
Abstract
Degenerative disc disease (DDD) is a primary contributor to low-back pain, a leading cause of disability. Progression of DDD is aided by inflammatory cytokines in the intervertebral disc (IVD), particularly TNF-α and IL-1β, but current treatments fail to effectively target this mechanism. The objective of this study was to explore the feasibility of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) epigenome editing-based therapy for DDD, by modulation of TNFR1/IL1R1 signaling in pathological human IVD cells. Human IVD cells from the nucleus pulposus of patients receiving surgery for back pain were obtained and the regulation of TNFR1/IL1R1 signaling by a lentiviral CRISPR epigenome editing system was tested. These cells were tested for successful lentiviral transduction/expression of deactivated Cas9 fused to Krüppel Associated Box system and regulation of TNFR1/IL1R1 expression. TNFR1/IL1R1 signaling disruption was investigated through measurement of NF-κB activity, apoptosis, and anabolic/catabolic changes in gene expression postinflammatory challenge. CRISPR epigenome editing systems were effectively introduced into pathological human IVD cells and significantly downregulated TNFR1 and IL1R1. This downregulation significantly attenuated deleterious TNFR1 signaling but not IL1R1 signaling. This is attributed to less robust IL1R1 expression downregulation, and IL-1β–driven reversal of IL1R1 expression downregulation in a portion of patient IVD cells. In addition, RNAseq data indicated novel transcription factor targets, IRF1 and TFAP2C, as being primary regulators of inflammatory signaling in IVD cells. These results demonstrate the feasibility of CRISPR epigenome editing of inflammatory receptors in pathological IVD cells, but highlight a limitation in epigenome targeting of IL1R1. This method has potential application as a novel gene therapy for DDD, to attenuate the deleterious effect of inflammatory cytokines present in the degenerative IVD.
Introduction
Low-back pain (LBP) and related pathological conditions are a major health care and socioeconomic concern, as LBP is the leading cause of disability worldwide, 1 and has indirect and direct costs of $100–200 billion/year in the United States alone. 2 Degenerative disc disease (DDD) is considered a primary contributor to LBP, 3 –7 which is associated with the breakdown of the extracellular matrix (ECM) of the intervertebral disc (IVD), decreased IVD height, and inflammation. 8 –10 Current clinical LBP treatments do not focus on resolving or slowing DDD, and simply aim to ameliorate pain, which often only provides a short-term solution. 11
Treatment strategies that target the mechanisms behind DDD have the potential to provide improved treatments for LBP associated with DDD. Although the exact mechanisms of DDD are incompletely understood, it is clear that inflammation is a major contributor. 12 –24 Therapeutics that slow DDD progression by providing regulation of inflammatory signaling are needed. This signaling is mediated by inflammatory cytokines, of which several have been identified in the degenerative IVD and shown to regulate DDD preclinically (TNF-α, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-17, interferon-γ [IFN-γ]). 9,19 –25
Two cytokines implicated as primary mediators of DDD are TNF-α and IL-1β. These cytokines signal apoptosis, propagation of proinflammatory signaling, and ECM degradation. 12,14,15,26 –28 This catabolic signaling is mediated by downstream activation of NF-κB, a transcription factor that induces expression of proinflammatory and catabolic target genes involved with IVD breakdown and cell survival. 29 –31 Therefore, inhibiting TNF-α/IL-1β signaling may slow or prevent DDD progression by decreasing proinflammatory and catabolic processes in the degenerative disc.
Inhibition of TNF-α and IL-1β signaling can be achieved by direct cytokine inhibition or in a more pathway-specific manner through receptor inhibition. Previous clinically tested methods have taken the approach of direct cytokine inhibition by monoclonal antibodies, 32 –34 but they have limitations. One limitation is that they only effectively block signaling for a short period after delivery, as they have a relatively short half-life in vivo. 35 This is problematic for their use as a DDD therapy as effective application requires epidural injections, which have to be performed in clinic, something not feasible to do on a weekly basis.
An additional limitation is that these therapies often broadly block cytokine action, which results in elimination of beneficial signaling functions, as it is specific receptors that mediate the negative effect of these cytokines. For example, TNF-α can exhibit proinflammatory, catabolic, and apoptotic signaling when binding TNFR1, but it confers antiapoptotic and regenerative effects when binding TNFR2. 36 In this regard, receptor inhibition methods that target specific inflammatory cytokine/receptor (e.g., TNF-α/TNFR1) interactions while protecting and promoting cell and ECM protective interactions (e.g., TNF-α/TNFR2) can specifically inhibit catabolic TNF-α and IL-1β signaling.
In this study, we investigate a gene therapy strategy to achieve long-term receptor-specific inhibition of TNF-α and IL-1β signaling in human degenerative IVD cells. This method uses Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) epigenome editing to downregulate TNFR1/IL1R1 expression. In brief, CRISPR epigenome editing performs sequence-specific epigenome alterations to regulate gene expression when targeted to gene regulatory elements. 37,38 In addition, this system has been demonstrated to have minimal off-target effects, and is continually being improved to minimize off-target effects to negligible levels. 39
The specific CRISPR epigenome editing system we are using utilizes the transcription repression domain Krüppel Associated Box (KRAB) to repress gene expression. We are delivering this system to cells through lentivirus, which provides an efficient means of gene delivery and provides an ability to achieve long-term signaling regulation, as lentivirally delivered genes integrate into the host cell genome. This provides the potential for one time delivery of CRISPR epigenome editing systems, to provide long-term effects.
We previously demonstrated successful use of these lentiviral CRISPR epigenome editing systems to engineer stem cells and neuron response to inflammatory environments. 40,41 In this study, we investigated whether these systems are able to regulate inflammatory signaling in human degenerative IVD cells. This was done to explore their potential as a single injection gene therapy that would be delivered into the IVD to achieve long-term inhibition of disc degeneration without need for repeated injections. Specifically, we investigated TNFR1/IL1R1 modulation by CRISPR epigenome editing systems in human nucleus pulposus cells (hNPCs) obtained from surgical degenerative IVD tissue.
We measured our ability to express the CRISPR epigenome editing systems in hNPCs, their ability to modulate TNFR1/IL1R1 expression and signaling, and to ameliorate the ECM degrading and apoptotic effects of TNFR1/IL1R1 signaling. Furthermore, we also investigated how TNF-α signaling was modulated by CRISPR epigenome editing-based downregulation of TNFR1 in hNPCs on a genome wide level using RNA-seq. In addition, we investigated limitations in IL1R1 signaling regulation revealed during experiments.
Overall this study demonstrates that lentiviral CRISPR epigenome editing systems can efficiently be delivered into human IVD cells, promote IVD cell survival, promote anabolic ECM gene expression, reduce NF-κB activity, and prevent catabolic gene expression under inflammatory conditions. This provides evidence for the potential use of CRISPR epigenome editing systems to regulate IVD cell response and be utilized as an injectable gene therapy to slow DDD progression.
Materials and Methods
Experimental overview
Experiments were conducted to test the ability of lentivirus encoding CRISPR-based transcriptional repressors targeting TNFR1 and IL1R1, to engineer human degenerative IVD cells, and to better thrive within the degenerative inflammatory environment. We transduced hNPCs with these lentiviral vectors to investigate the ability of this gene therapy to transduce native disc cells and perform targeted repression of TNFR1 and IL1R1 expression (verified through quantitative reverse transcriptase [qRT]-PCR). Effects of this targeted repression on NF-κB activity, apoptosis, and anabolic/catabolic gene expression under inflammatory conditions were tested to verify its functionality and determine its feasibility as a potential DDD gene therapy. This experimental overview is also graphically described in Supplementary Fig. S1.
hNPCs extraction and culture
Nucleus pulposus (NP) tissue was obtained from surgical tissue waste of deidentified patients undergoing surgery for DDD-related back pain (n = 5). All patients showed signs of DDD, as verified by MRI, and axial back pain. The NP was macroscopically dissected and immediately processed for cell isolation based on previously described methods. 42 In brief, NP tissue was rinsed twice with washing medium (Dulbecco's modified Eagle's medium [DMEM] high glucose [Gibco], 165 μg/mL gentamicin sulfate [Gibco], 100 μg/mL kanamycin sulfate [Sigma], 1.25 μg/mL amphotericin B [Gibco]), coarsely minced, and then digested enzymatically in washing medium containing 0.3% (w/v) collagenase type II (Worthington Biochemical), 0.2% (w/v) pronase (Sigma), and 5% fetal bovine serum (FBS) (Hyclone) for 2–3 h at 37°C with 5% CO2 under gentle agitation.
Cells were then passed through a 70 μm cell strainer and washed twice (400 g for 10 min). Cells were counted and plated at 10,000 cells/cm2 in base culture medium (DMEM high glucose with 10% FBS, 50 μg/mL gentamicin, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]) supplemented fresh with 2 ng/mL fibroblast growth factor 2 (FGF-2) (Peptrotech). The inclusion of FGF-2 in medium was done to aid in cell expansion, as previous studies demonstrated FGF-2 aids in NP cell expansion while maintaining their phenotype. 43,44 Cells were cultured in this medium at 37°C and 5% CO2 in a humidified atmosphere, and subcultured at 90% confluency.
Lentiviral vector constructs
The epigenome editing lentiviral vectors were built based on previous TNFR1/IL1R1 targeting screens 40 to contain the Streptococcus Pyogenes deactivated Cas9 (dCas9)-KRAB fusion protein, a gRNA that is complementary to the promoter of the target gene (TNFR1: 5′-CAGTGTTGCAACAGCGGGAC-3′, IL1R1: 5′-TGGGGTCCTTGGGCGACTGC-3′) or a nontargeting control (NTC) gRNA (5′-TTTTTAATACAAGGTAATCT-3′) with no complementary genomic sequence, and an enhanced green fluorescent protein reporter (general vector map shown in Fig. 1A).
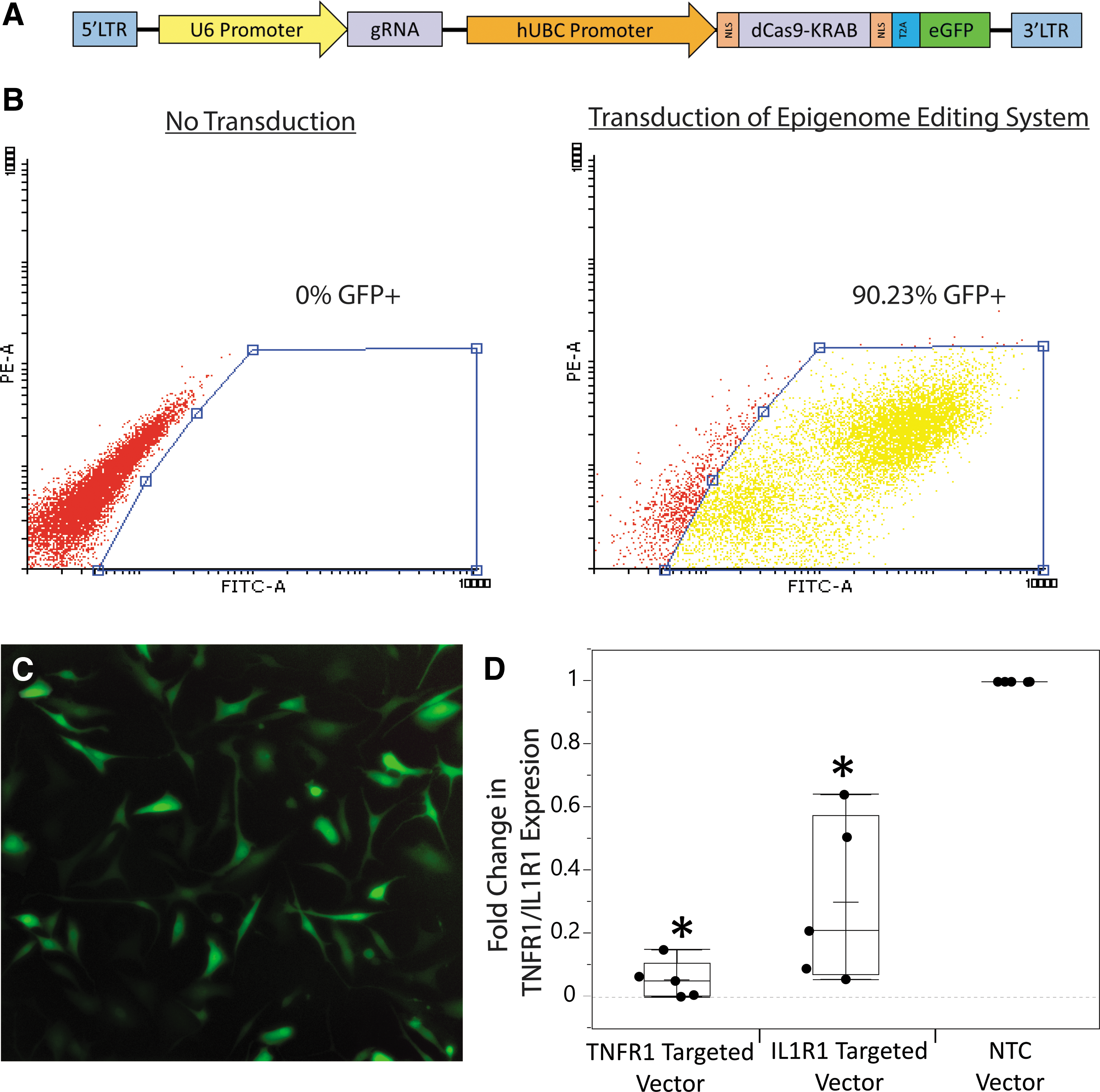
Epigenome editing system components, and transduction and gene regulation by it in hNPCs.
The gRNAs within these vectors are adjacent to the appropriate protospacer adjacent motif sequence within the genome (NGG) to ensure proper binding of this system to the targeted sequence. For measurement of NF-κB activity, a luminescent NF-κB reporter lentivirus was used (49335; Addgene). These constructs were used to produce lentivirus to allow for delivery of these systems through lentiviral transduction.
Lentivirus production
All lentivirus production utilized the following protocol. Lentivirus was produced in HEK293T cells (CRL-11268; ATCC). Before plating cells, six-well culture dishes were pretreated with 0.001% poly-
The next day, cells were cotransfected with 4 μg of transfer vector, 3 μg of psPAX2 (12260; Addgene), and 1.2 μg of pMD2.G (12259; Addgene) using Lipofectamine 2000 (Thermofisher). After 16–20 h, the transfection medium was replaced. Supernatant containing lentivirus was collected 24 and 48 h after removal of transfection medium and filtered through a 0.45 μm polyvinylidene fluoride filter. Lentivirus was concentrated 100 × (lenti-X; Takara) and stored at −80°C until use.
Gene delivery by lentiviral transduction
hNPCs (passage 2) were plated in six-well plates (180,000 cells/well) in culture medium overnight. The next day, cells were transduced with 1 mL of diluted lentiviral stock (100 × stock diluted 1:16 in culture medium for epigenome editing lentivirus, and 1:100 for NF-κB reporter lentivirus), supplemented with 8 μg/mL polybrene. After 20–22 h, virus was removed and cells were washed with PBS five times. After transduction, cells were then cultured for at least 1 week to allow for transgene expression, and were then sorted using flow-activated cell sorting (BD FACS Aria Cell Sorter) to select for successfully transduced cells and measure transduction efficiency. Sorted cells were expanded until enough cells were obtained for experiments.
Initial measurement of gene and signaling regulation
Quantitative RT-PCR
TNFR1/IL1R1 expression in hNPCs transduced with epigenome editing vectors was measured by qRT-PCR. Total RNA was harvested using Purelink RNA micro kit (Invitrogen). Complementary DNA (cDNA) synthesis was performed with purified RNA using the High-Capacity cDNA Reverse Transcription Kit with RNAse Inhibitor (Applied Biosystems). Subsequently, cDNA was used for quantitative PCR (qPCR) with Taqman assays (Applied Biosystems) for TNFR1 (Hs01042313_m1), IL1R1 (Hs00991002_m1), and GAPDH (Hs02758991_g1). TNFR1/IL1R1 expression was normalized to GAPDH expression, and analyzed as fold change in expression relative to the NTC group (ΔΔCt method).
NF-κB activity
The NF-κB activity of epigenome-edited hNPCs treated with TNF-α/IL-1β was analyzed through the lentivirally introduced luminescent NF-κB reporter to determine whether our vectors are able to modulate functional signaling of TNFR1/IL1R1. One day before dosing, hNPCs were plated (6,000 cells/well) in 96-well plates in 100 μL of culture medium. They were then treated with a range (0, 0.15, 1, and 10 ng/mL) of TNF-α (TNFR1-edited and NTC cells) or IL-1β (IL1R1-edited and NTC cells).
After 24 h, NF-κB activity (firefly luciferase luminescence assay 45 ) and number of cells (CCK8 assay; Dojindo) were measured. Induction of NF-κB activity was normalized to number of cells and analyzed as a fold change in luminescence relative to each untreated control (0 ng/mL) group (NTC, TNFR1 edited, or IL1R1 edited).
Verification of epigenome editing mediated signaling regulation
Annexin V/propidium iodide apoptosis assay
We measured the incidence of apoptotic cells post-TNF-α/IL-1β treatment using the Annexin V/propidium iodide (PI) flow cytometry assay in epigenome-edited hNPCs. 46 In brief, hNPCs were plated (150,000 cells/well) in 2.5 mL of base culture medium in six-well plates. The following day, cells were treated with 0, 1, or 10 ng/mL of TNF-α (TNFR1-edited and NTC cells) or IL-1β (IL1R1-edited and NTC cells) or 1 ng/mL of TNF-α and IL-1β (all cell types). After 24 h, cells were harvested and suspended in 1 × Annexin V Binding buffer (Thermofisher) (1.5 × 105 cells/mL) and 200 μL was incubated with 20 μL of Annexin V-Pacific blue readyflow reagent (Thermofisher) and 1 μL of 50 μg/mL PI (Thermofisher) for 15 min at room temperature.
Staining was immediately analyzed by flow cytometry (BD FACSCanto). Quadrant-based analysis was performed using Flowing Software 2.5.1 with gating based on unstained and untreated cells. The apoptotic cells were analyzed as Annexin V (+)/PI (−) and Annexin V (+)/PI (+) stained to include cells at both early and late stages of apoptosis. 46 Fold change in apoptotic cells was analyzed relative to each untreated control group (NTC, TNFR1 edited, or IL1R1 edited).
qRT-PCR postinflammatory challenge
Expression of ECM-related genes Aggrecan (ACAN) and MMP-3 was measured post-TNF-α/IL-1β treatment in epigenome-edited hNPCs. hNPCs were plated (30,000 cells/well) in 24-well plates in 500 μL of base culture medium. The following day, the medium was replaced with medium containing 0, 1, or 10 ng/mL of TNF-α (TNFR1-edited and NTC cells) or IL-1β (IL1R1-edited and NTC cells) or 1 ng/mL of TNF-α and IL-1β (all cell types). After 24 h, RNA was isolated and cDNA was synthesized as described in the previous Quantitative RT-PCR section.
The synthesized cDNA was used for qPCR with Taqman assays (Applied Biosystems) for ACAN (Hs00153936_m1), MMP-3 (Hs00968305_m1), and GAPDH (Hs02758991_g1). Expression of each gene was normalized to GAPDH expression and analyzed as fold change in expression (ΔΔCt method) relative to each untreated control group (NTC, TNFR1 edited, or IL1R1 edited).
RNA-seq analysis
Patient nucleus pulposus cells (NTC and TNFR1 edited, 26 years male) were analyzed by RNA-seq with and without TNF-α treatment to analyze gene regulation specificity and the pathways being modulated. In brief, hNPCs were plated (150,000 cells/well) in 2.5 mL of base culture medium, in six-well plates. The following day, cells were treated with 0 or 10 ng/mL of TNF-α. After 24 h, RNA was isolated as described in the previous quantitative RT-PCR section. Poly(A)-selected RNA-seq libraries were constructed with KAPA Stranded mRNA-seq kit (Kapa Biosystems) using 500 ng total RNA.
Sequencing reads were aligned to the hg19 build of the human genome using HISAT2. 47 Sam files were converted to bam format and sorted using SAMtools. 48 Reads mapping to UCSC known genes 49 were counted using featureCounts from the SubRead package. 50 Reads were normalized and differential analysis was done in a pairwise manner using DESeq2. 51
Gene Ontology biological processes modulated were determined from genes significantly upregulated and downregulated in each group comparison (i.e., NTC vs. NTC+TNF-α) using Enrichr. 52 –54 To remove noise, only biological processes with >15% overlap in associated genes were considered significantly modulated. Transcription factors associated with genes significantly upregulated and downregulated were determined using mirExTra 2.0. 55
Analysis of maintenance of TNFR1 and IL1R1 downregulation
Regulation of TNFR1 and IL1R1 expression was analyzed by qRT-PCR postinflammatory challenge to determine whether their downregulation was maintained or overcome postinflammatory challenge. hNPCs (TNFR1/IL1R1 edited and NTC) were plated and cytokine treated in the same manner as that described in the qRT-PCR postinflammatory challenge section. After 24 h, RNA was isolated and cDNA was synthesized as described in the previous Quantitative RT-PCR section. The synthesized cDNA was used for Taqman qPCR assays (Applied Biosystems) for TNFR1 (Hs01042313_m1), IL1R1 (Hs00991002_m1), and GAPDH (Hs02758991_g1). IL1R1 and TNFR1 expression was normalized to GAPDH expression, and analyzed as fold change in expression relative to the untreated NTC group (ΔΔCt method).
Statistics
Aside from RNA-seq data (details in RNA-seq Analysis section), all statistical analyses and graphing of results were performed using JMP Pro 13 (SAS). Owing to the presence of non-normal data, as tested for by the Shapiro–Wilk test, data were treated as non-normal and all statistics were performed on ranks. For all measurements, a Student's t-test with a Bonferroni correction was performed on ranks to determine significance (0.05/9, α = 0.0055).
Data from each experiment are all graphed as box plots showing each patient data point, mean, median, and interquartile range for each group. Spearman's rho correlations, with Bonferroni correction to account for multiple correlations, were performed to analyze correlations in measured TNFR1/IL1R1 expression downregulation to subsequent signaling measurements (0.05/4, α = 0.0125).
Results
Transduction and receptor expression regulation
TNFR1 and IL1R1 targeting epigenome editing vectors were able to successfully transduce hNPCs and downregulate TNFR1/IL1R1 expression (Fig. 1). Transduction of epigenome editing systems was efficient, reaching 44–90% transduction efficiency across different patients. Measurement of TNFR1/IL1R1 gene expression demonstrated significant downregulation by both TNFR1 and IL1R1 epigenome editing systems with robust (85–99%) downregulation of TNFR1 (p < 0.0001) and less robust (36–94%) downregulation of IL1R1 (p = 0.0008) relative to NTC (Fig. 1D).
NF-κB modulation
Measurement of NF-κB activity post-TNF-α dosing demonstrated that epigenome editing of TNFR1 in hNPCs significantly modulates TNFR1 signaling (Fig. 2A). At all doses of TNF-α, there was significant decreases in NF-κB induction compared with NTC (150 pg/mL: p = 0.0034, 1 ng/mL: p = 0.0038, 10 ng/mL: p = 0.0025). In addition, NF-κB induction was nonsignificant in TNFR1 epigenome-edited hNPCs for most groups, demonstrating maintenance of TNFR1 signaling close to baseline levels (150 pg/mL: p = 0.95, 1 ng/mL: p = 0.073, 10 ng/mL: p = 0.0007).
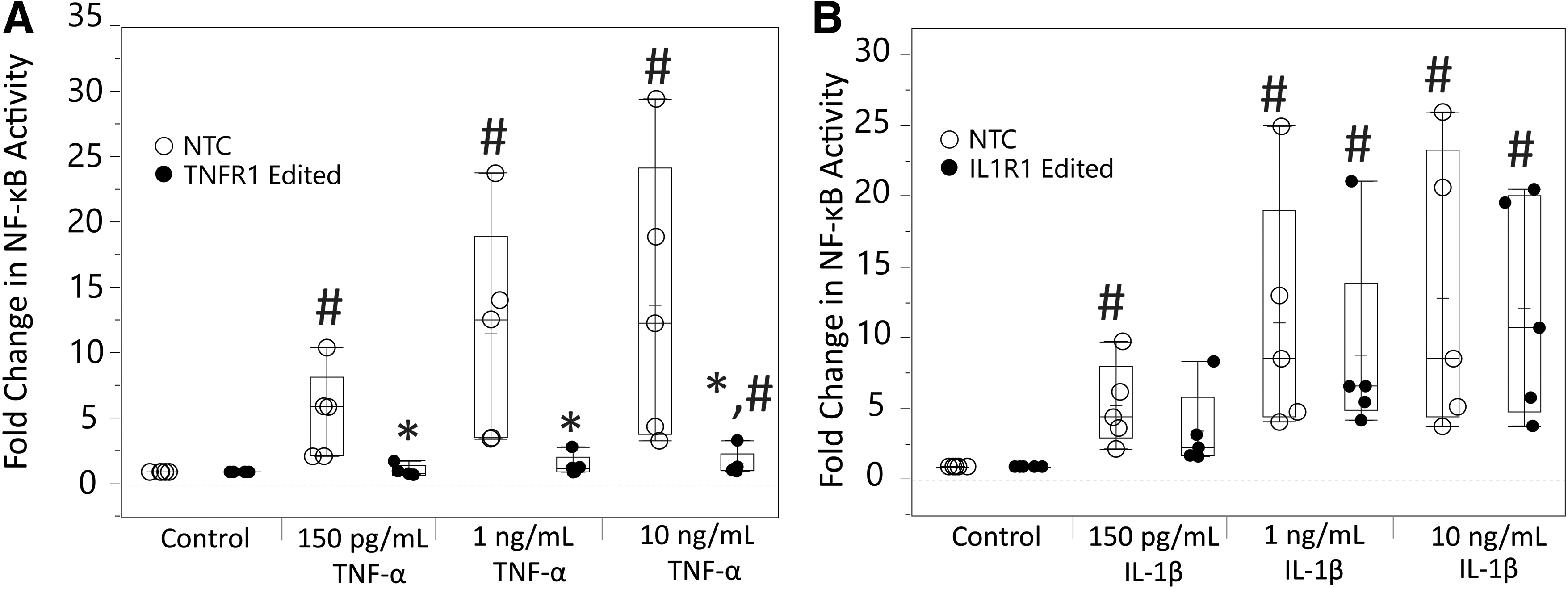
Measurement of regulation of TNFR1/IL1R1 signaling by epigenome editing systems in hNPCs through NF-κB activity assay.
IL1R1 epigenome editing in hNPCs overall did not show robust IL1R1 signaling modulation, with no significant decreases in IL-1β–mediated NF-κB induction compared with NTC (150 pg/mL: p = 0.286, 1 ng/mL: p = 0.78, 10 ng/mL: p = 0.97) (Fig. 2B). Although, at the 150 pg/mL IL-1β dose, NF-κB induction was nonsignificant within IL1R1 epigenome-edited cells (p = 0.011).
Apoptosis attenuation
Measurement of apoptosis by the Annexin V/PI assay post-TNF-α/IL-1β treatment demonstrated modulation of apoptosis by epigenome editing in hNPCs (Fig. 3). Consistent with other results, TNFR1 epigenome editing showed strong TNFR1 signaling modulation as apoptosis was significantly inhibited in TNFR1 epigenome-edited cells at both doses of TNF-α compared with NTC (1 ng/mL: p < 0.0001, 10 ng/mL: p = 0.0005). Epigenome editing of IL1R1 once again did not demonstrate strong IL1R1 signaling modulation, with no significant reduction in IL-1β–induced apoptosis in IL1R1 epigenome-edited hNPCs relative to NTC (1 ng/mL: p = 0.66, 10 ng/mL: p = 0.87).
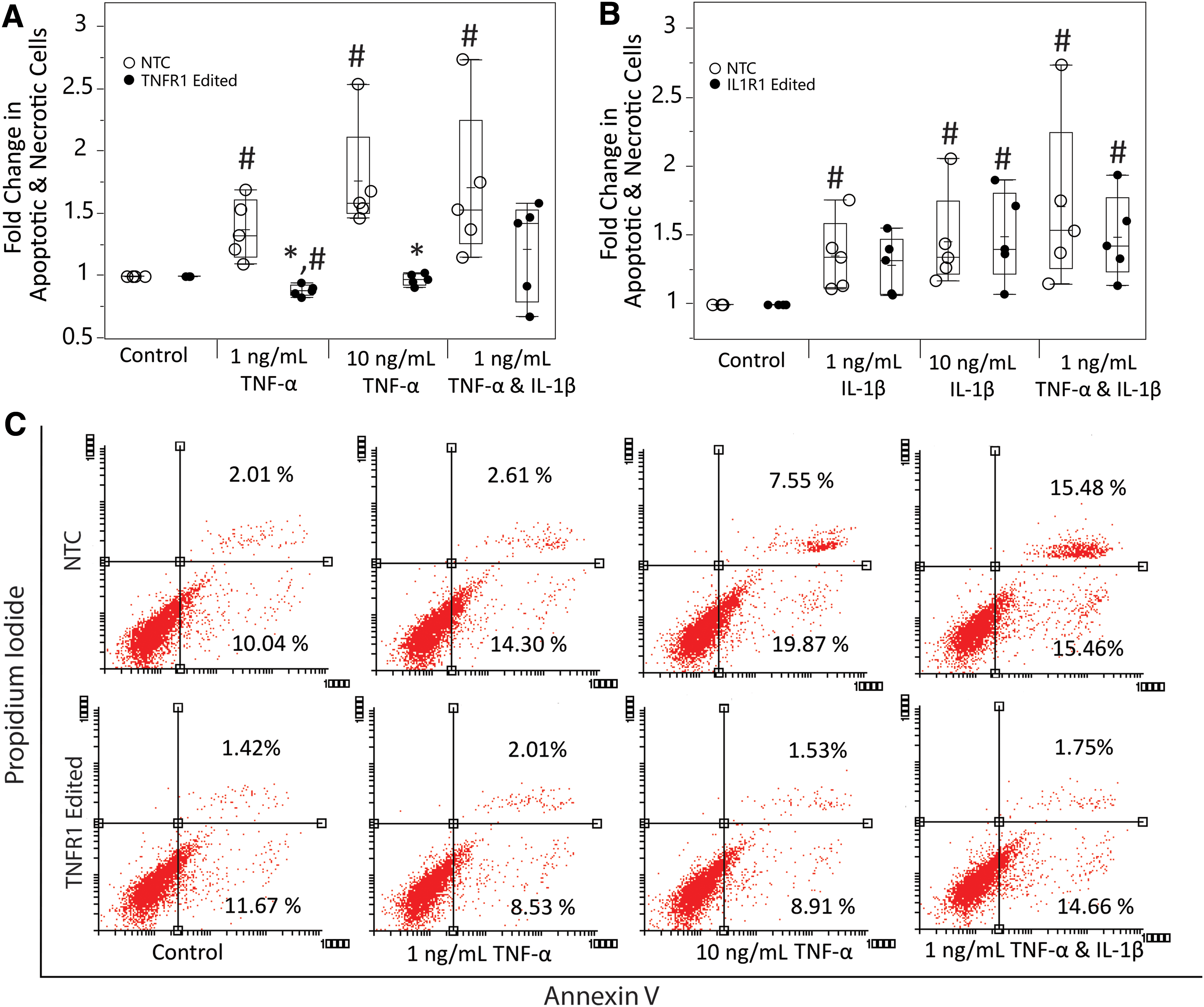
Measurement of inhibition of TNF-α–/IL-1β–induced apoptosis by epigenome editing systems in hNPCs through Annexin V/PI assay.
When looking at how apoptosis rates were affected in epigenome-edited cells in the presence of TNF-α and IL-1β together, TNFR1 epigenome editing alone decreased the mean apoptosis rate. In addition, the overall TNF-α– and IL-1β–induced apoptosis within these cells was nonsignificant (p = 0.52, compared with control), whereas it was significant in the NTC (p = 0.0007, compared with control). This indicates that TNFR1 epigenome editing may have the ability to provide protective effects even with IL-1β presence. This effect was not seen in IL1R1 epigenome-edited cells as there was significant apoptosis induction (p = 0.0006, compared with control).
ACAN and MMP-3 expression modulation
Epigenome editing demonstrated protection against cytokine-induced catabolic changes in gene expression. TNFR1 epigenome editing significantly protected against TNF-α–induced decreases in ACAN expression (Fig. 4A). ACAN expression significantly decreased only in NTC cells (NTC: 1 ng/mL: p = 0.0002, 10 ng/mL: p < 0.0001, TNFR1-edited cells: 1 ng/mL: p = 0.21, 10 ng/mL: p = 0.42, compared with control) and was significantly increased compared with NTC in TNFR1 epigenome-edited cells (1 ng/mL: p = 0.002, 10 ng/mL: p = 0.001). Similar to apoptosis data, ACAN expression decreases after TNF-α and IL-1β dosing was nonsignificant in TNFR1 epigenome-edited cells (p = 0.12, compared with control), whereas it was significant in NTC cells (p < 0.0001, compared with control).
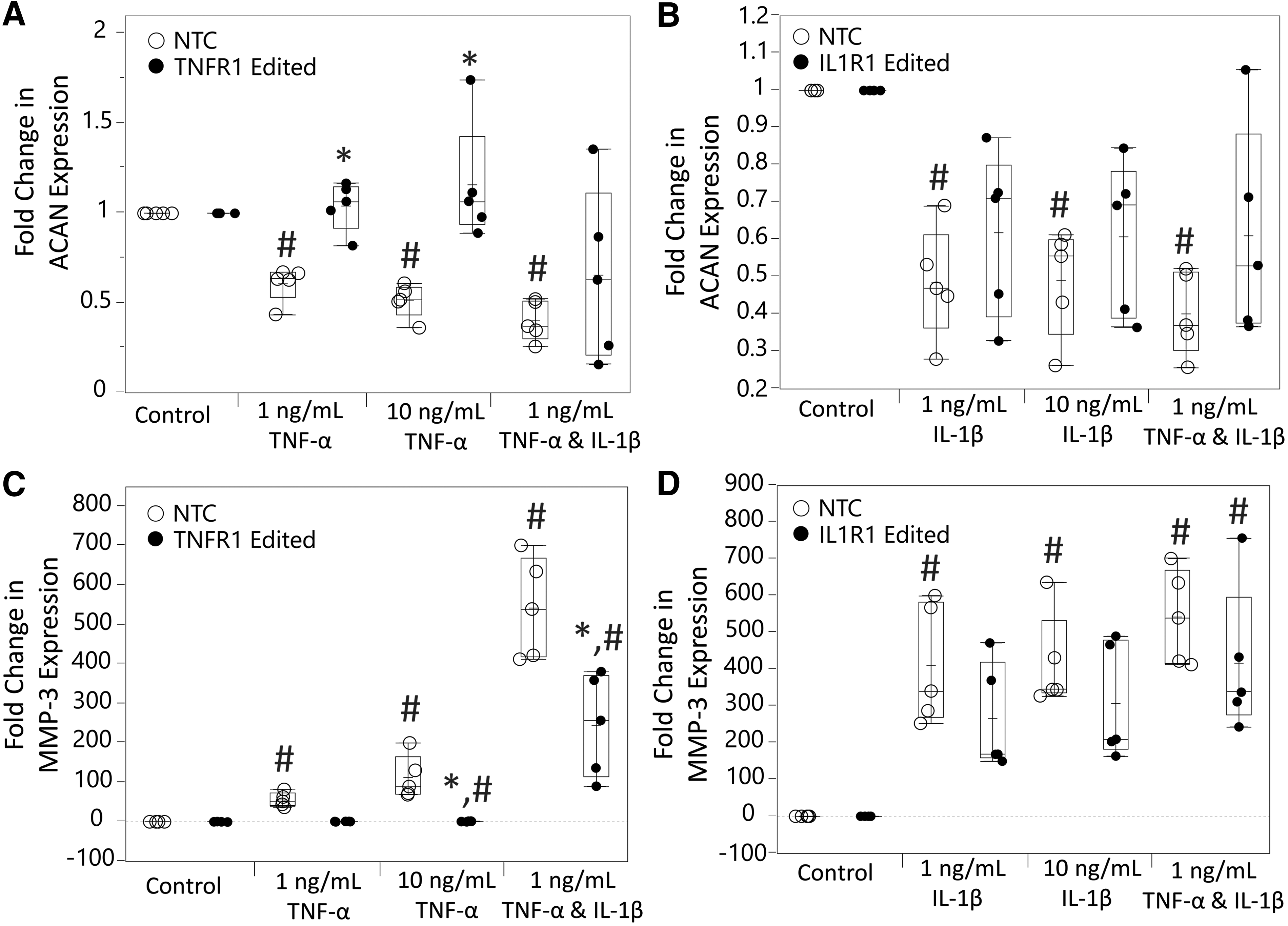
Measurement of inhibition of TNF-α–/IL-1β–induced changes in aggrecan (ACAN) and MMP-3 expression by epigenome editing systems in hNPCs through qRT-PCR.
IL1R1 epigenome-edited cells once again did not show robust effects, but there was some IL-1β signaling modulation, as ACAN decreases were nonsignificant in IL1R1 epigenome-edited cells compared with the untreated control (1 ng/mL: p = 0.13, 10 ng/mL: p = 0.0072, 1 ng/mL TNF-α and IL-1β: p = 0.038), whereas they were significant for all treatments in NTC cells (1 ng/mL: p = 0.0002, 10 ng/mL: p = 0.0006, 1 ng/mL TNF-α and IL-1β: p < 0.0001) (Fig. 4B).
Regarding MMP-3 expression induced by cytokine treatment, TNFR1 epigenome editing robustly protected against this as demonstrated by lowered TNF-α–induced MMP-3 induction in TNFR1 epigenome-edited cells relative to NTC (1 ng/mL: p = 0.0075, 10 ng/mL: p = 0.0004) and a significant overall decrease in MMP-3 induction with simultaneous TNF-α and IL-1β dosing (p = 0.0025) (Fig. 4C). IL1R1 epigenome editing did not significantly decrease TNF-α–/IL-1β–induced MMP-3 expression relative to NTC cells (1 ng/mL IL-1β: p = 0.25, 10 ng/mL IL-1β: p = 0.29, 1 ng/mL TNF-α and IL-1β: p = 0.17) (Fig. 4D).
Correlations of outcome measurements
Spearman's rho correlations of decrease in TNFR1 and IL1R1 expression resulting from epigenome editing, to all signaling measurement outcomes, demonstrated significant correlation (NF-κB activity: ρ = 0.0023, apoptosis: ρ ≤ 0.0001, ACAN expression: ρ ≤ 0.0001, MMP-3 expression: ρ = 0.0011). This indicates that gene downregulation by these systems directly correlates with the functional outcomes they deliver for both TNFR1 and IL1R1.
RNA-seq analysis
Gene expression analysis, by RNA-seq, of the effects of TNF-α dosing on hNPCs and how TNFR1 downregulation modifies those effects provided further evidence to support data from other assays and some additional insights into TNF-α signaling in human IVD cells. Differential expression analysis identified 3,706 genes that change expression in NTC cells when dosed by TNF-α, whereas in TNFR1 epigenome-edited cells only 11 genes were differentially expressed (Fig. 5A, B). Analysis of all genes differentially expressed after TNF-α treatment indicated several significantly modulated biological processes in NTC cells (top 10 biological processes for genes upregulated and downregulated shown in Fig. 5C).

RNA-seq analysis of changes in gene expression after TNF-α dosing in hNPCs with and without downregulation of TNFR1 by CRISPR epigenome editing.
Genes upregulated by TNF-α in NTC cells are related to inflammation (IL1B, IL6, IL6ST), ECM degradation (MMP3, MMP9), and apoptosis (CASP1, CASP8), whereas downregulated genes are associated with an anabolic cell phenotype (ACAN, GDF6, SOX9, TGFB1, TGFBR1, TIMP2) and cell survival in the hypoxic disc environment (HIF1A) (Fig. 5). Analysis of transcription factors associated with genes differentially expressed indicated 23 significantly associated transcription factors with the top 6 and their respective number of gene interactions shown in Fig. 5D.
Transcription factors associated such as STAT1, STAT3, and IRF1 have previously been associated with TNF-α signaling. 56,57 This is the first time IRF1 and TFAP2C have been associated with human IVD inflammatory signaling. Overall, all biological processes and transcription factor changes induced by TNF-α were inhibited by CRISPR epigenome targeting of TNFR1.
We also investigated potential off-target effects of our TNFR1 gRNA using RNA-seq data and the CRISPOR gRNA analysis tool. 58 Using CRISPOR, we identified 62 potential off-target genes. Only two of these potential off-target genes (NPTXR, Col21A1) were identified as significantly downregulated within RNA-seq data, indicating only two potential off-target genes being regulated by our TNFR1 epigenome-editing system. RNA-seq data is available on gene expression omnibus (GEO) database, under accession number GSE132958.
Analysis of maintenance of TNFR1 and IL1R1 downregulation
As correlations showed a significant relationship between TNFR1 and IL1R1 gene downregulation and signaling outcomes, but we saw little population level changes after IL1R1 epigenome editing, we looked deeper at expression of these receptors in epigenome-edited cells under inflammatory conditions. We measured TNFR1 and IL1R1 expression postcytokine dosing to investigate whether epigenome edits were maintained or reversed to help provide some reasoning behind why IL1R1 epigenome edits are not effective.
From this, we saw that TNFR1 downregulation was maintained with cytokine dosing (Fig. 6A) and that IL1R1 expression was being upregulated back to NTC levels in a subset of the patients after IL-1β dosing (Fig. 6B). We saw two patients maintained >30% downregulation of IL1R1 with IL-1β dosing and three patients whose IL1R1 levels returned to NTC levels after IL-1β dosing. The two patients who maintained IL1R1 downregulation had >70% downregulation of IL1R1 without IL-1β dosing. This indicates that there is a threshold of receptor downregulation that needs to occur in order for it to be maintained under inflammatory challenge.
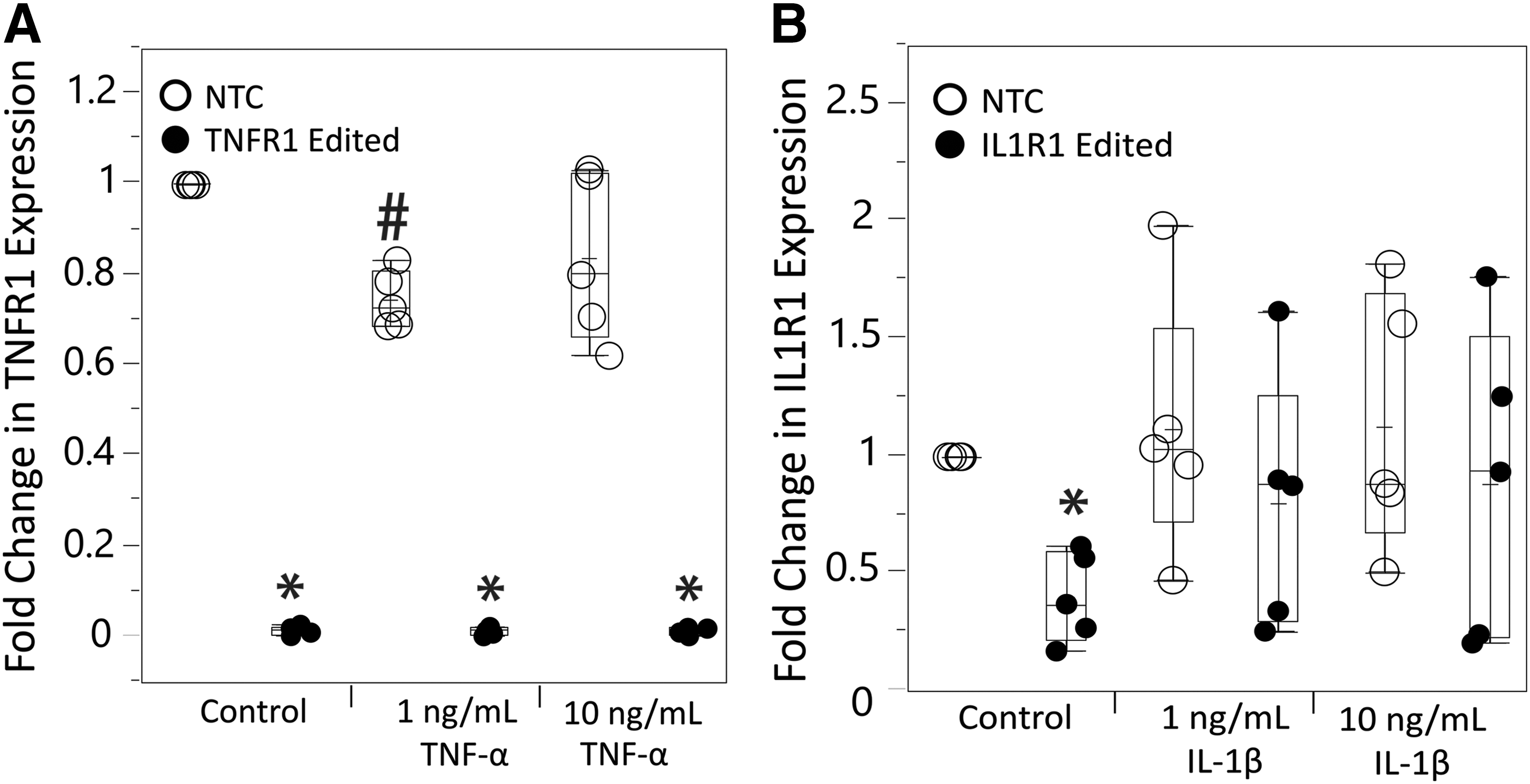
Measurement of changes in TNFR1
Discussion
The recent advent of CRISPR technologies has provided a highly useful set of gene regulation tools, which is being utilized to aid in treatment of a wide range of disease states. This study focuses on the application of CRISPR epigenome editing as a tool to modulate the response of human IVD cells to an inflammatory environment to prevent disease progression. Overall, we demonstrate an ability to regulate deleterious inflammatory signaling pathways through lentivirally delivered CRISPR epigenome editing machinery in clinically relevant pathological human IVD cells. This indicates that epigenome editing systems that provide control over endogenous gene expression have the potential to serve as a gene therapy for inhibition of disc degeneration.
This study obtained hNPCs from pathological IVD tissue and demonstrated an ability to successfully introduce epigenome editing systems into them and regulate expression of targeted inflammatory receptors. Transduction rates were up to 90% (Fig. 1), demonstrating an ability to successfully introduce these systems into IVD cells at high rates. This is of clinical importance as any gene therapy is only effective when it successfully transduces the cell at adequate efficiency. The method of entry was through lentivirus, which can be controversial due to potential carcinogenic effects of lentivirus induced by its nature of random integration of delivered genes in the genome.
Although this has been of concern in the past, it is less concerning with newer generations of lentivirus as their safety profiles have been improved to make them a more clinically viable gene delivery method. 59 This is evidenced by recent clinical trials utilizing lentivirus for direct gene delivery for treatment of Parkinson's and macular degeneration, which have not yet seen adverse effects related to the lentivirus. 60,61 Therefore, the idea of utilizing lentivirus for gene delivery to the IVD in a clinical setting is feasible.
Looking at gene regulation of TNFR1 and IL1R1 in transduced cells, there was significant gene downregulation for TNFR1 and IL1R1, but rates were more robust for TNFR1-edited cells (85–99%) than IL1R1-edited cells (36–94%) (Fig. 1). This demonstrates that our designed systems provide more consistent gene regulation of TNFR1 than of IL1R1, and that cell control over IL1R1 expression may be more complex and requires a more multifaceted approach.
Analysis of TNFR1/IL1R1 signaling through measurement of NF-κB activity demonstrated that receptor signaling was modulated by epigenome editing systems. Regulation of TNFR1 signaling was robust, with NF-κB induction significantly reduced at all doses of TNF-α and maintained close to baseline levels in TNFR1-edited cells (Fig. 2A). This demonstrates regulation of TNFR1 signaling with both low and high levels of TNF-α present. Although reported levels of TNF-α in the degenerative disc are generally in the picogram range (0.1–400 pg/mL), 19,20,62 this provides assurance that TNFR1 signaling will continue to be inhibited even if there are large local fluctuations in TNF-α, which can be observed after injury. 63
Regulation of IL1R1 signaling was insignificant with only slight decreases in mean NF-κB induction at 150 pg/mL IL-1β (Fig. 2B). Based on outcomes of gene regulation, this is not surprising as IL1R1 expression regulation was less robust; therefore, more IL1R1 was still present to allow for NF-κB activation. Overall, we demonstrate that these epigenome editing systems are able to regulate the signaling of these inflammatory receptors, when there is robust gene expression downregulation.
Cell death within inflammatory environments is known to aid in progression of DDD as it has been demonstrated that these environments induce apoptosis through signaling through inflammatory receptors, including TNFR1 and IL1R1. 17,18,28 We measured whether epigenome editing systems repressing TNFR1 or IL1R1 were able to attenuate cytokine-induced apoptosis. Our measurements indicate that our TNFR1 epigenome editing system successfully inhibits TNF-α–mediated apoptosis in pathological human IVD cells, whereas our IL1R1 epigenome editing system is not able to inhibit apoptosis in these cells (Fig. 3).
In addition, inhibition of TNFR1 expression by epigenome editing attenuated the apoptotic effects of TNF-α and IL-1β treatment combined. This shows that TNFR1 inhibition alone may help protect against the catabolic effects of IL-1β. This is potentially explained by an increased ratio of TNFR2/TNFR1 signaling that shifts TNF-α signaling to the TNFR2 pathway, which is considered antiapoptotic and regenerative and may counteract IL-1β signaling effects. 26,36
Additional mechanisms of TNFR1/IL1R1 signaling involvement in DDD are their induction of catabolic gene expression changes. 15,16,26,27,28 These include genes that mediate ECM synthesis and breakdown. Aggrecan is an important ECM component of the IVD and its decreased synthesis caused by inflammatory signaling plays a role in DDD. 10,14,15 Matrix metalloproteinases also play a large role in DDD as they mediate the breakdown of IVD tissue. 12,14,16,27 We measured expression of ACAN and a matrix metalloproteinase, MMP-3, postepigenome editing to determine whether catabolic changes in expression are attenuated.
Our measurements showed that TNFR1 epigenome editing strongly attenuated these changes, whereas IL1R1 epigenome editing showed no significant attenuation (Fig. 4). As was also shown in apoptosis measurements, in TNFR1-edited cells, catabolic gene expression changes induced by TNF-α and IL-1β dual presence were significantly attenuated. This further indicates that effective TNFR1 inhibition on its own may have a therapeutic effect. This idea is also supported by a recent study that demonstrates TNF-α inhibition protects against painful disc degeneration in an animal model, when anti-TNF-α is administered at initiation of degeneration. 25 This and our data support TNFR1 epigenome editing alone as a potential therapeutic strategy.
To further understand pathways being modulated by our CRISPR epigenome editing of TNFR1, we performed RNA-seq on hNPCs (NTC and TNFR1 edited) with and without TNF-α dosing. Our results demonstrated that TNFR1 epigenome editing effectively abolishes nearly all gene expression changes associated with TNF-α treatment (Fig. 5B). This includes increase in expression of catabolic genes (CASP1, CASP8, IL1B, IL6, IL6ST, MMP3, MMP9) and decrease in expression of anabolic genes (ACAN, GDF6, HIF1A, SOX9, TGFB1, TGFBR1, TIMP2). Therefore, TNFR1 downregulation inhibits TNF-α–mediated inflammation, cell death, and catabolism.
The biological processes involving genes downregulated by TNF-α are associated with ECM organization, cartilage development, chondrogenesis, and regulation of cell proliferation and apoptosis, indicating decrease in a phenotype that mediates anabolic effects and improves cell survival (Fig. 5C). The biological processes involving genes upregulated in NTC cells overall encompassed enhancement of inflammatory response, especially through IFN signaling (Fig. 5C). These processes involved with gene upregulation and downregulation associated with TNF-α signaling did not appear in analysis of TNFR1-edited cells, indicating that TNFR1 downregulation effectively abolishes activation of biological processes associated with TNF-α signaling.
Looking at the top six transcription factors associated with TNF-α treatment in NTC cells (Fig. 5), all but IRF1 and TFAP2C (also known as AP2-Gamma) have been studied in cells of the IVD and demonstrated relevance in disc degeneration. 64 –67 This indicates that IRF1 and TFAP2C could be targets of interest to pursue in future work in controlling response to TNF-α in IVD cells and in studying basic disc degeneration biology.
Analyzing the specificity of our TNFR1 downregulating, CRISPR epigenome editing system using the RNA-seq expression data indicated that there were 2 potential off-target genes significantly regulated by our CRISPR system out of 62 potential off-target genes, and their regulation was modest compared with the targeted gene. This limitation will be considered in future work by using CRISPR systems that have been modified to reduce off-target effects. 39
Overall, all of these data indicate that when robust gene regulation is achieved, epigenome editing of inflammatory receptors can modulate inflammatory signaling in pathological IVD cells. We confirmed this through significant Spearmans rho correlations of outcome measurements to TNFR1/IL1R1 downregulation. This along with the data demonstrating that IL1R1 expression downregulation was less robust helps explain why IL1R1 regulation was ineffective in these studies, but there was still the question of why there was no effect even with rates of up to 94% downregulation. To help answer this, we measured TNFR1 and IL1R1 expression under inflammatory challenge.
This experiment demonstrated that TNFR1 downregulation was maintained with cytokine dosing, and IL1R1 downregulation was only maintained in some patient cells with cytokine dosing. In the majority of patient IVD cells, IL1R1 expression went back to control cell levels (Fig. 6). This indicates that our epigenome editing systems are not always able to maintain IL1R1 gene downregulation, meaning our IL1R1 regulation approach requires improvement to be beneficial to the majority of patients.
The reversal of IL1R1 epigenome editing modifications may be due to IL1R1 gene expression being more heavily regulated by epigenome modifications than TNFR1. If this is the case, then epigenome modifications may be overcome by internal epigenome modifying machinery. Another mechanism may be IL1R1 expression activation at other transcription start sites as it has several transcription start sites that are ∼30–70 kb away from each other. 49 Regulatory elements of TNFR1 expression are all concentrated within one region as it has only one known transcription start site. 49 This may be why TNFR1 signaling downregulation was more robust.
Approaches to achieve better IL1R1 regulation can include use of CRISPR epigenome editing systems that methylate the DNA to provide a more stable modification, 68 multiplex targeting of IL1R1 regulatory elements, or use of CRISPR gene editing methods to knockout IL1R1 that have shown effectiveness in other cell types. 69,70 Future work will focus on novel methods of regulating IL-1β signaling, and multiplex regulation of IL1R1 and TNFR1 signaling will be of great interest. Although, our results and a recently published animal model 25 demonstrate that TNFR1 inhibition itself can provide protection in the context of disc degeneration. Therefore, investigating effects of TNFR1 downregulation within more dynamic in vivo settings will be of primary interest for future work.
Conclusion
In this study we demonstrate that CRISPR epigenome editing systems can be lentivirally introduced into pathological human IVD cells to regulate their expression of inflammatory receptors. This regulation results in attenuation of inflammatory pathways, which propagate the deleterious effects of inflammatory cytokines in the IVD. This was demonstrated by decreased NF-κB activation, decreased apoptosis, and inhibition of catabolic gene expression changes in TNFR1 epigenome-edited cells.
As seen with IL1R1, there is the limitation of gene downregulation levels being dependent on the genetic target, and reversal of targeted gene downregulation. This indicates that the epigenome editing approach may need to be tailored for each gene to result in successful regulation (i.e., use of DNA methylation systems, multiplex targeting of regulatory elements). Overall it was demonstrated that CRISPR epigenome editing can regulate signaling of inflammatory receptors in pathological IVD cells and is a promising gene therapy strategy for inhibition of disc degeneration.
Footnotes
Acknowledgments
We thank Jake Weston for cell culture support in this study, and Joshua Stover for helping build the epigenome editing plasmids used in this study. Research reported in this publication was supported by the National Institutes of Health (Grant Nos. R03AR068777 and 1S10RR026802-01); AO foundation startup grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.