
Abstract
Existing adenoviral vector systems have two drawbacks. It is labor-intensive and time-consuming to load a transgene in these systems, and transgene-harboring vectors are dead ends: they cannot be reused to construct a vector carrying another transgene or achieving new characteristics. To conquer these shortcomings, single plasmid-based adenoviral vector systems were constructed where a unique PmeI site was located at the position for insertion of the exogenous gene. The polymerase chain reaction (PCR) amplified transgene could be cloned into PmeI-linearized starting plasmids using one step of Gibson assembly to generate target adenoviral plasmids, which were then ready for virus rescue. This procedure was termed restriction assembly. To expand the application of these systems, two ClaI sites were created upstream and downstream of the fiber gene to generate an upgraded starting plasmid pKAd5f11pABR-EPG. The modified fiber gene, amplified by overlap extension PCR, could be used to substitute the original fiber in pKAd5f11pABR-EPG to generate an adenoviral plasmid with a new fiber by restriction assembly. On the other hand, pKAd5f11pABR-EPG was also a starting adenoviral plasmid for expressing other transgenes. In conclusion, easy-to-use and upgradable adenoviral vector systems are introduced here, which offer extensive versatility and can serve as a basic platform and functional component library for the synthetic biology of adenoviral vectors.
Introduction
Adenoviruses are non-enveloped icosahedral particles and contain a linear, double-stranded DNA genome 26–46 kb in length. According to the host range in vertebrates and organization of the genome, adenoviruses are classified into five distinct genera: Mastadenovirus, Aviadenovirus, Siadenovirus, Atadenovirus, and Ichtadenovirus. 1 Some of these have been reconstructed into gene delivery vehicles. Adenoviral vectors are widely applied in gene therapy, recombinant vaccine fields, as well as in basic research due to their outstanding biological properties, such as effective gene delivery and expression, the relative ease of manipulation, the ability to produce high titers, and their biosafety as non-integrating viruses. For these reasons, great efforts are being made to construct new adenovirus vectors or modify existed ones. 2 –4
Many approaches have been developed for generating adenoviral vectors. 5 In the early days, the linear viral genome was directly extracted from a cultured virus, digested with restriction enzyme, and used to construct recombinant adenovirus using the in vitro ligation method. After that, virus genomic DNA was integrated into a plasmid and could grow in bacteria culture. Homologous recombination between a shuttle plasmid, which mainly carries transgene, and a backbone plasmid, which contains most portion of viral genome, can be carried out to generate a recombinant virus in eukaryotic packaging cells. The efficiency can be further improved if homologous recombination takes place in bacteria; and tedious plaque purification can be avoided because adenoviral plasmids generated in bacterial colonies are homologous. The method of homologous recombination in bacteria has been very successful since it was established two decades ago, and it is still globally popular today. 6 –8 Recently, new methods, such as direct ligation after digestion with rare-cutter restriction enzyme (especially homing endonuclease), 9 site-specific recombination, bacterial artificial chromosome–based recombination, and even DNA assembly, have been used to construct adenoviral vectors. 10 –12
Several DNA assembly methods are used across the synthetic biology community, among which the most used method is isothermal or Gibson assembly (after Daniel Gibson who developed the technique). 13 –15 In a Gibson assembly reaction, T5 exonuclease, thermostable high-fidelity DNA polymerase, and Taq DNA ligase are combined in a cocktail with overlapping fragments of double-stranded DNA. Exonuclease chews back from the 5′ end to create 3′ overhangs, allowing the single-stranded homologous ends to anneal. The DNA polymerase fills in the gaps within each annealed fragment, and finally the ligase seals nicks in the assembled DNA. Gibson assembly is a sequence-independent seamless DNA assembly method and can ligate multiple fragments to form a plasmid in one step.
Although many methods are available, constructing adenoviral vectors is still highly skilled and tedious work for general molecular biology laboratories. By modifying a previous HAdV-5-based vector system, this study attempted to establish adenoviral vector systems that work under a method called restriction assembly. This method is much simpler and easier to use by combining restriction digestion and the Gibson assembly technique.
Methods
Plasmids and cells
The plasmids pAdEasyf11p, pFiber5-11p, and pAd5GXP were constructed previously. pAdEasyf11p was a fiber-modified adenoviral backbone plasmid carrying the fusion gene of HAdV-5 and HAdV-11p fibers (F5-11p). pFiber5-11p was a plasmid formed by self-ligation of the EcoRI-digested fragment of pAdEasyf11p (the short one carrying the F5-11p fusion gene). 16 pAd5GXP was a green fluorescent protein (GFP)-harboring E1/E3-deleted adenoviral plasmid, with the original PmeI site in the pIIIa gene being removed by site-directed mutation (there is no PmeI site in pAd5GXP). 17 Plasmids pLVX-TRE3G and pLVX-EF1a-Tet3G were purchased from Clontech Laboratories (Mountain View, CA). Plasmids pAdEasy-1, pShuttle, and pShuttle-CMV were purchased from Stratagene (La Jolla, CA).
A549 (ATCC CCL-185) and 293 (ATCC CRL-1573) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS; HyClone, Logan, UT) at 37°C in a humidified atmosphere supplemented with 5% CO2 and passaged twice a week. For virus rescue or amplification, confluent cells were split at a ratio of 1:2 or 1:3. When the cells reached 70–90% confluence, transfection or infection was performed. After incubation for the indicated period, the culture medium was replaced with fresh DMEM containing 2% FBS.
Construction of the starting adenoviral plasmid preloaded with human EF1a promoter
Information on polymerase chain reaction (PCR) primers, templates, and size of PCR products for plasmid construction is summarized in Supplementary Table S1. First, cytomegalovirus (CMV) promotor in the pShuttle-CMV plasmid was replaced with human EF1a promoter to generate the pSh5-EF1a shuttle plasmid, and the GFP coding sequence (CDS) was inserted into the multiple cloning sites (MCS) to generate the plasmid pSh5-EF1aGFP (Supplementary Fig. S1). Second, the homogenous recombination method was carried out by combining the backbone plasmid pAdEasyf11p and PmeI-linearized pSh5-EF1aGFP to generate the adenoviral plasmid pAd5f11p-EF1aGFP according to the instructions of the manufacturer of the AdEasy system; the fusion gene F5-11p in pAd5f11p-EF1aGFP was exchanged to the PmeI site-mutated adenoviral plasmid pAd5GXP to generate the adenoviral plasmid pAd5f11p-CGXP (Supplementary Fig. S2). In pAd5f11p-CGXP, features of mutation of the original PmeI site and F5-11p fusion gene were combined. Third, the fragment between the two BstZ17I sites in pAd5f11p-EF1aGFP (the short one that contained the GFP expression cassette) was fused with a short fragment (containing DNA sequence outside of the two BstZ17I sites in pAd5f11p-EF1aGFP) to generate pKAd5-EG-BstZ17I, and GFP CDS in pKAd5-EG-BstZ17I was replaced with a PmeI site (GTTTAAAC) to generate the pKAd5-EF1aBP plasmid (Supplementary Fig. S3). Finally, the fragment between the two BstZ17I sites in pAd5f11p-CGXP (the one short that contained the GFP expression cassette) was replaced with the corresponding fragment in pKAd5-EF1aBP to generate the adenoviral plasmid pKAd5f11p-EF1aP (Supplementary Fig. S4).
Construction of the starting adenoviral plasmid for carrying gene expression cassettes
EF1a promoter and GFP CDS were both deleted, and a PmeI site was added in pKAd5-EG-BstZ17I to generate the plasmid pKAd5ES-BP. Then, the fragment between the two BstZ17I sites in pAd5f11p-CGXP (the short one that contained the GFP expression cassette) was replaced with the corresponding fragment in pKAd5ES-BP to generate the adenoviral plasmid pKAd5f11pES-PmeI (Supplementary Fig. S5).
Combination of the Tet-On 3G inducible gene expression system into a single plasmid
The Tet-On 3G CDS and woodchuck hepatitis virus posttranscriptional regulatory element in pLVX-EF1a-Tet3G were linked together by overlap extension PCR with the primers 1306TetonP1–1306TetonP4 (Supplementary Table S1), and the PCR product was inserted into the two PstI sites in pLVX-TRE3G to generate the plasmid pLVX-TRE3G-Teton by restriction ligation cloning. GFP CDS was amplified using pAd5GXP as the template with the primers 1306TET-GFPF1 and 1306TET-GFPR1 (Supplementary Table S1), and inserted into the BamHI/EcoRI sites in pLVX-TRE3G-Teton to generate pLVX-TRE3G-TetGFP (Supplementary Fig. S6). pLVX-TRE3G-TetGFP contained two gene expression cassettes and had all the elements needed for inducible expression of the GFP reporter gene.
Construction of the adenoviral plasmid carrying the transgene using the restriction assembly method
Overlaps between neighboring fragments are prerequisites for Gibson assembly. The term “overlap” is used to refer specifically to the part of the oligonucleotide in the primers or double-stranded DNA in DNA fragments needed for Gibson assembly. To insert a single transgene to the PmeI site in pKAd5f11p-EF1aP, primers with 5′ overhangs were designed and synthesized (Supplementary Table S1). GFP CDS were amplified with the primers 1805F11p–EF1PmeGFP1/2 using pAd5GXP as the template (Q5 High-Fidelity DNA Polymerase; cat. no. M0491S; New England Biolabs, Ipswich, MA), and the overhangs finally became overlaps at both ends of the PCR product that satisfied the following DNA assembly reaction. pKAd5f11p-EF1aP plasmid was digested with PmeI and recovered from agarose gel (Zymoclean Large Fragment DNA Recovery Kit; cat. no. D4045; Zymo Research, Irvine, CA). PCR product (PmeI-GFP) and PmeI-linearized pKAd5f11p-EF1aP were combined with the NEBuilder HiFi DNA Assembly Master Mix (cat. no. E2621; New England Biolabs), and the sample was incubated at 50°C for 60 min according to the manufacturer's instructions. Four microliters of the assembled product was used to transform 100 μL top 10 competent Escherichia coli cells. The cells were spread on kanamycin-containing LB agar plates and incubated at 35°C for 1 day. The positive colonies were picked and grown in liquid LB medium containing kanamycin at 35°C with shaking. Plasmids were extracted and analyzed with restriction enzymes. The generated plasmid was termed pKAd5f11p-EPG.
pKAd5f11pES-PmeI was a starting adenoviral plasmid for harboring more exogenous genes or regulation elements. Tet-On 3G and GFP expression cassettes were amplified with the primers 1805F11p-TGteton1 and 1805F11p-TGteton2 (Supplementary Table S1) using pLVX-TRE3G-TetGFP as the template, and the PCR product was fused into PmeI-linearized pKAd5f11pES-PmeI to generate the plasmid pKAd5f11p-TGFPT according to the same procedure for constructing pKAd5f11p-EPG.
Construction of the adenoviral plasmid carrying the transgene with the help of a shuttle plasmid
The DNA fragment containing two overlaps and MCS with each PacI site on both ends was artificial synthesized and used to replace the original MCS of pUC19 by Gibson assembly to generate the shuttle plasmid pUC19-PM. The linked Tet-On 3G and GFP expression cassettes were excised from the pLVX-TRE3G-TetGFP plasmid by ClaI/KpnI digestion and inserted into the ClaI/KpnI site in pUC19-PM to generate pUC19-PTGFPW. The gene expression cassettes were excised from pUC19-PTGFPW by PacI digestion and fused into the PmeI site in pKAd5f11pES-PmeI to generate pKAd5f11p-TGFPW by DNA assembly, as described above.
Construction of the starting adenoviral plasmid for fiber modification
The fragment between the SacI and MfeI restriction sites of pFiber5-11p was modified and amplified by overlap extension PCR with the primers 1808F511pABR1–1808F511pABR6 (Supplementary Table S1). One ClaI site (atcgat) was added into the remnant sequence of the deleted E3 region, another ClaI site was created by one nucleotide point mutation downstream of the F5-11p CDS, and RGD4C coding sequence (tgtgactgccgcggagactgtttctgc) was added into the region coding the AB loop of the HAdV-11p fiber knob. 18 –21 The PCR product was cut with SacI/MfeI and used to replace the original fragment in pFiber5-11p to generate the pF5-11pABR plasmid. pF5-11pABR was digested with EcoRI, treated with CIP to remove 5′-P, and used to replace the corresponding fragment in pAdEasyf11p to generate pAdEasyf11pABR. The fragment containing the modified fiber gene was excised from pAdEasyf11pABR by BamHI digestion and used to replace the corresponding part in pKAd5f11p-EPG to generate pKAd5f11pABR-EPG (Supplementary Fig. S7).
Construction of a shuttle plasmid to facilitate the modification of the fiber gene
The fragment between the SacI and MfeI restriction sites of pFiber5-11p was modified and amplified by overlap extension PCR with the primers 1808F511pABR1/2/3 and 1808F511pABR6. One ClaI site (atcgat) was added into the remnant sequence of the deleted E3 region, and another ClaI site was created one nucleotide point mutation downstream of the F5-11p CDS. The 1,818 bp PCR product was used as a template to amplify a 1,349 bp PCR product with the primers 1808F511p-EC1 and 1808F511p-EC2, and the 1,349 bp fragment was further fused into the HindIII/EcoRI-digested pMD18-T plasmid to generate pMD-F511pEC by DNA assembly (Supplementary Fig. S8).
Rescue and purification of recombinant adenovirus
The adenoviral plasmid was digested with PacI, and the linearized plasmid DNA was recovered by ethanol precipitation and used to transfect 293 cells (Lipofectamine 3000; Thermo Fisher Scientific, Waltham, MA). GFP focuses or plaques occurred 5–10 days post transfection, suggesting successful rescue of the recombinant virus. The seed virus was propagated in 293 cells, and the amplified virus was purified with traditional CsCl ultracentrifugation. The viral particle (vp) unit titer was determined by measuring the genomic DNA content of the purified virus, where 100 ng genomic DNA is equivalent to 3.0 × 109 vp, since a 32 kb genome has a molecular mass of 2.0 × 107. 22
Restriction analysis of virus genomic DNA
Virus genomic DNA was extracted using the modified Hirt procedure. 23 Briefly, 293 cells grown in a T75 flask (90% confluent) were infected with purified virus at a multiplicity of infection (MOI) of 100 vp/cell. The virus was removed after incubation for 4 h, and the culture medium was changed to fresh DMEM +2% FBS. When complete cytopathic effect (CPE) started to occur (about 48 h post infection), the culture medium was discarded, and the round-up cells were washed once with phosphate-buffered saline. The cells were lysed at 50°C for 30 min in 3 mL lysis buffer containing 25 mM Tris-Cl, 0.5 mM EDTA, 0.8% sodium dodecyl sulfate, and 50 μg/mL proteinase K (pH 7.6). Cellular debris and chromosomal DNA were precipitated by the addition of 1.1 mL solution containing 3 M CsCl, 1 M potassium acetate, and 0.67 M acetic acid. The tube was gently mixed and placed in ice for 20 min. After centrifugation for 25 min at 15,000 g at 4°C, the supernatant (approximately 3.3 mL) was transferred to a new tube, and 6.6 mL cold absolute ethanol was added. After gentle mixing, the tube was placed in ice for a further 5 min, and viral DNA was precipitated by spinning at 3,000 g for 5 min. After washing once with 70% ethanol, DNA was dissolved in 200 μL TE buffer (10 mM Tris-Cl, 0.5 mM EDTA; pH 7.6) containing 50 μg/mL RNaseA. DNA could be further cleaned with the genomic DNA clean and concentration kit (cat. no. D4010; Zymo Research) if necessary. Virus genomic DNA was digested with restriction enzymes and resolved on 0.6% agarose gel containing ethidium bromide by electrophoresis.
Transduction of A549 cells with recombinant adenovirus
A549 cells were seeded onto a 12-well plate. When the cells reached 70% confluency, they were infected with virus at the indicated MOIs. The virus was removed 4 h later, and fresh DMEM +2% FBS with or without 1 μg/mL doxycline (Dox) was added. The expression of GFP was observed under a fluorescence microscope 48 h post infection.
Results
Single plasmid-based adenoviral vector system for expression of the transgene
Using overlap extension PCR, restriction ligation cloning, homologous recombination in E. coli, and DNA assembly, an adenoviral plasmid termed pKAd5f11p-EF1aP was constructed with the following features: it contained a E1/E3-deleted HAdV-5 genome; the HAdV-5 fiber gene was replaced with the fusion gene HAdV-5 and HAdV-11p fibers (F5-11p); the original PmeI site was removed by mutating gtttaaaC to gtttaaaT without changing the amino acid sequence encoded by the pIIIa gene; regulatory elements, including human EF1a promoter and SV40 polyA signal, were integrated into the E1 region; and a PmeI site (GTTTAAAC) was inserted between the EF1a promoter and the SV40 polyA signal (Fig. 1a). The sequence information around the PmeI site was needed to design PCR primers to create overlaps for the following DNA assembly, and this is shown in Fig. 1b. Digestion with EcoRV produced fragments in agreement with the predicted molecular weights, suggesting successful cloning of the pKAd5f11p-EF1aP plasmid (Fig. 1c). The process of constructing pKAd5f11p-EF1aP is shown in Supplementary Figs. S1–S4. The name pKAd5f11p-EF1aP was given to represent the plasmid containing Kanamycin-resistant gene and the genome of human Adenovirus 5 carrying the chimeric fiber of HAdV-11p and the EF1a promoter for transgene expression.

Construction of the starting adenoviral plasmid preloaded with human EF1a promoter. An adenoviral plasmid (pKAd5f11p-EF1aP) containing E1/E3-deleted HAdV-5 genome was constructed with the following features: the original HAdV-5 fiber gene was replaced with a fused fiber of HAdV-5 and HAdV-11p (F5-11p); the original PmeI site in the pIIIA coding sequence was removed through a synonymous mutation; and human EF1a promoter, SV40 polyA signal, and a PmeI site in between were integrated into the E1 region. The plasmid map of pKAd5f11p-EF1aP
The unique cut of PmeI on pKAd5f11p-EF1aP made it possible to transfer the gene of interest to the viral genome using DNA assembly. PCR primers for DNA assembly should contain at least two sequence components: a gene-specific sequence at the 3′ end, which should have a Tm of 50–60°C to ensure the specific amplification of target gene; a non-priming overlap sequence at the 5′ end, which was homologous to the 5′-terminal sequence upstream or downstream of the PmeI site in pKAd5f11p-EF1aP with a Tm between 50°C and 60°C. PCR primers were designed and synthesized, and GFP CDS with overlaps on both ends were amplified by PCR using high-fidelity DNA polymerase. The PCR product was combined with PmeI-linearized pKAd5f11p-EF1aP, and DNA assembly was carried out to generate pKAd5f11p-EPG where the GFP reporter gene was integrated between the EF1a promoter and the SV40 polyA signal (Fig. 2a). pKAd5f11p-EPG represented pKAd5f11p-EF1aP plasmid carrying EF1a promoter-controlled GFP gene. Plasmids from five kanamycin-resistant E. coli colonies were extracted and analyzed with HindIII digestion, and the results showed that all of the randomly selected colonies carried pKAd5f11p-EPG (Fig. 2b). The adenoviral plasmid pKAd5f11p-EPG was digested with PacI to release the genome of the recombinant virus, and the recovered DNA was used to transfect 293 cells, a cell line of constitutively expressed HAdV-5 E1 genes. Focuses comprising scores of GFP-positive cells could be found on the monolayer culture under a fluorescence microscope 5 days post transfection, indicating the rescue of the recombinant virus (Fig. 2c). The focuses kept growing and finally turned into plaques. The recombinant adenovirus HAdV5f11p-EPG was identified by restriction analysis of the virus genomic DNA (Fig. 2d). When A549 cells were infected with HAdV5f11p-EPG at a MOI of 50 vp/cell, nearly 100% cells expressed GFP 2 days post infection (data not shown). All of these facts suggested that pKAd5f11p-EF1aP worked effectively as a single plasmid-based adenoviral vector system to express the exogenous gene.
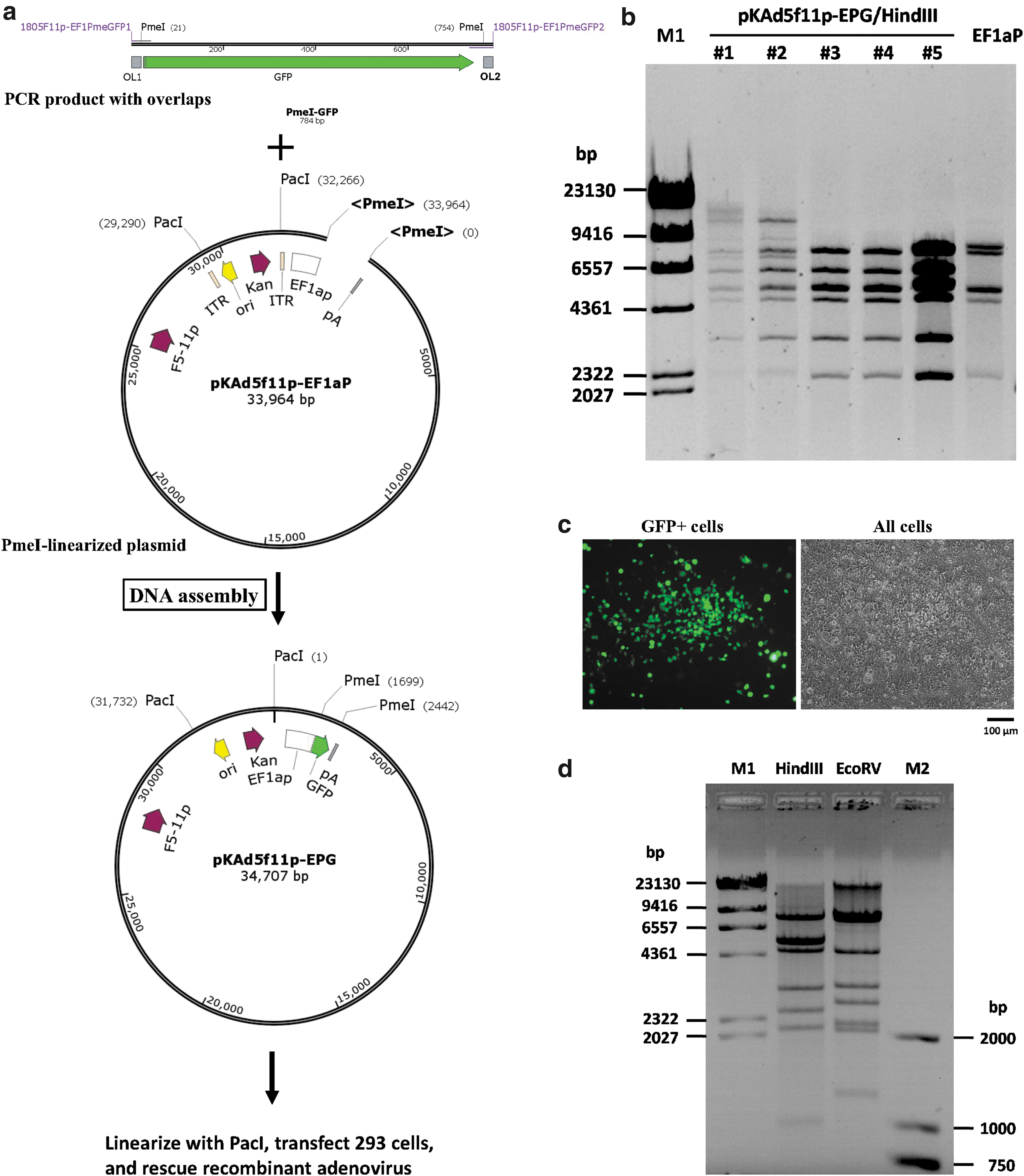
Construction of a recombinant adenovirus carrying the green fluorescent protein (GFP) reporter gene using the restriction assembly method.
Single plasmid-based adenoviral vector system for incorporation of the exogenous gene expression cassette
Commercial vector systems often provide limited options for choosing regulation elements to express the transgene. Here, an attempt was made to construct a starting adenoviral plasmid carrying no promoter for the transgene, giving researchers the freedom to assemble their own exogenous gene expression cassette. To this end, EF1a promoter was removed from pKAd5f11p-EF1aP to generate pKAd5f11pES-PmeI (Fig. 3a and Supplementary Fig. S5). pKAd5f11pES-PmeI represented the E1-deleted pKAd5f11p-EF1aP plasmid with the reserved encapsidation signal (ES) and PmeI site. The PmeI site was reserved for incorporation of exogenous DNA, and the sequence information around PmeI was needed for designing PCR primers for DNA assembly (Fig. 3b). The plasmid was identified by restriction analysis (Fig. 3c). For proof of principle, PCR was performed to amplify two linked gene expression cassettes using the plasmid pLVX-TRE3G-TetGFP as the template. pLVX-TRE3G-TetGFP was a plasmid constructed previously for inducible expression of target gene based on the Tet-On 3G system (Supplementary Fig. S6). The PCR product with overlaps on both ends was mixed with PmeI-linearized pKAd5f11pES-PmeI and subjected to DNA assembly reaction to generate the pKAd5f11p-TGFPT plasmid (Fig. 4a). Plasmids were extracted from five randomly selected colonies and digested with EcoRV, and the electrophoresis result showed that all five were correct pKAd5f11p-TGFPT plasmids (Fig. 4b). Recombinant HAdV5f11p-TGFPT was rescued, amplified, and purified according to the procedure described in the Methods. Restriction analysis results demonstrated that HAdV5f11p-TGFPT contained the predicted genome (Fig. 4c). The incorporated two exogenous gene expression cassettes in HAdV5f11p-TGFPT included complete elements needed for inducible expression of GFP. When Dox was added to the culture system of virus-infected A549 cells, GFP expression could be induced (Fig. 4d), suggesting functional HAdV5f11p-TGFPT.

Construction of the starting adenoviral plasmid for cloning the foreign gene expression cassette. An adenoviral plasmid (pKAd5f11pES-PmeI) containing E1/E3-deleted HAdV-5 genome was constructed with the same features as pKAd5f11p-EF1aP, except that the EF1a promoter was excluded. The plasmid map of pKAd5f11pES-PmeI

Construction of a recombinant adenovirus carrying two gene expression cassettes using the restriction assembly method.
Shuttle plasmid strengthened the application of pKAd5f11pES-PmeI
pKAd5f11pES-PmeI was suitable for incorporating gene expression cassette, which might contain many DNA elements. PCR or overlap extension PCR was needed to add overlaps or combine several DNA fragments together to satisfy the DNA assembly. In situations where PCR is not applicable, such as the secondary structure existed or the fragment was too long, a shuttle plasmid would be helpful. The shuttle plasmid pUC19-PM contained MCS where DNA element could be inserted through restriction ligation cloning, overlaps that were compatible with the flank sequences of the PmeI site in pKAd5f11pES-PmeI, and two PacI sites that were needed for separating the target fragment of the combined DNA elements and overlaps from the plasmid backbone of the ampicillin-resistant gene and pBR322 origin of replication (Fig. 5a). pUC19-PM represented plasmid pUC19 backbone with PacI-site-flanked overlaps and MCS. As a proof of principle, Tet-On 3G and GFP expression cassettes were excised from pLVX-TRE3G-TetGFP by digesting with ClaI and KpnI, and inserted into the ClaI/Kpn sites in pUC19-PM to generate pUC19-PTGFPW. The fragment including the cassettes and overlaps was isolated from the plasmid by PacI digestion, and combined with PmeI-linearized pKAd5f11pES-PmeI. DNA assembly was carried out to generate the adenoviral plasmid pKAd5f11p-TGFPW (Fig. 5b). All 10 randomly selected E. coli colonies contained the correct pKAd5f11p-TGFPW plasmid (Supplementary Fig. S9). The restriction analysis of the first five is shown in Fig. 5c. Recombinant virus was similarly rescued and amplified as mentioned above. The virus was identified by restriction analysis of the extracted genomic DNA (Fig. 5d), and it was functional, as the inducible expression of GFP could be observed (Fig. 5e).

Construction of a recombinant adenovirus with the help of a shuttle plasmid.
Single plasmid-based adenoviral vector system for fiber modification
The capsid of the adenovirus could be modified in order to change virus tropism or construct a vectored vaccine. 24,25 HAdV-5 based vector system has been thoroughly investigated and has become a versatile system for such purposes. In order to facilitate the modification of the fiber, the adenoviral plasmid pKAd5f11pABR-EPG was constructed based on pKAd5f11p-EPG. pKAd5f11pABR-EPG is different from pKAd5f11p-EPG in the following ways: the remnant E3 region of the HAdV-5 genome was shortened by 45 bp in the middle, and one ClaI site was added into this place; the “atcgtt” sequence, which was located 4 bp downstream of the stop codon of the original HAdV-5 fiber gene, was mutated to “atcgAt” to create another ClaI site; and the coding sequence of RGD4C was inserted into the fiber gene where the AB loop of the HAdV-11p Knob domain was encoded (Fig. 6a). 18,26 –28 ClaI is a dual cutter for pKAd5f11pABR-EPG. The 1.2 kb fragment containing the fiber gene in pKAd5f11pABR-EPG could be removed by ClaI digestion and restored conveniently by another fragment carrying the modified fiber gene through DNA assembly. pKAd5f11pABR-EPG represented the plasmid pKAd5f11p-EPG carrying a modified AB loop of the HAdV-11p fiber knob where the RGD4C coding sequence was inserted. To facilitate the generation of the fragment with modified fiber to replace the fiber gene in pKAd5f11pABR-EPG, the plasmid pMD-F511pEC was constructed with following features: the sequence between two ClaI sites in pMD-F511pEC was the same as that in pKAd5f11pABR-EPG, except that the RGD4C coding sequence was removed to restore an original F5-11p; fragments between EcoRV/ClaI on both sides could service as the overlaps for DNA assembly; and EcoRV sites were artificially added to help fragment excision (Fig. 6b). pMD-F511pEC represented the plasmid containing pMD18-T backbone, the chimeric fiber of HAdV-5 and HAdV-11p (F5-11p), and EcoRV/ClaI restricted overlaps. pMD-F511pEC could work as a shuttle or be used as a template for amplification of the fiber-modified fragment by overlap extension PCR. pKAd5f11pABR-EPG was successfully constructed (Fig. 6c). The recombinant virus HAdV5f11pABR-EPG was rescued from PacI-linearized pKAd5f11pABR-EPG-transfected 293 cells (Fig. 6d) and identified with restriction analysis (Fig. 6e). A series of fiber-modified viruses were successfully generated in the laboratory, suggesting the high efficiency of this system (data not shown).

Construction of a recombinant adenovirus with a modifiable fiber gene.
Verification of restriction assembly regions by sequencing
To evaluate reliability of the restriction assembly strategy, primers were synthesized and used to amplify fragments spanning restriction assembly sites using the genomic DNA of recombinant adenoviruses as the templates (Supplementary Table S2). The PCR products were recovered and sequenced. Eight restriction assembly sites were included in the six PCR products, which originated from HAdV5f11p-EPG, HAdV5f11p-TGFPT, HAdV5f11p-TGFPW, and HAdV5f11pABR-EPG. Fusion of fragments occurred as predicted, and no mutations were detected around the restriction assembly sites according to the sequencing results (Supplementary Fig. S10). The genomic DNA from the above-mentioned recombinant viruses, as well as that from another fiber-modified virus, was analyzed with next-generation sequencing (NGS), and no DNA assembly-related mutation was found (data not shown). These facts demonstrated that the restriction assembly strategy were reliable for the construction of adenoviral vectors.
Discussion
Generally, the process of transforming a wild-type adenovirus to a gene delivery vehicle harboring a target gene has three stages: constructing the infectious clone, establishing the vector system, and using vector system. In this study, single plasmid-based adenoviral vector systems for expressing foreign gene or modifying viral tropism are introduced. This report describes how to establish these systems and how they work—the latter two of the three stages.
Restriction ligation cloning alone is not adequate to reconstruct an adenovirus genome, which is usually >30 kb, to become a vector system. The following strategy was employed in this study: separate a short fragment from adenoviral plasmid to generate a small plasmid with the restriction ligation approach; perform site-directed mutations on the small plasmid by overlap extension PCR or DNA assembly; and restore the modified small plasmid back to the adenoviral plasmid by restriction ligation or DNA assembly. Some recent publications have described the construction of adenoviral vectors through direct DNA assembly of several PCR products. 29 –31 The strategy of direct assembly is brief, practical, and easy to understand. For the strategy followed in this study, it takes some effort to find the proper restriction sites and to program the whole procedure before construction begins. However, the experimental process is more cost-effective and time-saving. More plasmid fragments are used instead of PCR products, minimizing the cost and waiting time of sequencing verification and reducing the frequency of unwanted PCR mutation. Fewer and shorter fragments in DNA assembly increase the efficiency and success rate of the reaction, and the intermediate plasmids are sometimes useful for future purposes. The construction of infectious clones of adenovirus has been reviewed elsewhere, 4 and this process was substantially simplified using Gibson assembly. 32 If infectious clones of other adenoviruses are available, novel adenoviral vector systems can be developed with the strategy used here.
With respect to utilization, the adenoviral vector systems introduced here are simple, versatile, and upgradable.
It is simple to construct target adenoviral plasmid with these vector systems: linearize the starting plasmid at the insertion site by restriction digestion, and fuse the target DNA in by DNA assembly. The process was termed restriction assembly. It is labor- and time-saving when compared to the widely used method of homologous recombination in bacteria. One round of bacterial transformation is needed, and it takes only 2–3 days to go through the whole procedure of restriction assembly. For homologous recombination in bacteria, 1 week is needed for three rounds of transformation on the condition that everything goes smoothly. This method is even simpler than the improved in vitro ligation approach using rare-cutter endonuclease because constructing a shuttle plasmid is dispensable. 9,33,34 The restriction assembly method seems to be more efficient. Based on the authors' experience, there were fewer unwanted or unexplainable colonies. In the process of constructing four adenoviral vectors in this study, >20 colonies were randomly selected for identification with restriction analysis, and the results showed that all of them were correctly assembled (Figs. 2b, 4b, and 5c and Supplementary Fig. S9). This was further confirmed by sequencing the recombinant viruses (Supplementary Fig. S10).
The restriction assembly method provides more versatility. First, the incorporation of the target gene is free from the restriction of restriction endonuclease because Gibson assembly is sequence independent. For example, although the PmeI site exists in the starting adenoviral plasmids, the presence of the PmeI site within the target gene is allowed, since there is no step of PmeI cutting of the target gene in the restriction assembly procedure. The only restriction site that should not appear in the target gene is the PacI site because PacI is needed for linearizing the target adenoviral plasmid before virus rescue. Second, artificially introduced restriction sites do not necessarily exist in the target adenoviral plasmid. In the starting adenoviral plasmids, PmeI or ClaI sites were created. They are nothing more than entrances for the linearizing adenoviral plasmid at certain positions. If the introduced restriction sites affect the function of the desired recombinant virus, they can be removed, and the original sequence can be restored in the final target adenoviral plasmid with no extra effort. That is because the mismatching flap sequence at the end of overlaps (<10 nt) can be removed during Gibson assembly when proof-reading DNA polymerase is used (Supplementary Fig. S10). On the other hand, some extra sequences can even be introduced if they are included between the overlap and 3′ specific parts of the PCR primers. Third, there are many options for preparing fragment containing exogenous DNA before DNA assembly. PCR is the simplest and the most efficient method to clone a single gene. When the target gene is too long or too difficult to amplify by PCR, a shuttle plasmid can be used. For exogenous DNA that consists of parts that originate from various sources, overlap extension PCR is preferred, and the parts can also be added to the MCS of the shuttle plasmid successively by restriction ligation cloning when it is necessary. It is worth noting that direct assembly of multiple fragments can be another option, although this was not tested in this study. Finally, target adenoviral plasmids can be used as starting ones to construct new vectors if the introduced restriction sites are saved. For example, the target plasmid pKAd5f11p-EPG, which derived from the starting plasmid pKAd5f11p-EF1aP and carried the target gene GFP, can be used as a starting plasmid to construct new adenoviral vectors just as pKAd5f11p-EF1aP did. When being cut with PmeI, pKAd5f11p-EPG and pKAd5f11p-EF1aP produce the same DNA fragment needed for the following step of DNA assembly.
Another advantage of the introduced systems is upgradability. pKAd5f11p-EF1aP and pKAd5f11p-EPG are basic plasmids that can be used to express exogenous gene, while pKAd5f11pABR-EPG is an upgraded one that can be used to test the function of the modified fiber. The upgradability mentioned here means backward compatibility as well. pKAd5f11pABR-EPG is more than a system for testing the function of the modified fiber; it is also a starting adenoviral plasmid to express a foreign gene just as good as pKAd5f11p-EF1aP. If pKAd5f11pABR-EPG is to be used to express another transgene instead of GFP, it needs to be treated as the starting plasmid pKAd5f11p-EF1aP or pKAd5f11p-EPG by amplifying overlap-included CDS of the foreign gene by PCR and fusing it between the PmeI sites of pKAd5f11pABR-EPG by restriction assembly. No extra work is necessary. The secret of compatibility lies in the independence between the elements of the foreign gene CDS (defined by the PmeI sites) and the modified fiber (defined by the ClaI sites). It can be deduced that pKAd5f11pABR-EPG is also upgradable. Adenoviral vector systems with modifiable pIX or Hexon can be developed based on pKAd5f11pABR-EPG, and all these systems will work under the same strategy of restriction assembly and will be backward compatible. The compatibility and versatility can be further expanded if bigger fragments generated with dual-cutter are exchanged between these plasmids by restriction ligation cloning. For example, the feature of cloning the gene expression cassette of pKAd5f11ES-PmeI could be transferred to pKAd5f11pABR-EPG by exchanging the BstZ17I-digested fragment between them.
This work will help to broaden synthetic biology to the field of adenoviral vectors. Modularization and standardization are key properties of synthetic biology. 35 Adenoviral vector has been synthesized by assembling several parts of the virus genome. 29 –31 However, these parts are physical modules. Here, two functional modules (one for transgene cloning and the other one for fiber modification) and the strategy of restriction assembly for vector construction are provided. The starting, target, and intermediate plasmids could serve as a basic platform and component library for synthesizing more adenoviral vectors with novel properties.
In summary, traditional restriction ligation was combined with Gibson assembly to establish new HAdV-5-based vector systems. These systems work through a simple process of restriction assembly to incorporate exogenous DNA.
Footnotes
Acknowledgments
This work was supported by the National Science and Technology Major Project (2018ZX10711001). The adenoviral plasmids pKAd5f11p-EPG, pKAd5f11pES-PmeI, and pKAd5f11pABR-EPG will be available through Addgene.
Author Disclosure
The authors have no conflicts of interest.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.