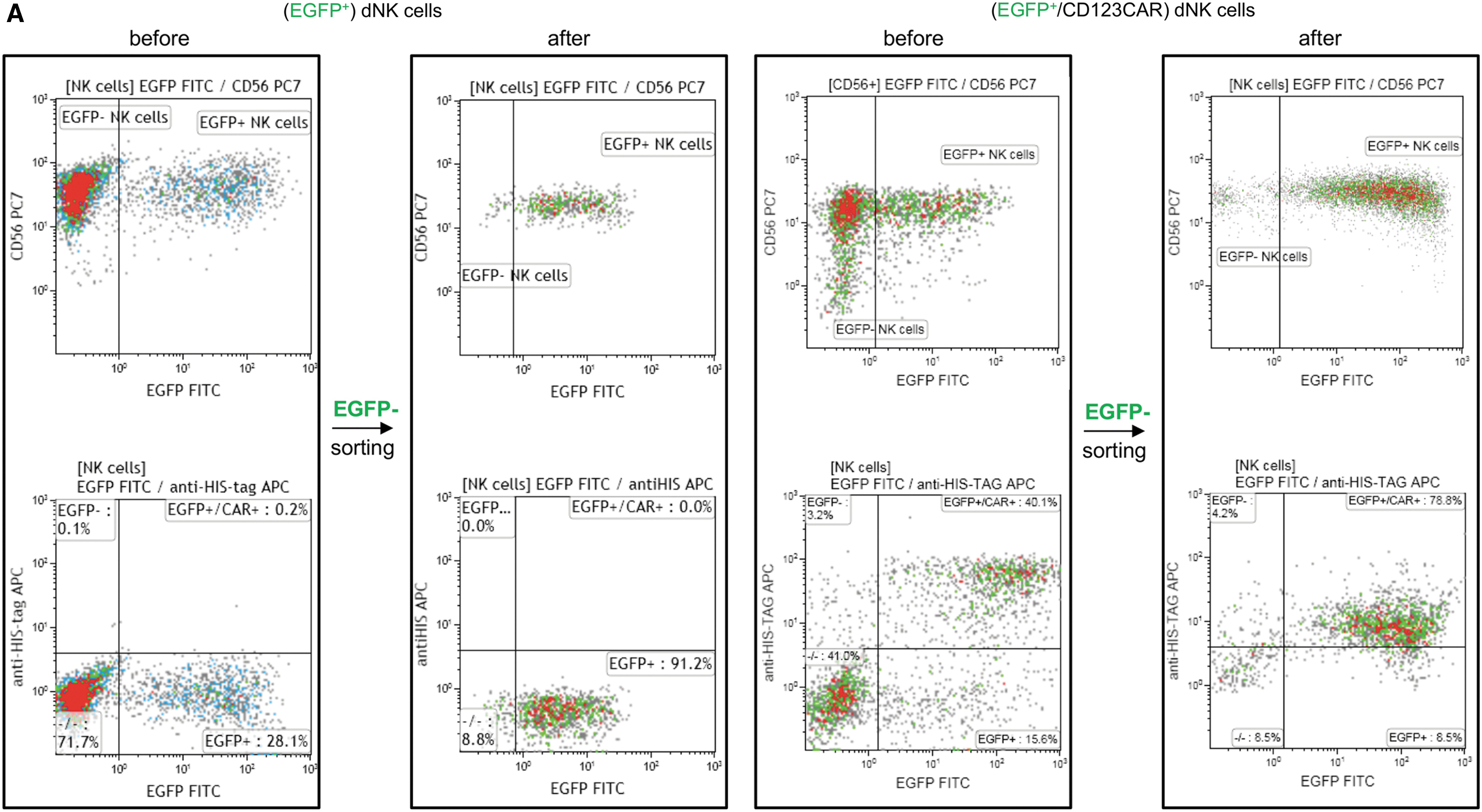
Enrichment of transduced primary NK cells by flow cytometric sorting. Primary donor NK (dNK) cells were sorted for EGFP expression 7 days after transduction with RD114/TR-pseudotyped alpharetroviral vectors designed to express EGFP alone or the CD123-CAR and EGFP via an IRES element.

CAR-NK cells exhibit improved anti-AML activity
After achieving enriched populations of CD123-CAR-NK-92 and CD123-CAR-dNK cells by sorting, the CAR-NK cells were compared for anti-leukemia activity using the CD123+ AML cell line KG-1a. At an E/T ratio of 5:1, both CAR-NK-92 and CAR-dNK cells significantly outperformed control NK cells at every time point examined (Fig. 3A). For example, after 18 h co-culture, unmodified NK or EGFP-expressing NK cells caused an approximately 30% reduction of KG-1a AML cells, while CAR-NK-92 and CAR-dNK cells eliminated >80% (median 88.0%, range 76.7–92.5%) and 60% (median 64.0%, range 59.7–70.5%) of AML cells, respectively. The specificity of the CAR-NK-92 and CAR-dNK cells was demonstrated by the loss of antileukemia activity upon performing double blocking in which effector cells were blocked by pre-incubation with a histidine-tagged complete CD123 protein to bind the CD123-CAR-scFv and the CD123 antigen on AML target cells was specifically blocked by addition of anti-CD123 antibody prior to the co-culture experiments (Fig. 3A).

Comparison of CAR-NK-92 and CAR-dNK cell cytotoxicity.
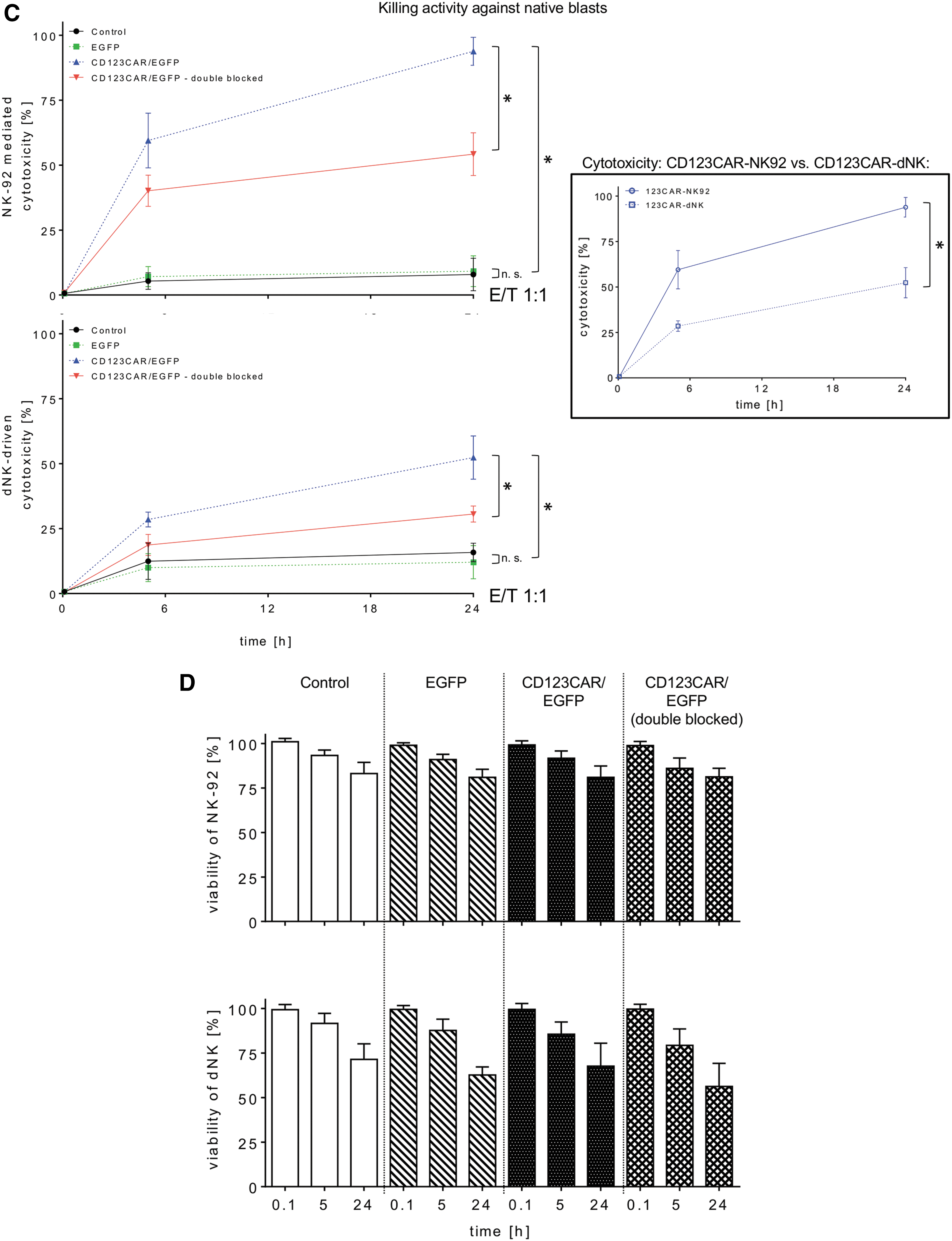

Unmodified and EGFP-modified primary dNK cells exhibited slightly more antileukemia activity than the respective NK-92 cells, but CAR-NK-92 cells were significantly better at eliminating KG-1a AML cells compared to CAR-dNK cells (Fig. 3A). While the viability of NK-92 cells remained relatively constant over the time course of these experiments, regardless of whether they were modified, the viability of primary dNK cells decreased significantly (Fig. 3B).
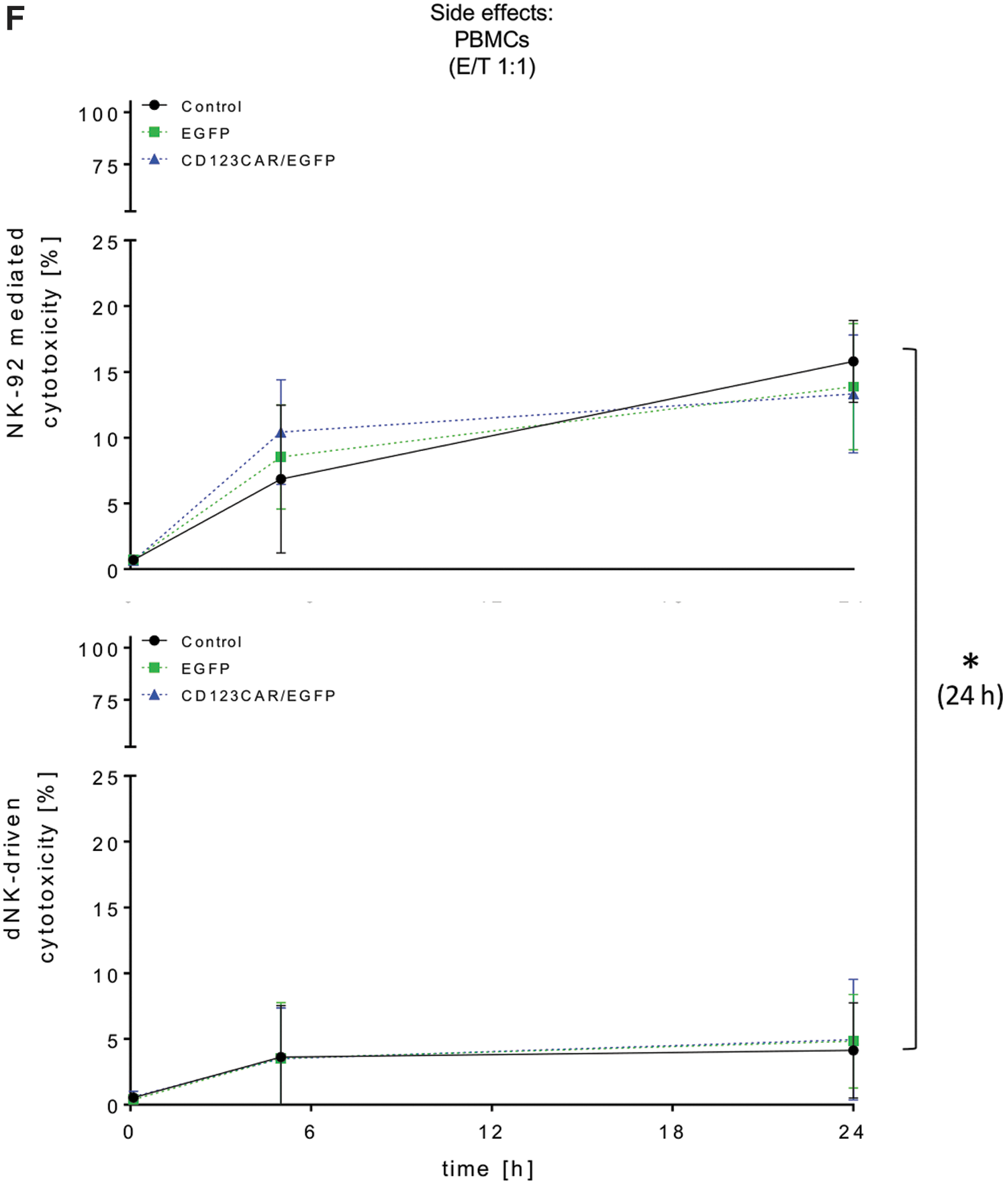
To test the activity of CAR-NK cells against primary AML blasts, samples from three different patients were tested for CD123 surface expression and were 99%, 84%, and 100% CD123+, respectively (Supplementary Fig. S2). CAR-NK-92 and CAR-dNK cells exhibited significantly greater elimination of primary AML cells versus unmodified or EGFP-modified NK cells after 24 h (Fig. 3C). As before, the specificity of the CAR-NK cell activity was shown as anti-CD123 antibody decreased elimination of AML cells, although not completely to levels determined for unmodified or EGFP-expressing control NK cells. Also in this setting, viability measurements showed that NK-92 cells were more stable than primary dNK cells, especially at later time points of co-culture with primary AML blasts (Fig. 3D). As additional evidence of improved target cell recognition, CAR-modified NK-92 (median 41.2%, range 27.5–57.8%) and primary human dNK cells (median 34.8%, range 21.4–39.7%) more efficiently formed E/T cell clusters compared to EGFP-modified NK cells (EGFP-NK-92: median 11.3%, range 1.3–15.6%; EGFP-dNK: median 8.1%, range 5.2–13.4%; Fig. 3E). Furthermore, CAR-NK cells did not exhibit greater nonspecific toxicity than unmodified or EGFP-expressing control NK cells as assessed in co-culture experiments with human PBMCs, HLMVECs, and HPLFs (Fig. 3F and Supplementary Fig. S3). Interestingly, primary dNK cells exhibited significantly lower nonspecific cytotoxicity than NK-92 cells (Fig. 3F and Supplementary Fig. S3). For example, CAR-NK-92 cells co-cultured with PBMCs at a ratio of 1:1 for 24 h exhibited more than twice the nonspecific toxicity compared to CAR-dNK cells under the same co-culture conditions (median 13%, range 8.9–15.7% vs. median 4.9%, range 0.2–9.9%; Fig. 3F).
Increased degranulation and cytokine secretion of CAR-NK cells
CD107a expression is a sensitive marker for NK cell activation and degranulation. 22 As for all other experiments, CAR-NK-92 and CAR-dNK cell populations were enriched by sorting for EGFP expression, and the purity of CAR-expressing cells was similar for CAR-NK-92 and CAR-dNK cells (Fig. 4A). Compared to EGFP-modified NK cells, CAR-NK-92 cells and primary CAR-dNK cells that were incubated with either KG-1a cells or primary human AML cells at two different E/T ratios (i.e., 0.5:1 and 1:1) for 5 h expressed higher levels of CD107a as determined by FCM (Fig. 4B and C). While CAR-NK-92 cells showed a tendency for attenuated increase of surface CD107a compared to higher expression levels on CAR-dNK cells upon co-culture with AML cells, this was not significant (Fig. 4B and C). The observation of reduced CD107a expression on CAR-NK cells co-cultured with AML target cells at an E/T ratio of 1:1 compared to 0.5:1 is consistent with previous studies that showed degranulation to be influenced by E/T ratios. 23,24 Analysis of supernatants from co-culture experiments containing EGFP-modified NK cells or CAR-NK cells and primary AML cells at an E/T ratio of 1:1 demonstrated significantly increased IL-6 secretion by CAR-NK-92 cells and significantly increased secretion of GraB, granulysin, and IL-6 secretion by CAR-dNK cells (Fig. 4D). Direct comparison of CAR-NK-92 and CAR-dNK cells showed higher GraA and IL-17A by CAR-NK-92 cells, while CAR-dNK cells secreted more TNF-α, IFN-γ, and granulysin (Fig. 4D).

Comparison of CAR-NK-92 and CAR-dNK degranulation and cytokine secretion.

CAR-NK cells exhibit increased interactions with AML target cells
Fluorescence-based microscopic tracking was used to monitor and assess the quality of specific interactions between CAR-NK-92 cells, CAR-dNK cells, and KG-1a cells in a time-dependent manner. As shown in time-lapse recorded images (every 30 s), green fluorescent CAR-NK cells made multiple contacts with KG-1a target cells, as marked with the dotted red circles (Fig. 5A). These interactions led to productive killing of KG-1a cells, as evidenced by blebbing of KG-1a cells. In contrast, EGFP-NK-92 cells did not interact specifically with KG-1a cells as often (Fig. 5B), which is consistent with the lower antileukemia effects observed in the cytotoxicity assays described above. Similarly, CAR-dNK cells also interacted more often and for a longer time period with KG-1a cells than EGFP-dNK cells did, and a greater amount of KG-1a blebbing was observed upon CAR-dNK contact with KG-1a cells (Fig. 5C and D; Supplementary Videos S1 and S2).

Fluorescence-based microscopic tracking to visualize the retargeted cytotoxicity of CAR-NK cells. KG-1a cells were intracellularly labeled with eFluor® 450 (blue target cells), while modified NK cells are green due to EGFP or CAR-EGFP expression. EGFP-NK and CAR-NK cells were co-cultured with KG-1a cells at an E/T ratio of 5:1, and retargeted or nonspecific cell contacts (time of selected cytolytic E/T interactions) were monitored for 8 h by time-lapse microscopy and analyzed using the Olympus scanR automated image and data analysis software. KG-1a cells were co-cultured with
Discussion
The concept of “off-the-shelf” cell therapy is an emerging field, and CAR-NK-92 cells are interesting candidates to drive further development of this important idea to generate improved cancer immunotherapy approaches. 3,25 Indeed, CAR-NK-92 cells have been tested in several Phase I/II clinical trials to target MUC1+ solid tumors (NCT02839954), CD19+ leukemia and lymphomas (NCT02892695), CD7+ leukemia and lymphoma (NCT02742727), and CD33+ AML (NCT02944162). 26 The three patients treated with CD33-CAR-NK-92 cells had relapsed and refractory AML. While no clinical benefit was shown in these patients with difficult-to-treat disease, safety and feasibility to deliver up to 5 × 109 irradiated CAR-NK-92 cells per infusion was demonstrated. 26 In a similar context, CAR-NK-92 (NK-92/5.28.z) cells are currently being deployed against ErbB2+ (HER2+) glioblastomas within a Phase I/II clinical study (NCT03383978) evaluating the safety, tolerability, and maximum tolerable/feasible doses. It was possible to expand NK-92/5.28.z cells to approximately 5 × 109 irradiated effector cells in a very short period of time. Similar to the present functional studies, modified NK-92/5.28.z cells also exhibited excellent viability, but a cytotoxic effect against ErbB2- malignant cell lines was also shown, indicating an elevated risk of off-target cytotoxic activity. 27 After contact with an ErbB2+ malignant cell line, modified NK-92/5.28.z cells secreted increased levels of GraB, IFN-γ, sFasL, IL-8, and IL-10, but release of IL-6 by these CAR-NK cells was not detected. 27 In contrast, IL-6 was the only cytokine that CD123-CAR-NK-92 cells were observed to secrete at significantly higher levels compared to EGFP-modified NK-92 cells.
NK cells can also be purified from PBMCs, and PBMC-derived CAR-NK cells targeting CD19+ B-cell acute lymphoblastic leukemia were evaluated in Phase I (NCT03056339) and Phase II (NCT01974479) clinical trials. In addition, optimization of the previously established X-VIVO 10/IL-2–based NK cell expansion protocol for unmodified dNK cells applied in an earlier clinical Phase I/II study resulted in a fully automated single process in a closed system using the CliniMACS Prodigy. 16,17,28
Cord blood–derived NK cells were used to generate CAR-NK cells that target CD19+ B-lymphoid malignancies and were tested in a Phase I/II study (NCT03056339). This CD19-CAR construct also encoded an IL-15 expression cassette for improved CAR-NK in vivo persistence and an inducible Caspase 9 as a safety feature to allow pharmacological removal of CAR-NK cells in the case of serious adverse events. CAR-NK cells derived from human induced pluripotent stem cells (iPSCs) were also recently demonstrated to exhibit similar antitumor activity as CAR-T cells in an ovarian cancer xenograft model but with lower systemic toxicity effects such as cytokine release syndrome. 29 Advantages of iPSC-derived CAR-NK cells and CAR-NK-92 cells include the possibility of generating enough CAR-NK cells to allow multiple infusions of high cell numbers into patients, the relatively homogenous inter- and intra-batch cell product, cost-effectiveness, and the opportunity to introduce additional genetic modifications that may improve in vivo CAR-NK cell activity. 27,30
As almost complete elimination of KG-1a and primary AML cells by CD123-CAR-NK-92 cells was observed, the data support continued investigation of the efficacy of CAR-NK-92 cells in AML treatment. Interestingly, CD123-CAR-dNK cells generated from primary human NK cells were significantly less effective in the eradication of AML cells. This may be a consequence of the decreased viability of CD123-CAR-dNK cells compared to CD123-CAR-NK-92 cells that was observed during the co-culture experiments. The slightly lower surface expression of the anti-CD123-CAR on sorted dNK cells may also have contributed to the decreased cytotoxic specificity toward CD123+ target cells. This was also evidenced by the marginally lower cell cluster formation by the CAR-dNK cells compared to the stronger cell–cell contacts observed by CAR-NK-92 cells interacting with KG-1a cells (Fig. 3E). Similarly, dNK cells exhibited significantly less nonspecific toxicity to non-target cells than NK-92 cells. This may be explained by the highly cytotoxic nature of NK-92 cells versus the more heterogeneous composition of primary dNK cells, which should contain an immune regulatory NK cell subpopulation as well. This was also observed by others who reported that anti-ErbB2-CAR (NK-92/5.28z) elicited similar potential side effects and toxicities against normal healthy and malignant ErbB2-negative target cells. 27,31 As the purity of the sorted CD123-CAR-NK-92 cells and CAR-dNK cells was similar, the differences in activity are likely due to distinct properties of the two types of NK cells.
In the cytokine secretion analyses, IL-17A was one of the most highly expressed cytokines detected after 24 h of contact of NK-92 or dNK cells with primary AML blasts (Fig. 4D). The observation of high levels of IL-17A secretion in CAR-NK-92/AML co-cultures corresponds well with the finding that two of three AML patients treated with CD33-CAR-NK-92 cells also had elevated IL-17A in their serum. 26 The results are in contrast with those of Nowakoska et al. who reported that CAR-NK-92 cells had elevated secretion profiles of GraB, IFN-γ, sFasL, and IL-10. 27 Interestingly, significantly increased secretions of granulysin by CD123-CAR-dNK cells compared to lower levels from EGFP+ dNK cells was detected during cytotoxic interaction against CD123+ target cells, which was not observed in cytotoxic assays against KG1a in previous studies. This may be due to differences between the current and earlier studies, for example the inclusion of IL-2 in the cytotoxicity assays in the current study. 17
In summary, the present results support the feasibility of NK cell redirection with molecules designed to target AML-specific antigens as a potential adoptive therapeutic approach to resolve AML. While both CD123-CAR-NK-92 cells and CD123-CAR-dNK cells significantly enhanced the antileukemia activity compared to unmodified or EGFP-modified NK cells, the CD123-CAR-NK-92 cells exhibited significantly higher activity against AML cells. However, CAR-dNK cells displayed significantly lower levels of cytotoxicity against cells from healthy tissues compared to CAR-NK-92 cells. Thus, to determine the optimal NK cell source, the advantages of increased anticancer activity have to be carefully evaluated against the risk of off-cancer toxicity. Advantages of using NK-92 cells may include the easier cultivation and expansion compared to dNK cells. Genetic optimization of NK-92 cells with gene-editing technologies such as CRISPR-Cas9 is also expected to be more efficient compared to gene deletion, addition, or correction in primary dNK cells. This may be an important point, as genome editing to prevent rejection of allogeneic cell line derived NK-CAR may be necessary for longer-term engraftment. Alternatively, the NK-92 cell line approach could be useful for repeat infusions in the context of lymphodepletion, with graft rejection being expected following immune recovery. However, as expansion and modification protocols for primary activated dNK cells continue to improve, the decreased off-cancer activity of dNK cells in particular makes them a good alternative NK source for future NK cell-based cell therapies. Additional factors that should be considered to help select the appropriate NK cell source to use for development of patient-individualized cell therapies include the type, location, and condition of the recurrently resistant tumor or leukemia cells. In addition to cell therapies using single cell types, one could also consider strategies to deliver multiple lymphocyte populations, such as NK, NK-like T, and T-cell subpopulations to induce a synergistic antitumor effect (e.g., via cell cross-talk through cytokine exchange).
Footnotes
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB738, Cluster of Excellence REBIRTH [EXC 62/1]), the Bundesministerium für Bildung und Forschung (BMBF, Joint Research Project IFB-Tx, PID-NET), From CARs to TRUCKs (Krebshilfe-Priority Program in Translational Oncology), and the European Union (FP7 projects PERSIST, CELLPID, and Horizon 2020 project SCIDNET). The authors thank D. Lenz and J. Wenzl for excellent technical support and Dr. F.L. Cosset (Lyon, France) for providing the expression plasimid for the RD114/TR envelope. We would like to acknowledge the assistance of the Cell Sorting Core Facility of the Hannover Medical School supported in part by Braukmann-Wittenberg-Herz-Stiftung and Deutsche Forschungsgemeinschaft.
Author Disclosure
The authors declare that they have no conflict of interest, except that A.S. is co-inventor on a patent application describing alpharetroviral SIN vectors.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Video S1
Supplementary Video S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.