
Abstract
Safe delivery of CRISPR/Cas endonucleases remains one of the major barriers to the widespread application of in vivo genome editing. We previously reported the utility of adeno-associated virus (AAV)-mediated CRISPR/Cas genome editing in the retina; however, with this type of viral delivery system, active endonucleases will remain in the retina for an extended period, making genotoxicity a significant consideration in clinical applications. To address this issue, we have designed a self-destructing “kamikaze” CRISPR/Cas system that disrupts the Cas enzyme itself following expression. Four guide RNAs (sgRNAs) were initially designed to target Streptococcus pyogenes Cas9 (SpCas9) and after in situ validation, the selected sgRNAs were cloned into a dual AAV vector. One construct was used to deliver SpCas9 and the other delivered sgRNAs directed against SpCas9 and the target locus (yellow fluorescent protein [YFP]), in the presence of mCherry. Both constructs were packaged into AAV2 vectors and intravitreally administered in C57BL/6 and Thy1-YFP transgenic mice. After 8 weeks, the expression of SpCas9 and the efficacy of YFP gene disruption were quantified. A reduction of SpCas9 mRNA was found in retinas treated with AAV2-mediated YFP/SpCas9 targeting CRISPR/Cas compared with those treated with YFP targeting CRISPR/Cas alone. We also show that AAV2-mediated delivery of YFP/SpCas9 targeting CRISPR/Cas significantly reduced the number of YFP fluorescent cells among mCherry-expressing cells (∼85.5% reduction compared with LacZ/SpCas9 targeting CRISPR/Cas) in the transfected retina of Thy1-YFP transgenic mice. In conclusion, our data suggest that a self-destructive “kamikaze” CRISPR/Cas system can be used as a robust tool for genome editing in the retina, without compromising on-target efficiency.
Introduction
Inherited retinal diseases are disabling disorders of visual function that affect millions of people worldwide. With the development of next-generation sequencing and better molecular diagnostic techniques, numerous genetic variants across many loci have been definitively associated with inherited retinal diseases. 1,2 Despite this increase in our understanding of genetic etiology and potential therapeutic targets, there remains no effective treatment for the majority of inherited retinal diseases. Although significant progress in gene therapy has been achieved over the last two decades, there are few sustained, safe, and effective ocular gene therapies for hereditary retinal diseases. 3 –5
Advances in genome editing techniques, in particular the recent advances in CRISPR/Cas technology, 6 have renewed excitement in ocular gene-based therapy. The CRISPR/Cas system has evolved in archaea and bacteria as a defence against viral intrusion and has been adapted to allow efficient editing of mammalian nuclear genomes. 6 CRISPR/Cas-based technology has proven to be a robust means for in vitro correction of genetic mutations in mammalian cells and is particularly attractive for treating inherited retinal diseases. 7 A number of in vivo studies in various animal models have yielded promising results for pre-emptive therapy for well-characterized monogenic ocular diseases. Bakondi et al. 8 and Latella et al. 9 report that successful ablation of the mutated rhodopsin gene prevented retinal degeneration in rodent models of severe autosomal dominant retinitis pigmentosa following electroporation of the CRISPR/Cas9 system into the retina. More recently, Yu et al. 10 demonstrated that CRISPR/Cas9-mediated disruption of a neural retina-specific leucine zipper protein (NRL) significantly improved rod survival and preserved cone function in a murine model of retinal degeneration. We were able to achieve high-efficiency genome editing in mouse retina using a dual AAV2-mediated CRISPR/Cas9 system. 11 However, potentially deleterious effects of prolonged overexpression of CRISPR/Cas endonuclease, including elevated off-target cleavage, 12,13 and cellular immune responses, 14 remain important safety hurdles to clinical application.
To address this, we have designed a self-destructive “kamikaze” CRISPR/Cas system that disrupts the CRISPR/Cas gene after active protein expression (Fig. 1). To determine the efficacy of in vivo genome editing by our “kamikaze” CRISPR/Cas construct, a Streptococcus pyogenes Cas9 (SpCas9) targeting sgRNA module, together with a yellow fluorescent protein (YFP) targeting sgRNA, was packaged into a dual AAV2 vector system for intravitreal delivery in Thy1-YFP transgenic mice.
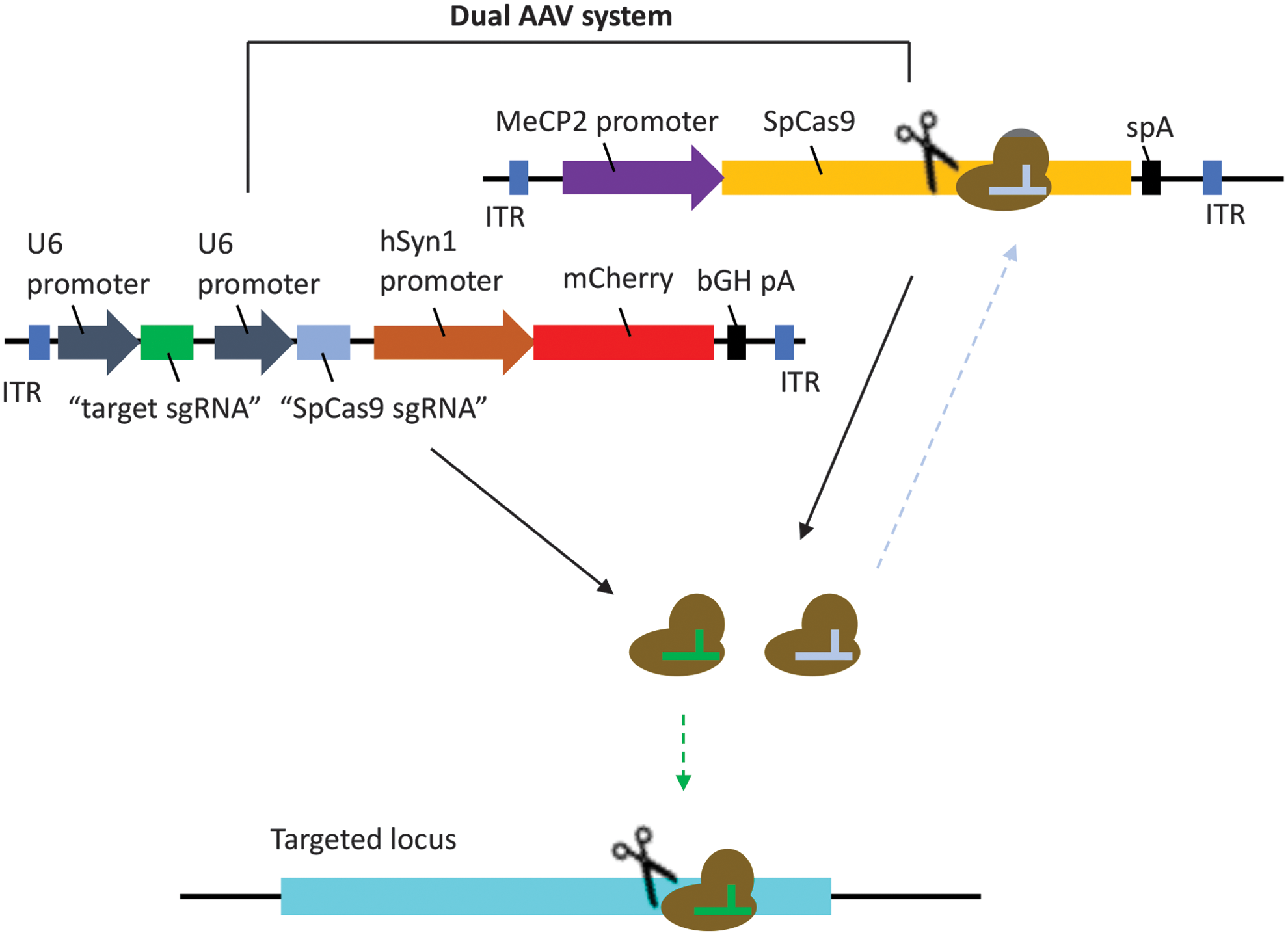
Schematics of kamikaze CRISPR/Cas system. A dual AAV vector system was used. One viral vector was used to deliver SpCas9 and the other delivered sgRNAs against SpCas9 and the target locus (YFP), in the presence of mCherry. AAV, adeno-associated virus; SpCas9, Streptococcus pyogenes Cas9; YFP, yellow fluorescent protein. Color images are available online.
Materials and Methods
Animals and housing
All procedures were conducted according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the requirements of the National Health and Medical Research Council of Australia (Australian Code of Practice for the Care and Use of Animals for Scientific Purposes). Ethics approval was obtained from the Animal Ethics Committees of the University of Tasmania (A14827) and St. Vincent's Hospital Melbourne (AEC 014/15). Thy1-YFP transgenic mice [B6.Cg-Tg(Thy1-YFP)16Jrs/J] were obtained from the Jackson Laboratory (mouse stock no. 003709; Bar Harbor, ME) and bred at the animal facility of the University of Tasmania (Hobart, Australia). C57BL/6 mice were purchased from the Animal Resources Centre (Perth, Australia). Mice were housed under standard conditions (20°C, 12/12-h light/dark cycle) with ad libitum access to food and water.
sgRNA design and vector construction
Four sgRNAs targeting the SpCas9 sequence were designed using a web-based CRISPR design tool (Zhang Lab, MIT). CRISPR/Cas in situ testing was carried out by incubating the individual synthetic SpCas9 sgRNA or LacZ sgRNA alone with the recombinant SpCas9 protein (catalog no. M0386S; New England Biolabs, Ipswich, MA) and the pX551 plasmid (SpCas9 construct; kindly provided by Feng Zhang, Addgene, catalog no. 60957). Samples were run on a 0.8% TAE agarose gel to visualize their cleavage efficiency for SpCas9. AgeI (catalog no. R0552S; New England Biolabs) digested pX551 plasmid was used as a positive control. Four SpCas9 sgRNAs were then cloned into a pX552-CMV-GFP plasmid (modified from Addgene, catalog no. 60958, by replacing the hSYN1 promoter with a CMV promoter) at the SapI restriction site for in vitro validation. Subsequently, the selected SpCas9 sgRNA (SpCas9 sgRNA4) was subcloned into an adeno-associated virus (AAV) package plasmid (pX552-hSYN1-mCherry-YFP sgRNA2, sgRNA6, or pX552-LacZ sgRNA) at the MluI (catalog no. R3198; New England Biolabs) restriction site to generate YFP or LacZ targeting kamikaze CRISPR/Cas9 construct. For in vitro validation, pX551-CMV-SpCas9 plasmid was modified from pX551 plasmid by replacing the MeCP2 promoter with a CMV promoter.
Cell culture and transfection
Stable YFP expressing HEK293A cells were generated using a lentivirus as previously described. 11,15 HEK293A-YFP cells were maintained in Dulbecco's modified Eagle's media (catalog no. 11965118; Life Technologies Australia, Scoresby, Australia) supplemented with 10% fetal calf serum (Sigma-Aldrich, St. Louis, MO), 2 mM glutamine (catalog no. 2503008; Life Technologies Australia), and 50 U/mL penicillin/streptomycin (catalog no. 15070063; Life Technologies Australia) in a humidified 5% CO2 atmosphere at 37°C. Transfection was undertaken with the FuGENE-HD transfection reagent (catalog no. E2311; Promega Australia, Alexandria, Australia) according to the manufacturer's instruction. Briefly, HEK293A-YFP cells were seeded onto a 6-well plate (2.5 × 105 per well) 24 h before transfection. A mixture of 7.5 μL FuGENE-HD transfection reagent with 1500 ng plasmid (750 ng/plasmid was used in dual-plasmid transfection) in 150 μL Opti-MEM (catalog no. 11058021; Life Technologies Australia) was added into each well. For in vitro validation of SpCas9 sgRNA, cells were collected for Western blot analysis at day 3 after transfection; for in vitro time course analysis, cells were harvested at day 1, 2, 3, 5, and 7 after transfection.
Western blot analysis
Cells were collected and lysed in ice-cold cell lysis buffer (catalog no. 89900; Thermo Fisher Scientific, Waltham, MA) and sonicated for 10 s by an ultrasonic cell disruptor (MISONIX Microson XL 2000; Qsonica, Newtown, CT). Total protein was quantified by a Bio-Rad protein assay (catalog no. 5000006; BIO-RAD, Hercules, CA) using a microplate reader (Infinite M1000 Pro; TECAN, Mannedorf, Switzerland). A total of 10 μg protein samples was separated using NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels (catalog no. NP0321BOX; Life Technologies Australia) and transferred to polyvinylidene fluoride membranes (catalog no. 162-0177; BIO-RAD) using the XCell II™ Blot Module (Life Technologies Australia). Membranes were blocked with 5% skim milk in TBS-T (10 mM Tris, 150 mM NaCl, and 0.05% Tween-20) at room temperature for 1 h and then incubated with the mouse monoclonal SpCas9 antibody (1:1,000 dilution, MAC133, lot number 2591899; Millipore, Billerica, MA) or mouse monoclonal β-actin antibody (1:2,000 dilution, MAB 1501, lot number 2722855; Millipore) at room temperature for 1 h. Membranes were washed, further incubated with the horseradish peroxidase-conjugated goat anti-mouse secondary antibody (1:5,000 dilution, catalog no. A-11045; Life Technologies Australia) at room temperature for 1 h, and developed using the Amersham ECL Prime Western Blotting Detection kit (catalog no. RPN2232; GE Healthcare Australia, Parramatta, Australia). The relative levels of SpCas9 protein of each sample were quantified using densitometry analysis (ImageJ software gel analysis) with normalization to β-actin.
YFP detection
YFP expressing HEK293A cells were trypsinized and harvested in PBS. Cells were stained with DAPI (5 μg/mL) to exclude the dead cells. The percentage of YFP-positive cells was then analyzed from a live-cell population with a Flow Cytometer (BD FACS Canto II; BD bioscience, Sparks, MD) and data were analyzed using FACS analysis software (FlowJo®; FlowJo LLC, Ashland, OR).
AAV production
Recombinant AAV2 was produced in HEK293D cells (kindly provided by Ian Alexander, Children's Medical Research Institute, Westmead, Australia), packaging either pX551 plasmid, containing SpCas9, or pX552-mCherry plasmid with the respective sgRNAs (SpCas9, YFP, or LacZ sgRNA), pseudoserotyped with the AAV2 capsid (pXX2), 16 and purified using an AAV2pro Purification Kit (catalog no. 6232; Clontech Laboratories, Mountain View, CA) as previously described. 11,15 Viral titer was determined by real-time quantitative PCR using a Fast SYBR Green Master Mix (catalog no. 4385612; Life Technologies Australia) with the pX551 or pX552 forward and reverse primers (Supplementary Table S1).
Off-target effect assessment
Off-target mutagenesis for the YFP targeting sgRNAs, YFP sgRNA2 and YFP sgRNA6, as well as SpCas9 targeting sgRNA, SpCas9 sgRNA4, was assessed using the online prediction tool (Cas-OFFinder) in the Mus musculus genome. Potential off-target effects were further investigated using the method described by Chen et al. 17 Briefly, 10 guide RNA sequences were designed to contain a single-nucleotide mismatch between the spacer and protospacer target at the YFP gene locus targeted by YFP sgRNA6, spanning from 5′ to the protospacer adjacent motif. These mismatch-containing guide RNAs (sgRNA M2–M20) were generated in conventional and kamikaze AAV-CRISPR/Cas9 systems and transfected in the YFP expressing HEK293A cells. Genomic DNA was extracted (catalog no. D4068; Zymo Research, Irvine, CA) at day 10 posttransfection, and T7E1 assay was performed at YFP targeting loci with forward and reverse primers (Supplementary Table S1) by using an EnGen Mutation Detection Kit (catalog no. E3321; New England Biolabs). Mismatch cleavages by T7E1 were analyzed by ImageJ. For Sanger sequencing, PCR amplicon (885 bp) from genomic DNA was purified using the DNA Clean & Concentrator kit (catalog no. D4033; Zymo Research) and sequenced in-house on the Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific) using YFP reverse sequencing primer (Supplementary Table S1). Sanger files were analyzed for insertions and deletions using the inference of CRISPR edits (ICE) tool (Synthego).
Intravitreal injection
For our in vivo time course analysis, a total of 76 C57BL/6 adult mice, aged between 12 and 14 weeks, were randomly separated into two groups, to receive either AAV2-SpCas9+AAV2-YFP sgRNA2 (n = 39) or AAV2-SpCas9+AAV2-SpCas9 sgRNA/YFP sgRNA2 (n = 37). For the YFP disruption experiments, a total of 49 Thy1-YFP transgenic mice, aged between 16 and 20 weeks, were randomly allocated into three groups: those receiving AAV2-SpCas9+AAV2-YFP sgRNA2 (n = 17), AAV2-SpCas9+AAV2-SpCas9 sgRNA/YFP sgRNA2 (n = 17), or AAV2-SpCas9+AAV2-SpCas9 sgRNA/LacZ sgRNA (n = 15). In addition, another 29 Thy1-YFP transgenic mice were used to test different YFP sgRNAs. These mice were randomly allocated into three groups: those receiving AAV2-SpCas9+AAV2-YFP sgRNA6 (n = 9), AAV2-SpCas9+AAV2-SpCas9 sgRNA/YFP sgRNA6 (n = 10), or AAV2-SpCas9+AAV2-SpCas9 sgRNA/LacZ sgRNA (n = 10).
Mice were anesthetized by intraperitoneal injection of ketamine (60 mg/kg) and xylazine (10 mg/kg). 11 Intravitreal injection was performed under a surgical microscope. After a small puncture was made through the conjunctiva and sclera using a 30-gauge needle, a hand-pulled glass micropipette connected to a 10 μL Hamilton syringe (Bio-Strategy, Broadmeadows, Australia) was inserted into the vitreous. A total of 1 μL dual-viral suspension (AAV2-SpCas9: 2.5 × 109 vector genomes [vg]/μL with AAV2-YFP sgRNA: 2.5 × 109 vg/μL, AAV2-SpCas9 sgRNA/YFP-sgRNA: 2.5 × 109 vg/μL, or AAV2-SpCas9 sgRNA/LacZ sgRNA: 2.5 × 109 vg/μL) was injected into one eye of each mouse using a UMP3-2 Ultra Micro Pump (World Precision Instruments, Sarasota, FL) at a rate of 200 nL/s. Any issues with the injection, including backflow upon removal of the needle and hemorrhaging of the external or internal vessels, retinal detachment was recorded and eyes were excluded from the study.
Electroretinography and optical coherence tomography
At 8 weeks following injection, mice underwent overnight dark adaptation (∼12 h), followed by electroretinography (ERG) assessment under fully dark-adapted conditions. Details for functional assessment have been outlined previously, 11,18 with the exception that the reference chloride silver electrode was placed around the outside of the eye. ERG analysis was as previously described 11,18 and returned the photoreceptor (a-wave)-, bipolar cell (b-wave)-, and ganglion cell-dominated (scotopic threshold response) components of the waveform. Group data are given as mean (± standard error of the mean [SEM]).
Following ERG recordings, retinal images were obtained using a spectral domain optical coherence tomography (OCT; Bioptigen, Inc., Morrisville, NC). Mice were positioned to capture optic nerve head (ONH)-centered 1.4-mm-wide horizontal B-scans (consisting of 1,000 A-scans). ImageJ software was used in a masked manner to quantify total retinal thickness (from the inner limiting to Bruch's membrane), retinal nerve fiber layer thickness (from the inner limiting membrane to the inner aspect of the inner plexiform layer), and outer retinal thickness (from Bruch's membrane to the outer plexiform layer) in each eye.
Retinal flat mount imaging and counting
Eyes were removed, fixed in ice-cold 4% paraformaldehyde for 1 h, and dissected under a dissecting microscope. After removing the cornea, iris, and lens, four equally spaced radial relaxing incisions, extending two-thirds of the way from the retinal periphery to the ONH, were made. The sclera and choroid were then removed along with residual vitreous and hyaloid vessels, leaving only the retina. The fully dissected retina was stained with NucBlue™ Live ReadyProbes™ reagent (catalog no. R37605; Life Technologies Australia) as a nuclear counterstain. Retinal images were captured by a fluorescence microscope (Zeiss Axio Imager Microscope; Carl-Zeiss-Strasse, Oberkochen, Germany) equipped with a charge-coupled digital camera (AxioCam MRm; Zeiss) and image acquisition software (ZEN2; Zeiss) as previously described. 11
The efficiency of YFP disruption was quantified using individual fluorescent images captured at × 400 magnification. A total of 24 images from three flat-mounted eyes treated with SpCas9 sgRNA/LacZ sgRNA, 36 images from five flat-mounted eyes treated with SpCas9 sgRNA/YFP sgRNA2, and 36 images from five flat-mounted eyes treated with YFP sgRNA2 were quantified manually using ImageJ v1.49 by an experienced grader (F.L.), masked to treatment status. For the second experiment with YFP sgRNA6, a total of 16 images from three flat-mounted eyes treated with SpCas9 sgRNA/LacZ sgRNA, 38 images from five flat-mounted eyes treated with SpCas9 sgRNA/YFP sgRNA6, and 36 images from six flat-mounted eyes treated with YFP sgRNA6 were quantified. Efficiency for each treatment group was determined as the proportion of YFP-negative cells relative to mCherry-expressing cells as previously described. 11
Statistical analyses
All statistical analyses were performed using Prism 7 software (GraphPad Software, Inc., La Jolla, CA). Group data are represented as mean ± SEM unless otherwise noted. Mean data were analyzed with unpaired t-tests and one-way or two-way analysis of variance followed by post hoc analysis (GraphPad Prism 7.0). A value of p < 0.05 was taken to be statistically significant.
Results
Generation and validation of kamikaze CRISPR/Cas construct in vitro
We first validated four sgRNAs (Fig. 2A) for SpCas9 targeting using an in situ cleavage assay. Robust cleavage of the SpCas9 plasmid (pX551) was found when each of the four designed SpCas9 sgRNAs was introduced to recombinant SpCas9 protein (Fig. 2B). We further confirmed the efficacy of SpCas9 gene perturbations by transfection of the SpCas9 expression construct (pX551-CMV-SpCas9) together with SpCas9 targeting CRISPR/Cas constructs carrying different SpCas9 sgRNAs (pX552-SpCas9 sgRNA1-4) into HEK293A cells. SpCas9 sgRNA4 had a clear destructive effect on SpCas9 (Fig. 2C), reduction of SpCas9 protein, as well as having a lower off-target score against the human genome as predicted by a web-based CRISPR design program (Zhang Lab, MIT). A time course analysis showed that SpCas9 protein was progressively reduced in cells following the transfection of selected SpCas9 targeting CRISPR/Cas construct (pX552-SpCas9 sgRNA4) compared with LacZ sgRNA control (pX552-LacZ sgRNA; p < 0.05; Fig. 2D, E).

Design and validation of SpCas9 sgRNA.
We next re-engineered our kamikaze CRISPR/Cas construct with YFP targeting sgRNAs or an LacZ targeting sgRNA (Fig. 3A), and the efficacy of YFP gene disruption in the YFP-expressing HEK293A cells was assessed. Two days after transfection, we observed a reduction of SpCas9 protein in cells that had received the kamikaze CRISPR/Cas construct compared with those cells that had received the conventional CRISPR/Cas construct (Fig. 3B). In terms of efficiency, the percentage of YFP-expressing cells was significantly reduced in cells transfected with the YFP targeting kamikaze CRISPR/Cas constructs (SpCas9 sgRNA/YFP sgRNA2: 10.0% ± 1.0% and SpCas9 sgRNA/YFP sgRNA6: 5.6% ± 0.4% respectively), compared with LacZ targeting kamikaze (SpCas9 sgRNA/LacZ sgRNA: 85.9% ± 1.3%) or LacZ targeting (LacZ sgRNA: 86.0% ± 1.4%) CRISPR/Cas construct at 10 days after transfection (Fig. 3C, D). Similarly, a lower percentage of YFP expressed cells were also found in cells transfected with the YFP targeting CRISPR/Cas construct (YFP sgRNA2: 16.7% ± 2.7% and YFP sgRNA6: 7.4% ± 1.4%, respectively; Fig. 3C, D).
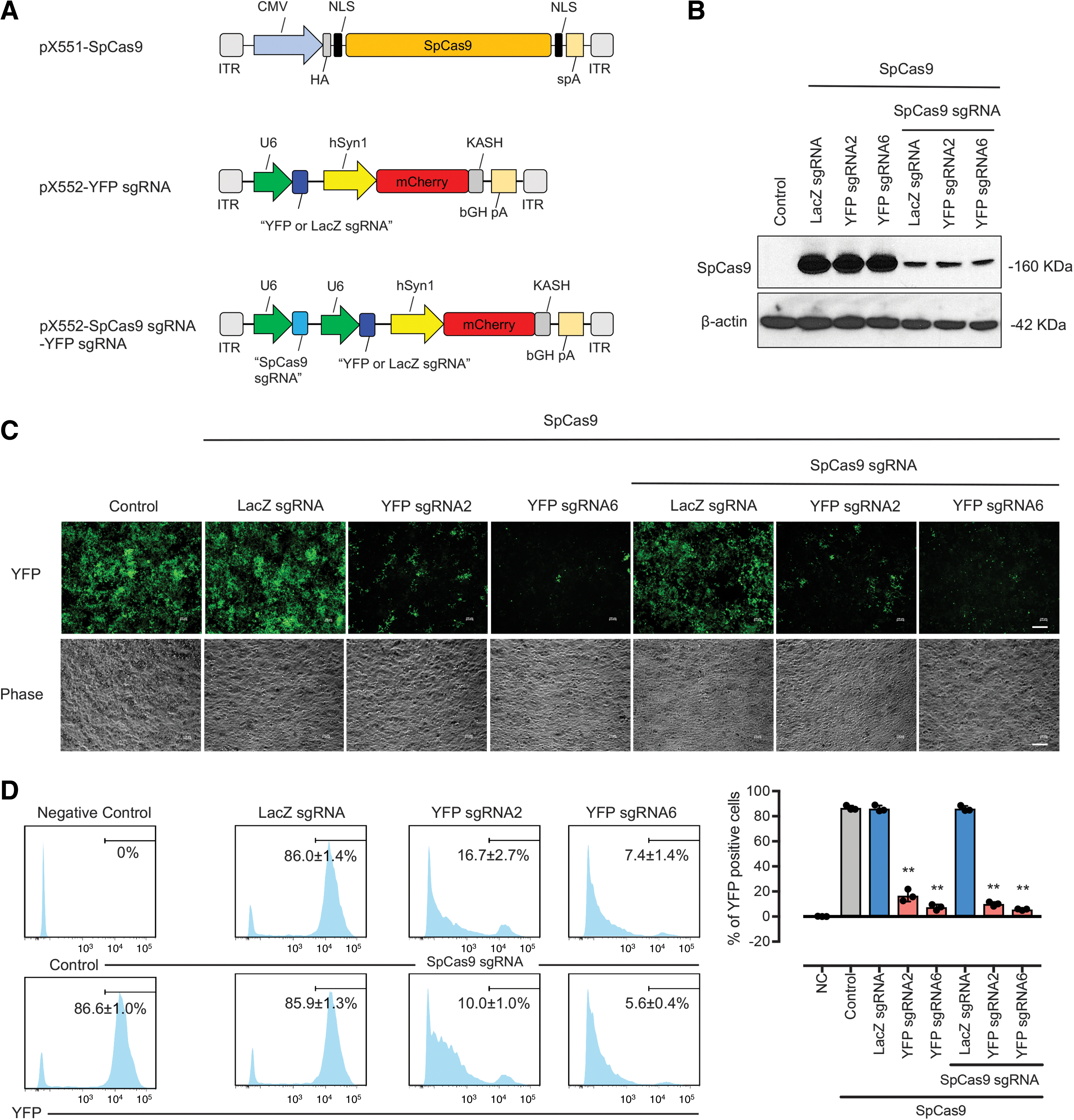
In vitro validation of kamikaze CRISPR/Cas construct.
In silico prediction of off-target sites for YFP sgRNA6 and SpCas9 sgRNA4 did not produce any significant candidates for testing, as both YFP and SpCas9 sequences were not found endogenously in the mouse genome. Next, we created a series of putative off-target sites by introducing a single-nucleotide mismatch along the YFP sgRNA6 sequence and compared the editing activity of conventional and kamikaze CRISPR/Cas systems (Fig. 4A). A similar frequency of indels was induced by conventional or kamikaze CRISPR/Cas systems for knockout of YFP (Supplementary Fig. S1). Almost all guide RNAs with single-nucleotide mismatches displayed editing activity at day 10 (Fig. 4B) with the highest editing produced by a mismatch located distal to the PAM (M18) at day 10. A statistical reduction of editing activity at M2–M8 and M20 mutant guide RNA positions was observed with our kamikaze CRISPR/Cas system compared with the conventional CRISPR/Cas system (Fig. 4C).
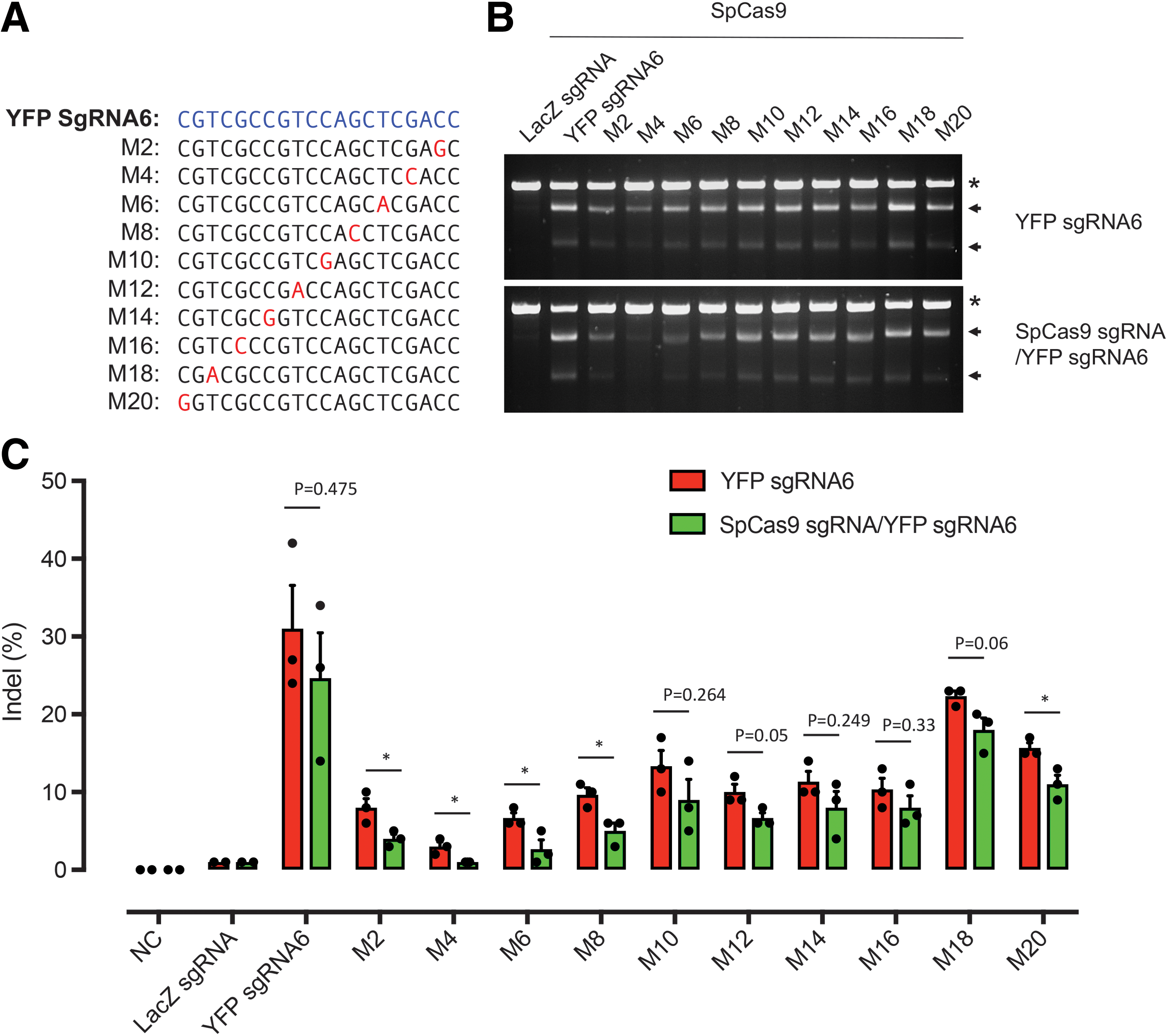
Kamikaze CRISPR/Cas reduced potential off-target in vitro.
In vivo delivery of kamikaze CRISPR/Cas construct in the mouse retina
To evaluate whether the reduction of SpCas9 expression by the kamikaze CRISPR/Cas construct compromises on-target editing efficiency, Thy1-YFP mice received a single intravitreal injection of a dual viral suspension of AAV2-SpCas9 along with the YFP targeting kamikaze CRISPR/Cas construct (AAV2-SpCas9 sgRNA/YFP sgRNA) or the LacZ targeting kamikaze CRISPR/Cas construct (AAV2-SpCas9 sgRNA/LacZ sgRNA) or a single YFP targeting CRISPR/Cas construct as a positive control (AAV2-YFP sgRNA2). The procedures for our in vivo study are shown in Fig. 5A. Eight weeks following treatment, images from the retinal flat mounts showed that there were fewer YFP-positive cells among mCherry-positive cells in mice that had received either AAV2-SpCas9 sgRNA/YFP sgRNA or AAV2-YFP sgRNA2 compared with AAV2-SpCas9 sgRNA/LacZ sgRNA (Fig. 5B). Specifically, the proportion of retinal YFP/mCherry-expressing cells was reduced to 5.5% ± 1.4% in AAV2-SpCas9 sgRNA/YFP sgRNA2-treated retina and 7.3% ± 1.3% in AAV2-YFP sgRNA2-treated retina, compared with 38.2% ± 1.7% in AAV2-SpCas9 sgRNA/LacZ sgRNA-treated eyes. Overall, there was an 85.5% (95% confidence interval [CI]: 78.4–92.6) and 80.9% (95% CI: 74.3–87.5) reduction in YFP-positive cells in AAV2-SpCas9 sgRNA/YFP sgRNA- and AAV2-YFP sgRNA2-treated retinas, respectively, compared with AAV2-SpCas9 sgRNA/LacZ sgRNA-treated eyes (Fig. 5C). No significant difference in the percentage of YFP disruption was found in between AAV2-YFP sgRNA2- and AAV2-SpCas9 sgRNA/YFP sgRNA2-treated retinas (p = 0.62; Fig. 5C). This was confirmed by using an alternate YFP targeting sgRNA (YFP sgRNA6), where the proportion of retinal YFP/mCherry-expressing cells was 17.0% ± 1.3% in AAV2-SpCas9 sgRNA/YFP sgRNA6-treated retina and 20.6% ± 1.2% in AAV2-YFP sgRNA6-treated retina, compared with 40.8% ± 2.0% in AAV2-SpCas9 sgRNA/LacZ sgRNA-treated eyes. This represents a relative reduction of 49.5% (95% CI: 43.5–55.5) and 58.3% (95% CI: 56.4–62.0) in AAV2-SpCas9 sgRNA/YFP sgRNA6- and AAV2-YFP sgRNA6-treated retinas compared with those that had received AAV2-SpCas9 sgRNA/LacZ sgRNA, respectively (Supplementary Fig. S2).

Kamikaze CRISPR/Cas-mediated genome editing of retinal cells in vivo.
Furthermore, SpCas9 gene perturbations in vivo were confirmed by qPCR. A time course analysis showed that SpCas9 mRNA was significantly reduced in the retinas following the treatment of AAV2-SpCas9 sgRNA/YFP sgRNA2 compared with AAV2-YFP sgRNA2 (p < 0.05 at week 8; Supplementary Fig. S3).
Retinal function and structure assessment by ERG and OCT
To evaluate the effect of our “kamikaze” CRISPR/Cas construct on retinal function and structure, ERG and OCT were performed at 8 weeks after intravitreal injection of viral suspensions in Thy1-YFP mouse. Group-averaged waveforms elicited using bright and dim flashes of light along with the group-averaged data from eyes injected with YFP targeting kamikaze-CRISPR/Cas constructs (AAV2-SpCas9 sgRNA/YFP sgRNA2, Fig. 6A, B) and YFP targeting CRISPR/Cas constructs (AAV2-YFP sgRNA2, Fig. 6E, F) suggest that both treatments affected retinal function when compared with the contralateral control eyes (Fig. 6A–F and Supplementary Figs. S4, S5, S6). LacZ targeting kamikaze-CRISPR/Cas construct (AAV2-SpCas9 sgRNA/LacZ sgRNA)-treated eyes retained normal retinal function (Fig. 6C, D). OCT analysis suggests that none of the CRISPR/Cas constructs negatively impacted retinal structure, as there were no significant differences in retinal nerve fiber layer and total retinal thickness between the vehicle and viral-injected eyes of all three groups (Fig. 6G–I).

Effect of AAV2-mediated CRISPR/Cas administration on retinal function. Averaged ERG waveforms at selected intensities for control (black traces) and SpCas9 sgRNA/YFP sgRNA2 (n = 4, red traces;
Discussion
This study builds on our recent work using AAV2-mediated CRISPR/Cas to edit genes in mouse retina. 11 While CRISPR/Cas9-mediated genome editing has shown promise for correcting disease-causing mutations, the potential for genotoxic effects with prolonged expression of CRISPR/Cas9 poses a significant barrier to the clinical utility of this technology. Several strategies have been used in an attempt to avoid off-target cleavage, including improved guide RNA design, 19,20 or modification of Cas9 enzymes. 21,22 Such approaches do not avoid accumulation of Cas9, which can increase the overall chance of off-target cleavage and immunological response. Our approach was to use a self-destructive CRISPR/Cas system that disrupts the CRISPR/Cas enzyme itself after the active protein has been expressed. Unlike other approaches, most of which act to control the activity of the CRISPR/Cas system via chemical, 23,24 and biophysical 25,26 modulation of Cas9, our kamikaze CRISPR/Cas system can significantly reduce accumulation of Cas9 protein in vitro and disrupt AAV-Cas9 construct in vivo, without dramatically compromising the efficiency of on-target editing. This approach is similar to that used by Merienne et al. 27 who demonstrated that progressively inactivating the nuclease using a Cas9 self-inactivating editing system resulted in a lower frequency of off-target cleavage in human iPSC-derived neurons in vitro and in mouse brains via lentiviral-mediated in vivo delivery. Chen et al. have also tested a similar self-restrictive CRISPR/Cas system in vitro. 17 While these previous works have shown the feasibility of a self-destructive CRISPR/Cas system, our study highlights the effectiveness of an AAV-mediated self-destructive CRISPR/Cas system for in vivo genome editing in the retina.
We observed similar efficiencies in YFP gene perturbation between the conventional and kamikaze CRISPR/Cas system, but also found differences between in vitro and in vivo models, especially in SpCas9 sgRNA/YFP sgRNA6 construct (% YFP reduction in vitro: 93.5% vs. in vivo: 49.5%) and YFP sgRNA6 construct (% YFP reduction in vitro: 91.4% vs. in vivo: 58.3%). This difference may be due to a dual AAV2 vector system being used to deliver the kamikaze CRISPR/Cas construct in vivo. We and others have recently demonstrated that CRISPR/Cas9 delivered using a dual AAV2 vector can effectively edit the genome in a number of organs in adult mice. 11,28 –30 However, expression of the CRISPR/Cas9 machinery requires the receipt of both Cas9 and sgRNA expression cassettes from two separate viral vectors, which may significantly reduce editing efficiency. Although a single viral vector system using Cas9 orthologs such as SaCas9 31 or CjCas9 32 may provide better in vivo editing efficiency, dual vector systems may still be required for mutation correction as they enable delivery of donor templates and appropriate promoter elements.
An unexpected reduction in retinal function was observed 8 weeks after injection of AAV2-SpCas9 sgRNA/YFP sgRNA2 or AAV2-YFP sRNA2. Interestingly, retinal function was unaffected in mice treated with AAV2-SpCas9 sgRNA/LacZ sgRNA, and therefore, deficits in retinal function are not related to the SpCas9 sgRNA construct per se, but may be related to either off-target effects of YFP targeting sgRNA or accumulation of nonfunctional fluorescent proteins resulting from CRISPR/Cas9 editing. To further explore this possibility, first, we tested a different YFP sgRNA (sgRNA6 that targets another region of the YFP sequence) in vivo. However, a significant decrease in retinal function was still present in AAV2-SpCas9 sgRNA/YFP sgRNA6- and AAV2-YFP sgRNA6-treated mice (Supplementary Figs. S7, S8, S9). We then searched the mouse genome for potential off-target sites for two YFP sgRNAs and SpCas9 sgRNA by in silico prediction (Cas-OFFinder) and performed whole-exome sequencing on the treated mouse retina. No significant candidate genes for the off-target sites were found by in silico prediction or by whole-exome sequencing (see Result in Supplementary Data). Therefore, it may be more likely that the reduction of retinal function arises from accumulation of mutated fluorescent proteins. Although fluorescence proteins such as GFP and YFP have been widely used in neuroscience research, 33 accumulation of nonfunctional proteins resulting from on-target deletions (indel) may lead to a deleterious effect on retinal protein homeostasis. 34 Moreover, a recent study also indicated that large on-target deletions could lead to potential genotoxicity. 29 Whether such mechanisms account for the functional deficits observed in our study requires further investigation. Although no retinal toxicity was observed by overexpression of Cas9 enzyme or through delivery of our self-destructive CRISPR/Cas system, this study was conducted over a relatively short period of time (8 weeks). As such, the long-term safety profile and whether the potentially deleterious effects caused by prolonged overexpression of truncated gene products in the retina, as potentially caused by our self-destructive CRISPR/Cas system, require further investigation.
In summary, we describe and characterize a self-destructive “kamikaze” CRISPR/Cas system for in vivo genome editing in the retina. This self-destructive kamikaze CRISPR/Cas system can effectively reduce the expression of SpCas9 in the mouse retina, without substantially sacrificing on-target editing efficiency. Therefore, our AAV2-mediated self-destructive CRISPR/Cas may be a useful tool for genome editing in the retina, especially when combined with high-fidelity forms of CRISPR/Cas.
Footnotes
Acknowledgment
CERA receives operational infrastructure support from the Victorian Government.
Author Contributions
Conceptualization: F.L., S.S.C.H., A.W.H., and G.-S.L. Methodology: F.L., S.S.C.H., B.V.B., A.W.H., and G.-S.L. Formal Analysis: F.L., S.S.C.H., B.V.B., A.W.H., and G.-S.L. Investigation: F.L., S.S.C.H., M.K.N.M.K., J.-H.W., V.C., V.H.Y.W., V.S., L.T., K.W., J.A.B., R.C.B.W., B.V.B., A.W.H., and G.-S.L. Resources: A.E.K., A.L.C., A.W.H., and G.-S.L. Writing—Original Draft: F.L., A.W.H., and G.-S.L. Writing—Review and Editing: S.S.C.H., M.K.N.M.K., J.-H.W., A.P., A.E.K., A.L.C., R.C.B.W., and B.V.B. Visualization: F.L., A.W.H., and G.-S.L. Supervision: A.W.H. and G.-S.L. Project Administration: F.L., A.W.H., and G.-S.L. Funding Acquisition: A.W.H. and G.-S.L.
Author Disclosure
The authors declare no competing financial interests exist.
Funding Information
This work was supported by funding from a Bayer Global Ophthalmology Award, the Ophthalmic Research Institute of Australia, an Australian National Health and Medical Research Council (NHMRC) grant (no. 1123329), an NHMRC Practitioner Fellowship (A.W.H., no. 1103329), an NHMRC Senior Research Fellowship (A.P., no. 1154389), and an Australian Research Council Future Fellowship (A.P., FT140100047).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.