
Abstract
Familial hemophagocytic lymphohistiocytosis (FHL) is a group of life-threatening, autosomal recessive disorders of severe hyperinflammation. FHL type 3 (FHL-3) accounts for about 30% of FHL cases. It is characterized by mutations in the UNC13D gene that give rise to functionally impaired or absent Munc13-4 protein, resulting in impaired secretion of lytic granules by cytotoxic lymphocytes. Etoposide-based therapy is currently used as the standard of care that results in around 60% 5-year survival, illustrating the need for novel treatment approaches. Key problems include treatment toxicity and failure to induce or maintain remission of the hyperinflammation. Instead of immunosuppression, transplantation of autologous gene-corrected T cells can be envisaged as an approach to restore the impaired immune reaction. This study established a protocol that enabled hyperactivated, FHL-3 patient-derived T cells to be cultured and a codon-optimized UNC13D expression cassette to be delivered by either alpha- or gamma-retroviral gene transfer. The data demonstrate that the established protocol can be applied to FHL-3 patient cells with various genetic backgrounds and that gamma-retroviral UNC13D transfer restored expression of functional Munc13-4, as well as degranulation capacity and cell-mediated cytotoxicity of those patient-derived CD8+ T cells. Furthermore, the study shows that the co-introduction of a truncated low-affinity nerve growth factor receptor coding sequence enabled the therapeutic effect to be optimized by enriching transduced cells in a Good Manufacturing Practice–compliant manner. In conclusion, this study lays the foundation for an adaptive immune cell therapy approach aiming at immunological stabilization of FHL-3 patients with autologous, immune-competent T cells prior to hematopoietic stem-cell transplantation.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) encompasses a genetically heterogenous group of systemic hyperinflammatory disorders associated with immunopathology. The phenotype is caused by an impairment of cytotoxic T lymphocyte (CTL) and natural killer (NK) cell function. 1 This entails failure to lyse antigen-presenting cells (APCs) upon chronic or continuous antigen stimulation 2 and results in an uncontrolled immune activation marked by an excessive production of interferon gamma (IFN-γ), macrophage hyperactivation, tissue infiltration, and multiple organ damage, with a fatal outcome if left untreated. 3,4 HLH either arises from a genetic predisposition (primary or familial HLH [FHL]) or is acquired (secondary HLH) in the context of other immunological triggers, such as an Epstein–Barr virus infection. 3,5 It manifests regardless of sex or ethnicity at all life stages. 6 The identification of the genetic basis of inherited HLH forms has significantly improved the understanding of the disease pathogenesis. 7,8 This is vital, since >80% of FHL patients present at the clinic during infancy, in many cases without an identifiable trigger that could be linked to the manifestation. 9 Given the rapidly fatal progression of the disease, especially in these young patients, fast diagnosis and treatment are crucial. Yet, due to the unspecific initial symptoms, diagnosis still proves difficult, despite the fact that diagnostic criteria have been established. 10,11 FHL type 3 (FHL-3) is the most abundant form of primary HLH in Germany and accounts for about 30% of all FHL cases worldwide. 12 It is characterized by homozygous or compound heterozygous loss-of-function mutations in the UNC13D gene, which translate into absent, truncated, or aberrant Munc13-4 protein variants. Munc13-4 is crucial for priming perforin-containing cytotoxic vesicles in CTLs and NK cells, which then fuse with the plasma membrane at the immunological synapse. In the absence of functional Munc13-4, cytotoxic vesicles are not processed properly, and degranulation is impaired. The compromised killing of target cells, including APCs, leads to excessive T-cell and macrophage stimulation and ultimately to the characteristic HLH phenotype. 4 Until now, hematopoietic stem-cell (HSC) transplantation (HSCT) poses the only long-term curative option for FHL-3. Since donor availability is a limiting factor for HSCT, gene therapy in autologous HSC has been proposed for different types of FHL to bypass this issue. 13,14 An important factor for successful HSCT is the stabilization of HLH patients prior to transplantation by controlling the ongoing hyperinflammation. The state-of-the-art treatment regimen upon admission of a patient to the clinic during the acute phase of the disease is to control hyperinflammation rapidly while defining the HLH subtype and screening for a potential infectious trigger. 9 Standard of care chemo-immunotherapy prior to HSCT includes a combination of etoposide, dexamethasone, cyclosporine A, and, occasionally, intrathecal therapy with methotrexate and corticosteroids. 15 More recent clinical protocols also suggest the use of anti-thymocyte globulin or Campath (anti-CD52 antibody). 16 This aggressive regimen contributes to the overall low survival rate of just 60%. 11 To stabilize the immune system before HSCT, in particular to normalize the cytokine release profile, it has been suggested to perform transfer of genetically corrected autologous CTLs prior to transplantation. 13 However, the limitations in culturing and transducing highly activated T cells rendered this approach moot. This study set out to restore expression of functional Munc13-4 in T cells obtained from FHL-3 patients during an acute HLH episode. Furthermore, the study co-integrated an inert surface receptor that enabled those CTLs that were successfully transduced to be selected and enriched, 17 and established that the corrected T cells were fully functional and able to mediate cell-mediated cytotoxicity. 9 In conclusion, a robust protocol has been established to culture and transduce hyperactivated FHL-3 patient-derived peripheral blood mononuclear cells (PBMCs) as the basis for an immune cell therapy approach to stabilize FHL-3 patients prior to HSCT.
Methods
Cell culture
Human HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) with 10% fetal calf serum (FCS; PAA), 1% penicillin/streptomycin 100 × (P/S; Sigma–Aldrich), and 5% sodium pyruvate (GE). Human K562 cells were cultivated in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco) with 10% FCS and 1% P/S. Mouse L1210 cells were cultivated in Iscove's Modified Dulbecco's Medium (IMDM; Life Technologies) with 10% FCS and 1% P/S. PBMCs of healthy donors were isolated from leukoreduction system (LRS) chambers that were kindly provided by the Blood Donation Center of the authors' Medical Center (donor consent, anonymized). In brief, blood was retrieved from the LRS chamber, washed with phosphate-buffered saline (PBS) supplemented with 2 mM EDTA (Sigma–Aldrich), and subjected to Biocoll Separation Solution (Biochrom GmbH) density gradient centrifugation according to the manufacturer's instructions. After removal of the top plasma layer, white blood cells were washed and frozen in cryoprotective solution (FCS with 10% DMSO; Sigma–Aldrich). PBMCs of primary and secondary HLH patients were obtained from the biobank of the Center for Chronic Immunodeficiency (donor consent, anonymized, approval of ethics committee). All primary cells were cultured in RPMI1640 medium supplemented with 10% FCS, 1% P/S, and 100 IU/mL of recombinant human interleukin 2 (IL-2; PreProTech). For stimulation of cells, IL-2 was increased to 1,000 IU/mL and PBMCs cultured for 3 days in the presence of magnetic beads conjugated with antibodies against CD2, CD3, and CD28 beads (Miltenyi Biotech) at a 2:1 cell-to-bead ratio. PBMCs were cultured for up to 3 months.
Gene transfer with retroviral vectors and enrichment of transduced cells
Both enhanced green fluorescent protein (GFP) and truncated low-affinity nerve growth factor receptor (ΔLNGFR)-P2A-UNC13D expression cassettes were cloned into an α- or γ-retroviral backbone (plasmids pAlpha.SIN.EFs.EGFP.wPRE and pES.12-6[g]ps, kindly provided by Axel Schambach, Hannover Medical School, and Rainer Löw, BioNTech IMFS, respectively) under the control of the elongation factor 1 alpha short (EFS) promotor. For production, 4.5 × 106 HEK293T cells on a 75 cm2 plate were transfected with a vesicular stomatitis virus G (VSV-G; plasmid pMD2.G), a gag/pol (plasmids pcDNA3.alpha.gag/pol.CO or pCsGPpA), and a vector plasmid (α-GFP, α-UNC, γ-GFP, or γ-UNC), respectively, using polyethylenimine, as previously described. 18 Supernatants were harvested 36 and 60 h after transfection and passed through a 0.45 μm filter. Pooled fractions were ultracentrifugated, the pellet re-suspended in 100 μL of pre-cooled PBS, and stored at −20°C. Biological titer (TU/mL) was determined by transduction of K562 cells and determination of the fraction of GFP or LNGFR (CD271)-positive cells 3 days later by flow cytometry. For transduction, PBMCs were seeded in poly-D-lysine pre-coated 48-well plates at a density of 2 × 105 cells/well and transduced with 100 TU/cell of the respective vector by spinoculation (centrifugation for 2 h at 600 g at 37°C). CD271-positive cells were selected from 1 × 107 of γ-UNC transduced cells using the CD271 MicroBead Kit (Miltenyi Biotech) according to the manufacturer's instructions.
Flow cytometry
For flow cytometric analyses, 2–5 × 105 cells were harvested and re-suspended in 50 μL FACS-Buffer [PBS (PAN-Biotech) supplemented with 5% FCS (PAA), 1 mM EDTA (Serva), and 0.1% sodium azide (Sigma–Aldrich)] containing either 0.3 μL (anti-CD8-APC, anti-CD8-FITC, and anti-HLA-DR-FITC; all BD Pharmingen) or 1 μl of antibodies (anti-CD25-PE [Miltenyi Biotech]; anti-CD107a-PE [BD Pharmingen], and anti-CD271-APC [Miltenyi Biotech]). Cells were incubated for 30 min at 4°C in the dark, washed, and then re-suspended in 150 μL FACS buffer. For Annexin V and 7-AAD staining, cells were washed with PBS and the pellet re-suspended in 100 μL Annexin V buffer (BD Pharmingen) supplemented with 5 μL Annexin V-APC (BD Pharmingen) and 1 μL 7-AAD (AppliChem). Samples were analyzed on a FACS Canto II (BD Biosciences) or BD Accuri (BD Biosciences). Data were analyzed using the FlowJo v10 software (FlowJo LLC) and the embedded flowAI application. 19
Western blot analysis
Transduced patient cells (1 × 106) were harvested at the indicated time points. Cell pellets were lysed, as described previously, 20,21 and the protein concentration in the supernatant was determined using the Lowry assay (BioRad). Protein lysate (35 μg) was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis before being blotted to a polyvinylidene difluoride (PVDF) membrane. 20,21 Staining for Munc13-4 and β-actin was performed separately using either 2 μg/mL goat anti-Munc13-4 (Everest; Biotech) or a 1:1,000 dilution of rabbit anti-β-actin (Cell Signaling Technology). Protein expression was visualized with horseradish peroxidase–conjugated secondary antibodies, anti-goat (1:500; Santa Cruz Biotechnology), or anti-rabbit (1:2,000; Dianova Hamburg), respectively, and West Pico (β-actin) or Femto (Munc13-4) chemiluminescence substrates (Thermo Fisher Scientific), respectively. For quantification of Munc13-4 expression, the background was subtracted from each band and normalized to the respective control β-actin band using ImageJ.
Degranulation assay
A degranulation assay was performed, as described previously. 22 After 2 days of pre-stimulation with 1.25 μg/mL phytohemagglutinin (PHA; Remel Europe Ltd.), 2 × 105 CTLs were washed to remove residual IL-2, re-suspended in 200 μL supplemented RPMI1640 medium, and incubated for 3 h in the presence of 1 μL anti-CD107-PE (BD Pharmingen) with or without 2 μL CD2/CD3/CD28 beads (Miltenyi Biotech), respectively. Then, cells were harvested and stained with 0.3 μL anti-CD8-APC (BD Pharmingen) and 1 μL anti-CD107-PE for flow cytometry analysis.
Cytokine release assay
PBMCs were seeded at a density of 1.5 × 105 cells in 200 μL culturing medium without IL-2 per well. After 9 h of incubation in the presence or absence of 1.25 μg/mL PHA, 100 μL of supernatants were harvested for analysis by cytometric bead array (CBA; BD Biosciences) to measure the concentration of granzyme B (gB), IFN-γ, and tumor necrosis factor according to the manufacturer's instructions. Flow cytometry results were analyzed using FlowJo v10.
Cytotoxicity assay
Antibody-dependent cellular cytotoxicity (ADCC) was determined, as previously described. 23 In brief, L1210 target cells were labeled with 51Chromium (51Cr) and effector cells loaded with CD3 antibody. Cell killing capacity of transduced patient cells was analyzed by incubating effector and target cells at various ratios, and measuring 51Cr release in a liquid scintillation counter (TopCount NXT; PerkinElmer).
Statistical analysis
Statistical analyses were performed with an unpaired or paired Student's t-test using Microsoft Office Excel 2016 or GraphPad Prism v7 (GraphPad Software, Inc.). Statistical evaluation were carried out using GraphPad Prism v7 (GraphPad Software, Inc.).
Results
UNC13D gene transfer to FHL-3 patient-derived T cells
In order to correct FHL-3 patient-derived T cells, retroviral gene transfer cassettes were designed, consisting of a codon optimized UNC13D gene under the control of an EFS promotor. For selection of transduced cells, the coding sequence for ΔLNGFR was included. 17 These constructs, as well as the respective GFP control vectors, were packaged into two different retroviral vector platforms: one derived from avian sarcoma leukosis virus (α-retroviral) 24 and the other from murine leukemia virus (γ-retroviral). 25 Since α- and γ-retroviral vectors only transduce dividing cells sufficiently, first an activation protocol was established that allowed hyperactivated FHL-3 patient-derived cells with various genetic backgrounds to be cultured and efficiently transduced. PBMCs of three FHL-3 patients and one secondary (2°) HLH patient were collected from individuals in the course of full-blown disease either before or during treatment (Table 1). PBMCs of three healthy donors (HD) served as controls and were used alongside patient cells to identify the best activation and transduction conditions. The optimized protocol consisted of 3 days of culturing of the freshly thawed PBMCs in the presence of CD2/CD3/CD28 beads and human IL-2 followed by transduction and subsequent cultivation with IL-2 supplemented medium (Fig. 1a) that promoted the proliferation of CD8+ CTLs (Supplementary Fig. S1). As shown for cells derived from FHL-3 patient C, two distinctive cell populations based on cell size and granularity were identified (Fig. 1b). While the population in gate 2 (G2) represented live cells, gate 1 (G1) contained a high fraction of pre-apoptotic and apoptotic cells, which were particularly numerous in samples derived from FHL-3 patient C (Fig. 1c and Supplementary Fig. S2a). In general, compared to normal PBMCs, PBMCs isolated from HLH patients contained a higher fraction of CD8+ cells (Fig. 1d and e), which were highly activated (CD25+, HLA-DR+; Fig. 1e and Supplementary Fig. S2b) and apoptotic (Fig. 1c).
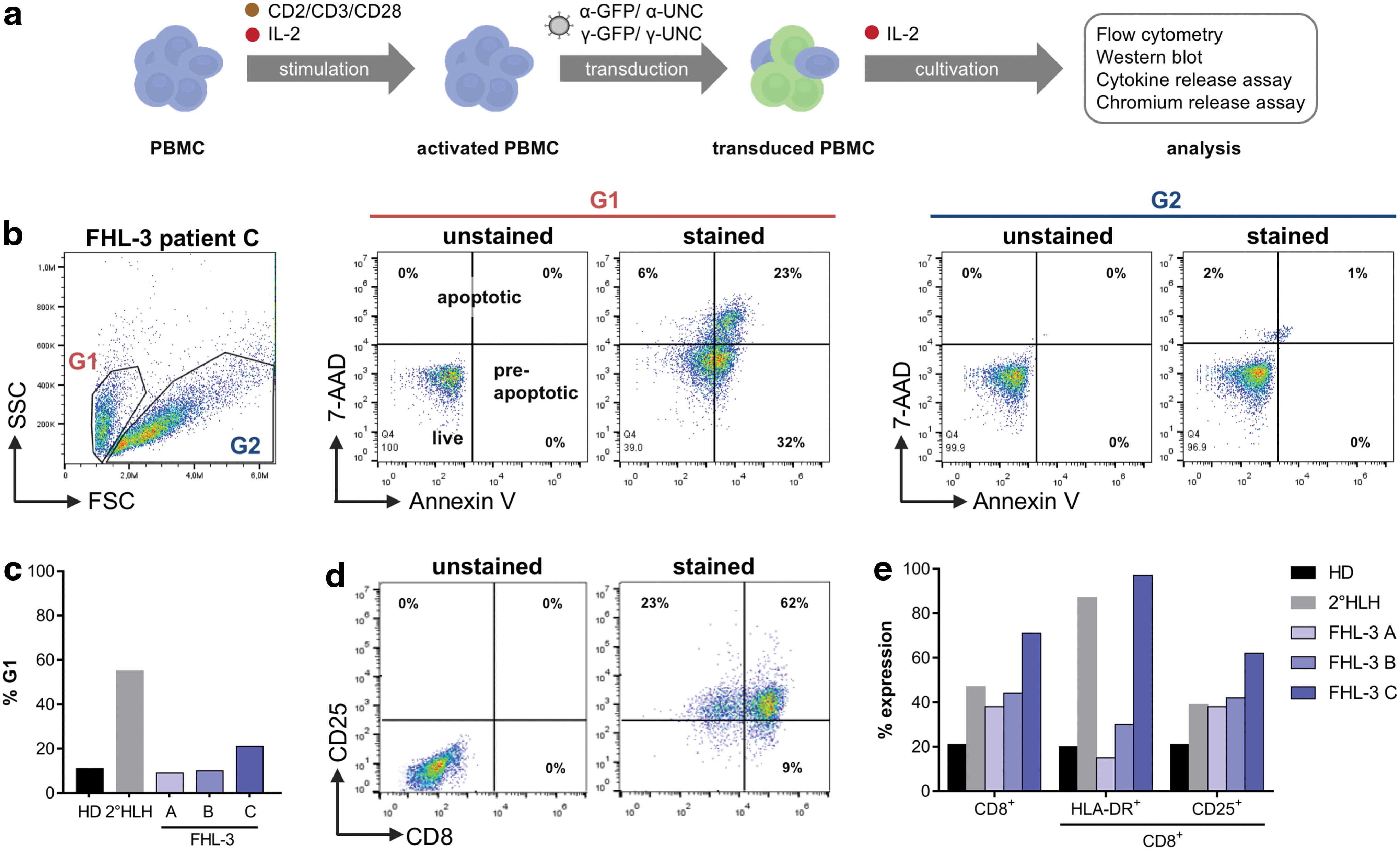
Characterization of FHL-3 patient cells.
Patient characterization
HLH, hemophagocytic lymphohistiocytosis; FHL, familial hemophagocytic lymphohistiocytosis; n/a, not applicable or not available; n/d, not determined; CNS, central nervous system; EBV, Epstein–Barr virus.
After successful activation, PBMCs were transduced with either α- or γ-retroviral UNC13D expression vectors (α-UNC, γ-UNC) or the corresponding GFP control vectors (α-GFP, γ-GFP; Fig. 2a). Depending on the platform, up to 74% of cells for α-UNC and α-GFP and 93% of cells for γ-UNC and γ-GFP constructs could be transduced, respectively (Fig. 2b). In order to monitor the stability of transgene expression, transduced cells were cultured for 2 weeks under conditions promoting the propagation of CD8+ T cells, with >90% of cells being CD8+ after 2 weeks (Supplementary Fig. S1). When analyzing transgene expression in those CTLs, significant differences (p < 0.05) between the percentages of α- and γ-retrovirally transduced cells (Fig. 2c) as well as transgene expression levels (Fig. 2d) were noticed, with γ-retroviral vectors clearly outperforming the α-retroviral platform. Importantly, cell expansion of γ-retrovirally transduced patient cells was not different from α-retrovirally transduced cells and transduced cells of HD (Fig. 2e). In conclusion, conditions were established that allowed highly activated T cells of FHL-3 patients to be efficiently transduced with the γ-retroviral vector.
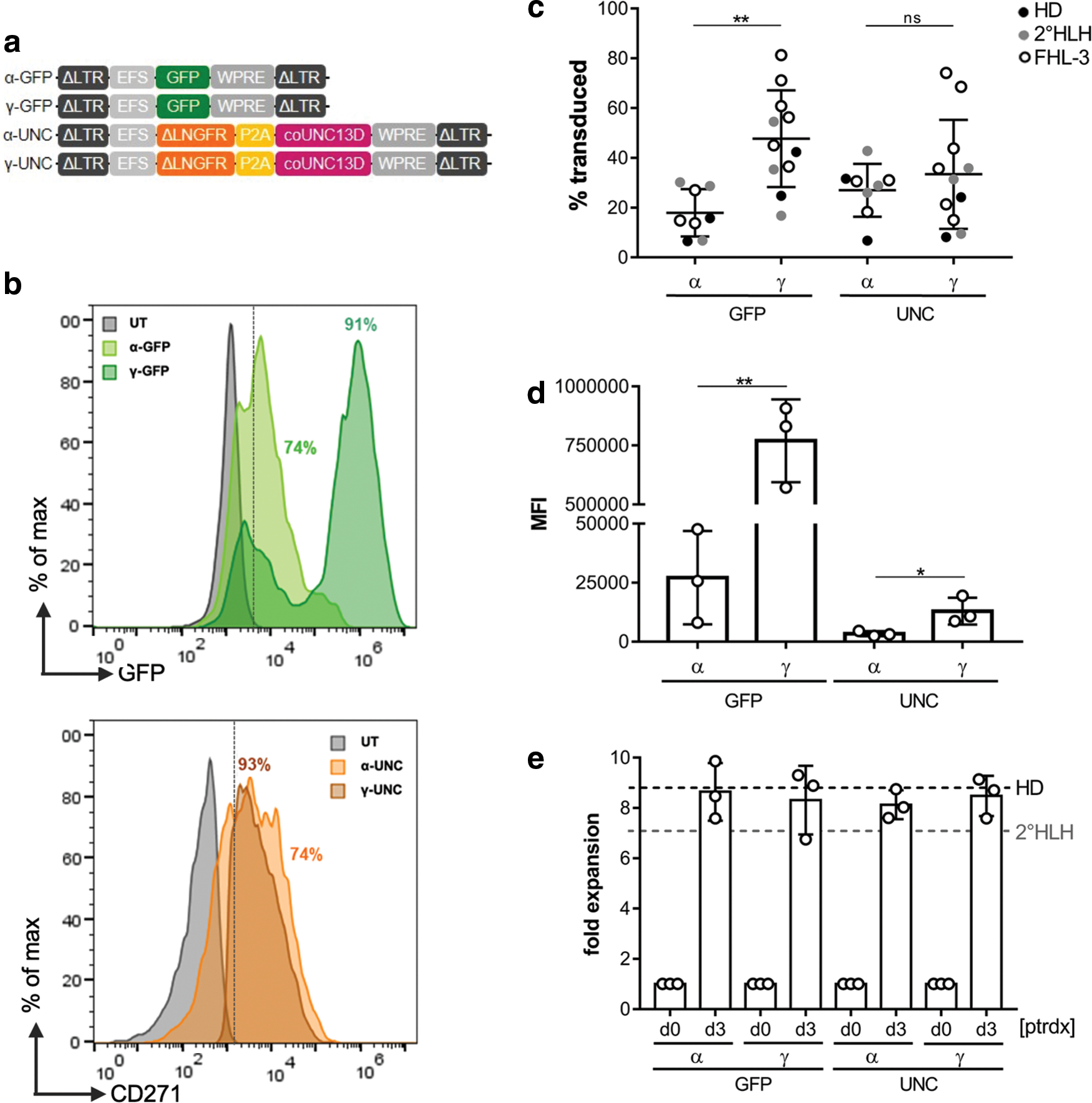
Retroviral gene transfer.
Restoration of cellular phenotype of FHL-3 patient cells
To assess whether gene transfer of the UNC13D cassette restored the cellular phenotype, the study investigated the degranulation capacity of γ-GFP- and γ-UNC transduced patient cells. To this end, stimulated and non-stimulated transduced FHL-3 patient T cells were incubated with an antibody against CD107a, which is located in the membranes of cytolytic vesicles and is only detectable on the cell surface upon stimulation of CTLs if the vesicles can be transported to and merged with the cell membrane. A control experiment with CD2/CD3/CD28 bead-stimulated CTLs of a HD revealed a prominent increase of CD107a surface staining compared to unstimulated CTLs (Fig. 3a). In contrast, untransduced (UT) and γ-GFP transduced CTLs of FHL-3 patient B showed no degranulation in response to the activating stimulus. On the other hand, when the patient cells were transduced with γ-UNC, bead stimulation resulted in a clear CD107a shift, similar to the one observed for HD cells. Likewise, cells derived from FHL-3 patients A and C could be rescued successfully by transduction with the γ-UNC vector (Supplementary Fig. S3). In sum, the degranulation activity of all patient cells that were transduced with the γ-UNC vector was comparable to the activity of HD cells (Fig. 3b). In addition, the study measured two representative factors that are either secreted by activated T cells, IFN-γ, or released by cytotoxic vesicles, gB. Notably, gB can only be released by CTLs if functional Munc13-4 is expressed. Supernatants of PHA-stimulated and unstimulated samples were collected and assayed by CBA. gB levels in the supernatant of stimulated UT and γ-GFP transduced patient cells were comparable to unstimulated samples, implying that gB was not released (Fig. 3c). In contrast, UNC13D gene transfer restored gB release in FHL-3 patient-derived cells upon PHA stimulation, suggesting a corrective effect of transduction with γ-UNC. As a control, IFN-γ levels were measured in the supernatants of all samples and were found to be comparable for all PHA-treated cells. In conclusion, these results demonstrate that UNC13D gene transfer restores functional degranulation of cytotoxic vesicles in FHL-3 patient-derived T cells.
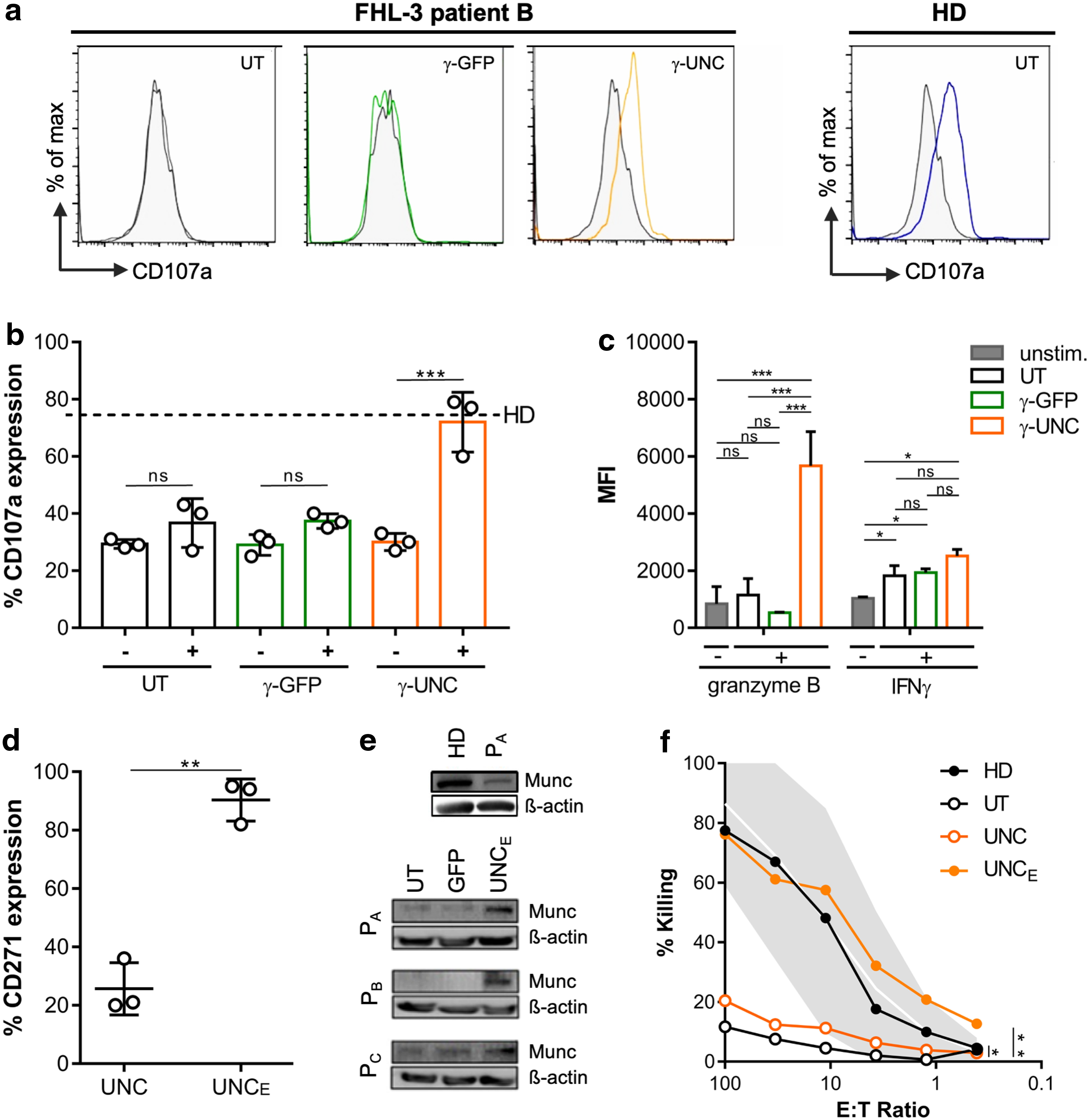
Functional assessment of genetically corrected FHL-3 patient cells.
For therapeutic application, it may be beneficial to infuse highly enriched UNC13D-positive cells. To facilitate Good Manufacturing Process (GMP)-compatible sorting of transduced cells, the γ-UNC vector also coded for ΔLNGFR (Fig. 2a), an inert cell surface marker that has already been employed in the clinic for cell enrichment. 17 LNGFR is also known as CD271. One round of selection using a CD271 selection column allowed the fraction of γ-UNC transduced cells of all three FHL-3 patients to be increased from about 20% (UNC) to 80–95% (UNCE; Fig. 3d). Since this selection was based on the ΔLNGFR surface expression, Munc13-4 protein expression levels in UT, γ-GFP transduced, and enriched γ-UNC transduced patient samples were determined. Whereas distinct Munc13-4 levels were detected in healthy donor cells or enriched γ-UNC transduced samples, UT and γ-GFP transduced patient CTLs showed, as expected (Table 1), no or only weak expression of endogenous Munc13-4 (Fig. 3e). Quantification of Munc13-4 protein expression in enriched γ-UNC transduced CTLs of all patients revealed that transgene expression levels were comparable to endogenous Munc13-4 in CTLs obtained of HD (Supplementary Fig. S4). Finally, the study sought to investigate whether transduction of FHL-3 patient CTLs with γ-UNC restores cell-mediated cytotoxicity. γ-UNC transduced cells of patient C before (UNC) and after enrichment (UNCE), respectively, were used in an ADCC assay using 51Cr-labeled L1210 cells as a target (Fig. 3f). Regardless of the effector-to-target (E:T) ratio, UT patient cells failed to mediate target cell killing at any dilution. Non-enriched γ-UNC transduced patient cells showed a weak but significant improvement in cytotoxicity relative to untreated cells (p < 0.05). In contrast, the CD271-enriched patient cells (UNCE) were able to kill the target cells at comparable levels to the HD control at all E:T ratios and at levels significantly above background (p < 0.01). Determination of the vector copy number (VCN) in these cells revealed that enrichment of CD271-expressing cells coincided with about a threefold increase of the average VCN in those cell populations (Supplementary Fig. S5). Collectively, these assays confirm that γ-retroviral transfer of a UNC13D expression cassette in PBMCs of FHL-3 patients restored expression of functional Munc13-4 protein, as well as degranulation and cytolytic activity of FHL-3 patient-derived CTLs.
Discussion
Impairment of Munc13-4 function, as observed in cells of FHL-3 patients, abolishes CTL and NK cell-mediated killing of APCs, ultimately leading to a severe and potentially lethal immunopathology. This study demonstrates that cell-mediated cytotoxicity of CTLs derived from three individual FHL-3 patients with different genetic backgrounds and different stages of HLH-related treatment protocols can be restored in vitro after retroviral transfer of an UNC13D expression cassette.
The functional correction of Munc13-4 deficient T cells was shown before in a setup using lentiviral gene transfer. 13 Because transduction of T cells with VSV-G pseudotyped lentiviral vectors is rather challenging, 13 a decision was made to integrate the UNC13D expression cassette into either an α- or a γ-retroviral backbone. Both α- and γ-retroviral vectors are attractive for clinical translation: both systems are available in a so-called self-inactivating (SIN) configuration, that is, they contain deletions in the long terminal repeat (LTR) U3 regions that inactivate the viral promotor activity. Preclinical studies have demonstrated that these deletions correlate well with decreased genotoxicity. 26,27 Moreover, as opposed to γ-retroviral vectors, which preferentially integrate proximal to transcription start sites, α-retroviral vectors are known to integrate into the host genome in a random pattern that further reduces genotoxicity. 27,28 On the other hand, a packaging cell line is available for the γ-retroviral vector system, 25 which will significantly decrease production costs once entering the clinical phase. Importantly in this context, the safety of SIN γ-retroviral vector systems has been demonstrated in several clinical trials, including the therapy of X-linked severe combined immunodeficiency (SCID-X1) in HSC or the generation of chimeric antigen receptor (CAR) T cells. 29,30 The comparison of the two retroviral systems showed that although comparable initial transduction efficiencies were reached, transgene expression levels in PBMCs transduced with the γ-retroviral constructs were considerably and consistently higher than those achieved with the α-retroviral backbone, a characteristic that was also observed for transduction of K562 cells. Given the limited number of patient cells that were available, a decision was made to proceed with the γ-retroviral vector for the proof-of-concept experiments.
After successful activation and transduction of patient T cells, the impact of UNC13D gene transfer on their cellular phenotype was analyzed. The assessment of the cellular functionality of γ-UNC transduced FHL-3 patient CTLs showed restoration of degranulation capacity, as well as normalized release of granzyme upon stimulation of the CTLs. The absence of detectable differences in IFN-γ secretion was expected and explained by the fact that IFN-γ is released over a Munc13-4 independent pathway. 3,5 Transplantation of functional, gene-corrected T cells can be envisaged as an approach to restore the impaired immune reaction in FHL patients in lieu of or in combination with an immunosuppression strategy. Unpublished data in a FHL-3 mouse model demonstrate that the adoptive transfer of functional T cells during an acute HLH with its hyperinflammatory environment cured the mice from disease and protected them from relapse. Accordingly, three clinical situations might benefit from adoptive immunotherapy with gene-corrected autologous T cells: (1) rescue therapy in a situation where standard-of-care remission-inducing therapy is not successful; (2) treatment of relapse in the phase between induction of remission and HSCT; and (3) prevention of relapse in the phase between induction of remission and HSCT. It is expected that patients will remain on some form of maintenance immunosuppression after achieving remission. Data on long-term disease evolution in transplanted FHL patients with mixed chimerism indicate that around 20% of functional donor cells are needed to prevent HLH relapse, 31 and some mouse and human data indicate that this threshold is also relevant for T-cell chimerism. Obviously, this level of chimerism can only be achieved if the corrected T cells have a proliferation advantage or if the defective T cells are eliminated. Since current understanding suggests that HLH is an antigen-driven immune reaction, these considerations refer to the antigen-specific T cells and not the total T-cell pool. Remission-inducing therapies, such as etoposide or Campath, effectively target T cells and antigen-activated T cells in particular. In that situation, adoptive transfer followed by in vivo expansion of corrected T cells in a lymphopenic environment may lead to the desired level of chimerism among the T cells specific for the (in most cases unknown) antigen.
To optimize outcome, this study demonstrates for the first time that γ-UNC transduced cells can be enriched using a GMP-compliant magnetic sorting system that enables the selection of CD271-positive cells and that such CD271-selected cells maintain cell-mediated cytotoxicity properties comparable to CTLs of healthy donors. It may also be envisaged to confer Campath resistance to the transfused cells by knocking out CD52. 32 It is, however, not clear yet whether the ex vivo manipulation of highly activated T cells from a donor with acute HLH—notably enriched for T cells with specificity for the relevant antigen—will impact on or limit their in vivo expansion potential.
Taken together, this study demonstrates successful γ-retroviral UNC13D gene transfer to and UNC13D transgene expression in CTLs obtained from three different FHL-3 patients during acute disease, as well as a GMP-compliant enrichment protocol that preserves functionality of the genetically corrected T cells. These data provide sufficient evidence to support further development of an autologous T cell–based immunotherapy approach for FHL-3 patients. The infusion of sufficient numbers of functionally corrected CTLs could interrupt the hyperinflammatory feedback loop during an acute HLH and hence serve as a means to stabilize these patients immunologically before HSCT.
Footnotes
Acknowledgments
We thank Alexandra Nieters and the CCI biobank team for PBMCs of HLH patients; the Blood Donation Center of our Medical Center for providing LRS chambers; Axel Schambach, Hannover Medical School, and Rainer Löw, BioNTech, for providing retroviral vector plasmids; Christien Bednarski and Beate vom Hövel for advice in setting up retroviral vector production; Jamal Alzubi and Nils Craig-Müller for help with flow cytometry; Gianni Monaco for flowAI assessment; Philipp Wolf and his team for support in establishing Munc13-4 Western blot; and Saskia König, Valentina Pennucci, and Laura Mosti for help with ddPCR. This work was supported by grants of the German Research Foundation (SFB 1160 P17 to T.C., P.A., and S.E.) and the German Federal Ministry of Education and Research (BMBF-01EO0803 to T.C. and S.E.).
Author Disclosure
Research in the lab of T.C. is supported by Cellectis and Miltenyi Biotec. T.C. is a consultant to TRACR Hematology. None of the other authors reports any conflicts of interest.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Materials and Methods
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.