
Abstract
Back pain is the leading cause of disability worldwide and contributes to significant socioeconomic impacts. It has been hypothesized that the degenerative intervertebral disc (IVD) contributes to back pain by sensitizing nociceptive neurons innervating the IVD to stimuli that would not be painful to healthy patients. However, the inflammatory signaling networks mediating this sensitization remain poorly understood. A better understanding of the underlying mechanisms of degenerative IVD-induced changes in nociception is required to improve the understanding and treatment of back pain. Toward these ends, a novel in vitro model was developed to investigate degenerative IVD-induced changes in dorsal root ganglion (DRG) neuron activation by measuring DRG neuron activity following neuron seeding on human degenerative IVD tissue collected from patients undergoing surgical treatment for back pain. Lentiviral clustered regularly interspaced palindromic repeat (CRISPR) epigenome editing vectors were built to downregulate the inflammatory receptors TNFR1, IL1R1, and IL6st in DRG neurons in single- and multiplex. Multiplex CRISPR epigenome editing of inflammatory receptors demonstrated that degenerative IVD tissue drives thermal sensitization through the simultaneous and redundant signaling of interleukin (IL)-6, tumor necrosis factor alpha (TNF-α), and IL-1β. This work elucidates redundant signaling pathways in neuron interactions with the degenerative IVD and suggests the need for multiplex targeting of IL-6, TNF-α, and IL-1β for pain modulation in the degenerative IVD.
Introduction
Back pain is among the most prevalent medical conditions worldwide, with low back pain ranked as the leading cause of disability 1 and ranked third in terms of disease burden according to disease-adjusted life years. 2 In addition, the treatment of low back pain and loss of productivity due to low back pain generates an estimated $100 billion socioeconomic cost in the United States alone. 3 Despite the prevalence of back pain and its significant impact on society, knowledge of the underlying mechanisms of back pain and treatment strategies remain limited.
Back pain is multifactorial in origin, with life-style factors, aging, genetic predisposition, injury, and disc degeneration—the most prevalent factor—demonstrated to be associated with elevated risks of back pain. 4 –8 Disc degeneration can be characterized by a cascade of events leading to observable changes in the intervertebral disc (IVD), including loss of disc height, 9 breakdown of the IVD extracellular matrix (ECM), 10,11 elevated inflammatory cytokine levels, 12 –17 and alterations in innervation of the IVD. 18 –21 Despite the association of these observed changes in the IVD with discogenic pain, 22 –24 the interactions between nociceptive neurons and human degenerative IVD tissue are largely speculative and poorly understood, and methods to modulate these interactions are limited.
It is hypothesized that multiple inflammatory cytokines present in the degenerative IVD contribute to discogenic pain by altering nociceptive thresholds of neurons innervating the IVD, thus sensitizing neurons to stimuli that are not painful in healthy patients. To test this hypothesis, an in vitro model was developed to study the interactions between human degenerative IVD tissue and neurons in direct contact with this tissue, and to test for the ability of the degenerative IVDs to alter sensory neuron activity in response to nociceptive stimuli. Additionally, clustered regularly interspaced palindromic repeat (CRISPR) epigenome editing of sensory neurons was utilized to identify the primary signaling pathways involved in these interactions and to develop novel treatment strategies for the treatment of discogenic back pain.
The healthy IVD is a largely aneural organ, with innervation limited to the peripheral lamellae of the annulus fibrosus (AF) and the cartilage endplates by neurons whose cell bodies reside in the dorsal root ganglion (DRG). 25 –28 The majority of these neurons are small, peptidergic, nociceptive neurons that co-express the neuropeptides substance P and calcitonin gene-related peptide (CGRP) 28 –33 and the acid and thermal sensitive ion channel TRPV1. 29 The degenerative IVD exhibits alterations in innervation, including an increase in the number and density of nociceptive neurons innervating the IVD. 34 –36 In addition, nociceptive neurons expressing the neuropeptides CGRP and substance P extend into the aneural regions of the IVD, including the inner AF and the nucleus pulposus (NP). 18,20,25,26,36 –38 Together, these changes suggest the degenerative IVD is potentially sensitive to nociceptive stimuli.
Nociceptive neurons innervating the degenerative IVD are exposed to multiple factors capable of altering the nociceptive thresholds of neurons, including pathologically high levels of tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6, as well as other pro-inflammatory cytokines 39,40 and pathologic, acidic pH levels. 41,42 Inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, have been demonstrated to sensitize nociceptive neurons to thermal stimuli, 43 –45 and induce mechanical allodynia and thermal hyperalgesia 46 –48 in models of peripheral neuropathy. The presence of these sensitizing factors in the degenerative IVD may contribute to discogenic pain by driving enhanced nociception of neurons innervating the IVD in response to stimuli that are not painful in healthy patients. In order to elucidate the underlying mechanisms of discogenic pain, an in vitro model was developed to investigate the ability of the degenerative IVD to alter nociception and utilized CRIPSR epigenome editing of sensory neurons to identify the primary factors and mechanism that mediate degenerative IVD-induced enhanced nociception. Furthermore, this study demonstrates that CRISPR epigenome editing of sensory is a potential neuromodulation strategy of nociceptive neuron interactions with the degenerative IVD.
The objectives of the present study were to develop an in vitro model to test the hypothesis that the degenerative IVD contributes to discogenic pain by inducing elevated nociceptive neuron activity in response to noxious stimuli, and to elucidate the primary factors and mechanisms within the degenerative IVD that mediate these changes in neuron activity. This work elucidates multiple neuron sensitization pathways in the degenerative IVD and identifies potential therapeutic targets and strategies for the treatment of discogenic pain.
Methods
Experimental overview
Experiments were conducted to investigate the role of multiple inflammatory cytokines, potentially contained in pathologic IVD tissue, to induce altered sensory neuron activity in response to nociceptive stimuli and to identify the primary factors and mechanisms that mediate these degenerative IVD-induced changes in nociception. First, a novel in vitro model was developed to investigate the response of sensory neurons directly seeded onto human pathologic IVD tissue, modeling the interactions between neurons and painful human degenerative IVD tissue, as would be observed in disc degeneration. 20,25 In the pathologic IVD tissue seeding experiments, rat DRG neurons were seeded directly onto healthy and pathologic IVD tissue, and subsequently subjected to thermal stimuli during imaging of heat-evoked calcium transients to test the ability of pathologic IVD tissue, in direct contact with afferent neurons, to alter neuron activity in response to nociceptive stimuli present in the IVD.
After developing the in vitro model, this model of interactions between sensory neurons and the degenerative IVD, along with CRISPR epigenome editing of DRG neurons, was utilized to investigate the potential roles of the IL-6, IL-1β, and TNF-α signaling pathways in pathologic IVD-induced elevated nociception. In these experiments, the expression of the cytokine receptors for IL-6, IL-1β, and TNF-α, (IL-6st, IL-1R1, and TNFR1) were downregulated independently in sensory neurons seeded directly onto pathologic IVD tissue, and neuron activity was measured via imaging of calcium transients. Lentiviral constructs expressing CRISPR epigenome editing systems that targeted the IL-6st, IL-1R1, or TNFR1 gene promoters were designed, built, and tested for their ability to regulate target gene expression in sensory neurons via quantitative polymerase chain reaction (qPCR). Following validation, CRISPR epigenome editing vectors targeting the IL6-st, IL-1R1, and TNFR1 gene promoter were delivered, independently of each other, to DRG neurons seeded onto pathologic AF tissue and tested for their ability to modulate pathologic IVD-induced elevated neuron activity in response thermal stimuli.
Once the IL-6, IL-1R1, and TNF signaling pathways were determined to be mediators of pathologic IVD triggered neuron sensitization, multiplex epigenome editing experiments were conducted to investigate the combined/redundant roles of IL-6, IL-1β, and TNF-α signaling pathway activation in mediation of pathologic IVD-induced enhanced neuron activity in response to nociceptive stimuli.
Pathologic IVD tissue
Pathologic IVD tissue was obtained from three female and two male patients undergoing surgical intervention for axial back pain, degenerative disc disease, and lumbar spondylosis. Patients showed signs of degenerative disc disease on magnetic resonance imaging, with degeneration severity ranging from mild to moderate degeneration, and reported axial back pain. (Additional patient information is reported in Supplementary Table S1.) IVD tissue was collected, transferred into a glass petri dish, washed twice with washing medium (Dulbecco's modified Eagle's medium [DMEM] high glucose [Life Technologies] supplemented with 1% genatmycin [Gibco], 1% kanamycin [Sigma–Aldrich], and 1% Fungizone [Gibco]), and the AF tissue was separated from the NP tissue. The AF tissue was cut into tissue constructs (∼1 cm2) and frozen at −20°C until use. Healthy IVD tissue was obtained from bovine caudal discs and processed as described above.
At the time of the experiments, healthy and pathologic AF tissue was thawed at room temperature. After thawing, AF tissue was incubated in washing media for 48 h under standard cell culture conditions (37°C, 5% CO2). Following incubation, media were replaced with washing media supplemented with 10% fetal bovine serum and cultured for 48 h prior to seeding with DRG neurons.
AF tissue culture seeded with DRG neurons
All procedures were performed with approval of the University of Utah Institutional Animal Care and Use Committee. Adult (200–250 g) Sprague–Dawley rat DRG were explanted, cleaned of anterior roots, posterior roots, and connective tissue, and dissociated using a previously described procedure. 49 Rat DRG neurons were seeded onto healthy and pathologic AF tissue at a density of 25,000 cells/cm2, and cultured in SATO− media (DMEM/F12 supplemented with 2.2% SATO− mix, 1% transferrin [Sigma–Aldrich], 2% insulin [Sigma–Aldrich], 1% GlutaMAX [Invitrogen], 0.5% gentamycin, and 2.5S nerve growth factor [10 ng/mL; Worthington Biochemical]) for 3–4 days until the experiments were conducted.
Imaging of heat-induced calcium transients in DRG neurons
Rat DRG neurons were loaded with the calcium indicator dye Fluo-4AM (4 μM; Molecular Probes) and incubated in the dark at 37°C for 1 h. Fluorescent calcium measurements were performed using a multi-photon microscope (Prairie View; Bruker; excitation 810 nm, emission 545, 0.5 Hz). DRG neuron-seeded AF tissue was incubated at 33°C for 15 min to establish a baseline calcium signal and then exposed to thermal stimuli of 35°C, 37°C, 38°C, 39°C, 40°C, 42°C, and 44°C for 2 min while calcium imaging was conducted. Cells were returned to baseline temperature for 5 min between exposures to elevated temperatures. 49
Image analysis was conducted using Fiji software 50 and the ΔF/F method, as previously reported. 49 Briefly, the ΔF/F, baseline mean, and standard deviation (SD) of ΔF/F were calculated for each neuron at 33°C. Neurons were considered to exhibit calcium transients in response to thermal stimuli if the ΔF/F for the cell was three SDs greater than the mean baseline value (33°C) in response to thermal stimuli. 49,51 Calibration was conducted to account for changes in calcium binding affinity of Fluo-4AM with changes in temperature.
Pathologic IVD tissue exposure
DRG neurons were seeded onto healthy and degenerative AF tissue (n = 5 patients; Supplementary Table S1) and cultured as described in the section on DRG neuron-seeded AF tissue culture. Media were replaced with 3 mL of fresh SATO− media supplemented and incubated for 2–3 days (37°C, 5% CO2) before imaging experiments were conducted. Following incubation, neurons were loaded with calcium indicating dye, as described in the section on imaging of heat-induced calcium transients in DRG neurons.
Lentiviral CRISPR epigenome editing vector construction
CRISPR epigenome editing vectors were created that co-express dCAS-KRAB-T2A-GFP and guide RNAs (gRNAs) that target the IL-6st, IL-1R1, or TNFR1 promoter regions using a previously described method. 49,52 Briefly, the promoter region of each target gene (IL-6st, TNFR1, or IL-1R1) was screened for gRNA target sequences with the necessary adjacent protospacer adjacent motif (-NGG) and selected based on minimizing off-target binding sites. Four guides were selected and screened for the promoter region of each target gene. The nontarget guide oligonucleotide was designed as a scramble DNA sequence that does not match the rat genome. Oligonucleotides were obtained (University of Utah DNA/Peptide Synthesis Core), hybridized, phosphorylated, and cloned into gRNA expressing plasmids (addgene plasmid 47108) using BbsI sites. To produce a lentiviral vector that co-expresses dCAS-KRAB-T2A-GFP and gRNA, gRNA cassettes were cloned via PCR and inserted into third-generation lentiviral transfer vector that expresses dCas-KRAB-T2A-GFP under the control of the human UbC promoter via BsmBI sites. Epigenome editing vector constructs were produced in HEK293T cells using a previously reported method, 53 concentrated, and stored at −80°C until use to produce transduction media (DMEM). For transduction of rat DRG neuron-seeded AF tissue, media were removed and replaced with transduction media supplemented with polybrene (8 μg/mL), and cells were cultured for 24 h. Following transduction, viral media were removed and replaced with fresh SATO− media. Transduced DRG neuron AF tissue constructs were cultured in SATO− media until the time of the experiments.
qPCR experiments
Seven days following transduction, cells were harvested for total RNA using the PureLink RNA microscale kit (Life Technologies; n = 3 independent transductions). cDNA synthesis was conducted using the High Capacity cDNA RT kit (Life Technologies). Reverse transcription qPCR using TaqMan Universal PCR Master Mix (Life Technologies) was performed with the TaqMan Gene Expression Detection Assay (Life Technologies) with oligonucleotide primers for IL-6st, IL-1R1, TNFR1, and GAPDH. Results are expressed as the fold increase in mRNA expression of IL-6st, IL-1R1, or TNFR1 normalized to GAPDH expression using the ΔΔCt method.
Epigenome-edited neuron pathologic AF tissue exposure
After verifying regulation of target gene expression via epigenome editing, DRG neurons transduced with verified epigenome editing lentiviral constructs targeting the IL-6st, TNR1, or IL1R1 gene promoter region were seeded onto pathologic or healthy AF tissue and cultured in SATO− medium for 6 days following removal of the lentivirus. Successful transduction was verified via fluorescent imaging of green fluorescent protein (GFP) in transduced neurons. Following incubation, neurons were loaded with the calcium indicator dye rhod-2 AM (4 μM; Molecular Probes) and incubated in the dark at 37°C for 1 h. Fluorescent measurements of calcium were performed using a multiphoton microscope (Prairie View; Bruker; excitation 1,105 nm, emission 585 nm, 0.5 Hz) as described in the section on imaging of heat-induced calcium transients in DRG neurons.
Multiplex epigenome-edited neuron degenerative AF tissue exposure
To allow for multiplex targeting of multiple receptors simultaneously, lentiviral CRISPR epigenome editing vectors were created that co-express dCAS-KRAB-T2A-GFP and IL-6st gRNA, dCas-KRAB-T2A-E2Crimson and IL-1R1 gRNA, and dCas-KRAB-T2A-tagBFP and TNFR1 gRNA (Supplementary Fig. S1). The efficacy of multiplex gene regulation in DRG neurons co-transduced with IL-6st, TFNR1, and IL-1R1 was confirmed via qPCR, as described in the section above on qPCR experiments, and expression of transgene was confirmed via fluorescence-activated cell sorting (BD Canto). 52 DRG neurons seeded onto AF tissue were co-transduced with epigenome editing vectors targeting the IL-6st, TNFR1, and IL-1R1 gene promoter regions in the following combinations (IL6st + IL-1R1, IL6st + TNFR1, IL1R1 + TNFR1, and IL6st + IL1-R1 + TNFR1) and cultured as described above. Successful transduction was verified via fluorescent imaging of GFP (IL-6st), blue fluorescent protein (TNFR1), and E2 Crimson (IL-1R1) in transduced neurons. Following incubation, neurons were loaded with the calcium indicator dye rhod-2 AM (4 μM; Molecular Probes) and incubated in the dark at 37°C for 1 h. Fluorescent measurements of calcium were performed using a multiphoton microscope (Prairie View; Bruker; excitation 1,105 nm, emission 585 nm, 0.5 Hz) as described in the section on imaging of heat-induced calcium transients in DRG neurons.
Curve fitting of neuron calcium transients
Data from experiments were fit to the sigmoidal Boltzman equation, with the percentage of neurons exhibiting heat-induced calcium transients plotted as a function of temperature. 49 Each trial was individually fit to produce values for T50 and Tmax. T50 was defined as the temperature at which 50% of the maximum response occurs. Tmax was defined as the maximum percentage of neurons exhibiting heat-induced calcium transients predicted by the curve fitting.
Statistical analysis
Degenerative AF tissue exposure experiment data were analyzed by two-way analysis of variance (ANOVA) on repeated measures with Tukey's post hoc test, treating tissue condition and temperature as factors. qPCR experiments were analyzed by one-way ANOVA with Tukey's post hoc test, treating CRISPR epigenome editing vector treatment as the factor. Significance was tested at α = 0.05 for all statistical analyses.
Results
Pathologic IVD tissue induces elevated DRG neuron activity in response to thermal stimulation
The percentage of neurons exhibiting heat-induced calcium transients in DRG neurons seeded onto pathologic AF tissue was significantly elevated over the percentage of neurons exhibiting heat-induced calcium transients (Fig. 1A) in DRG neurons seeded onto healthy AF tissue at temperatures as low as 33°C (p < 0.05). In addition, for ΔF/F, the maximum calcium transients (Fig. 1B) of neurons seeded onto pathologic AF tissue were significantly elevated over those of neurons seeded onto healthy tissue (p < 0.05).

Pathologic annulus fibrosus (AF) tissue triggers increased thermal nociception in sensory neurons.
The percentage of DRG neurons exhibiting heat-induced calcium transients as a function of temperature was well defined by a Boltzman equation for DRG neurons seeded onto pathologic AF tissue (r 2 = 0.96 ± 0.02) and healthy AF tissue (r 2 = 0.98 ± 0.01; Fig. 1A). From the Boltzman curve equation fits of each trial, T50—the temperature at which half of the maximum heat-induced calcium transient occurred—and Tmax—the maximum percentage of neurons exhibiting calcium transients—were calculated. The T50 of DRG neurons seeded onto pathologic AF tissue (34.94 ± 0.12°C) was significantly lower than the T50 of DRG neurons seeded onto healthy AF tissue (40.08 ± 0.17°C; p < 0.05; Fig. 1C). Additionally, the Tmax of neurons seeded onto pathologic AF tissue (58.06 ± 5.8%) was significantly elevated over the Tmax of neurons seeded onto healthy tissue (45.15 ± 4.2%; p < 0.05; Fig. 1D).
IL-6, TNF, and IL-1 signaling mediate pathologic AF tissue-induced increases in DRG neuron activity in response to thermal stimuli
gRNAs were designed, targeting the endogenous promoters of IL-1R1, TNFR1, or IL-6st to direct dCas9-fused KRAB to specific loci within the promoter via lentiviral vectors (Fig. 2A). Transduction of DRG neurons with CRISPR epigenome editing vectors targeting IL-6st, TNFR1, and IL-1R1 (Fig. 2B) demonstrated robustly downregulated gene expression of the target genes at the RNA level. For each gene, the maximum regulation of target gene expression in CRISPR epigenome-edited neurons was significantly downregulated when compared to DRG neurons transduced with nontarget lentiviral vectors (<10% of nontarget RNA expression; p < 0.05; Fig. 2B).
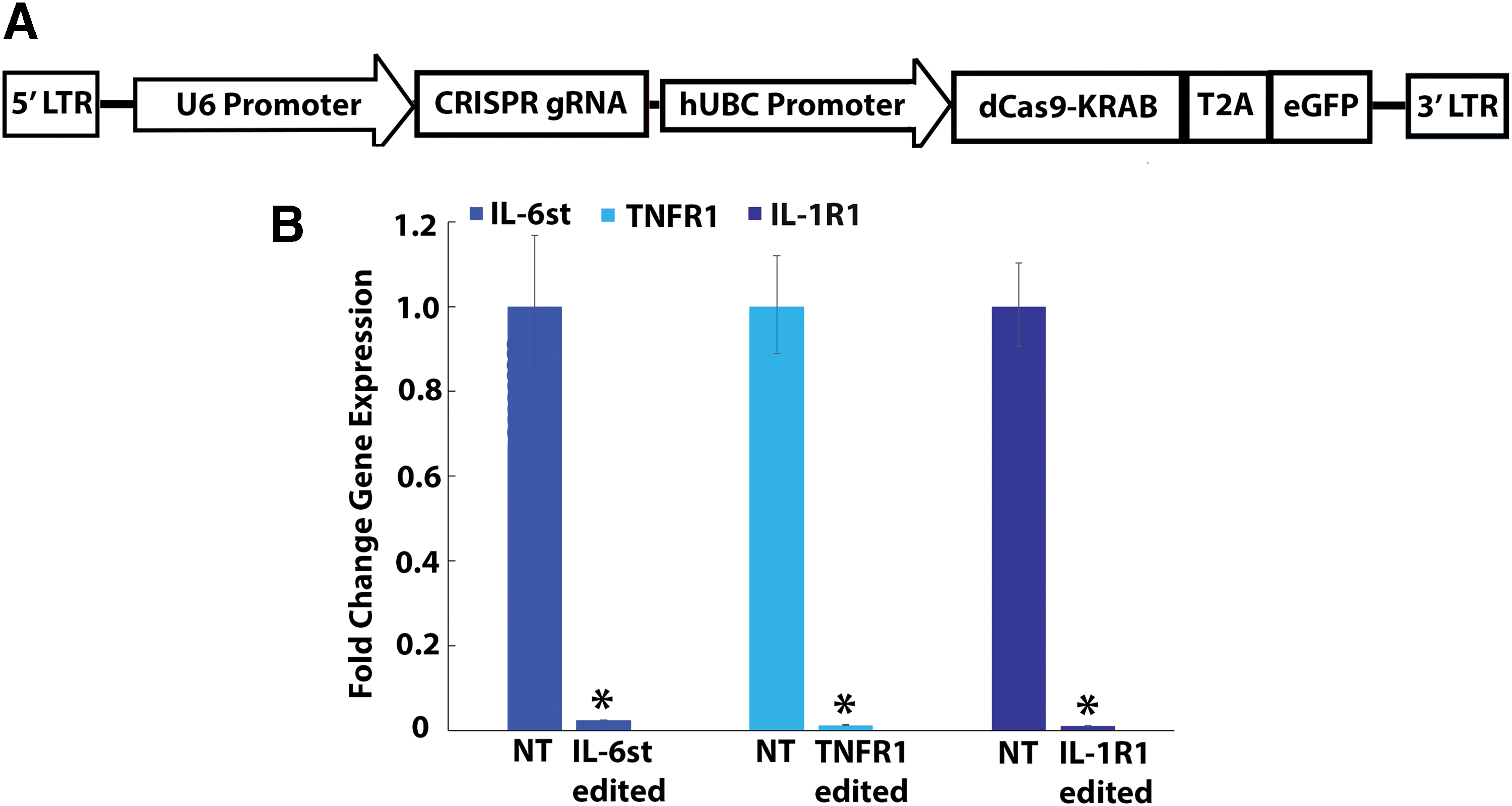
Epigenome editing of DRG neurons regulates IL-6st, IL-1R1, and TNFR1 gene expression in vitro.
The percentage of neurons exhibiting heat-induced calcium transients in singleplex IL-1R1, TNFR1, and IL-6st epigenome-edited neurons seeded onto pathologic AF tissue was significantly decreased (p < 0.05) compared to naïve (non-transduced) neurons seeded onto pathologic AF tissue (p < 0.05; Fig. 3A). However, the percentage of neurons exhibiting calcium transients in singleplex epigenome neurons seeded onto pathologic AF tissue remained significantly elevated over the percentage of neurons exhibiting calcium transients in naïve (non-transduced) neurons seeded onto healthy AF tissue (p < 0.05; Fig. 3A). Additionally, the percentage of neurons exhibiting heat-induced calcium transients in nontarget epigenome-edited neurons was not significantly different from the percentage of neurons exhibiting heat-induced calcium transients in naïve (non-transduced) neurons (Supplementary Fig. S2A). Moreover, for ΔF/F, the maximum calcium transients of naïve (non-transduced) neurons, as well as IL-1R1 and TNFR1 epigenome-edited neurons seeded onto pathologic AF tissue, were significantly elevated over the maximum calcium transients of neurons seeded onto healthy AF tissue (p < 0.05; Fig. 3B). Yet, the maximum calcium transients (ΔF/F) of IL-6st epigenome-edited neurons seeded onto pathologic AF tissue were not significantly elevated (p = 0.07) over the maximum calcium transients of naïve neurons seeded onto healthy AF tissue (Fig. 3B). Furthermore, nontarget epigenome editing of neurons had no effect on the maximum calcium transients for DRG neurons seeded onto healthy or pathologic AF tissue (Supplementary Fig. S2B).
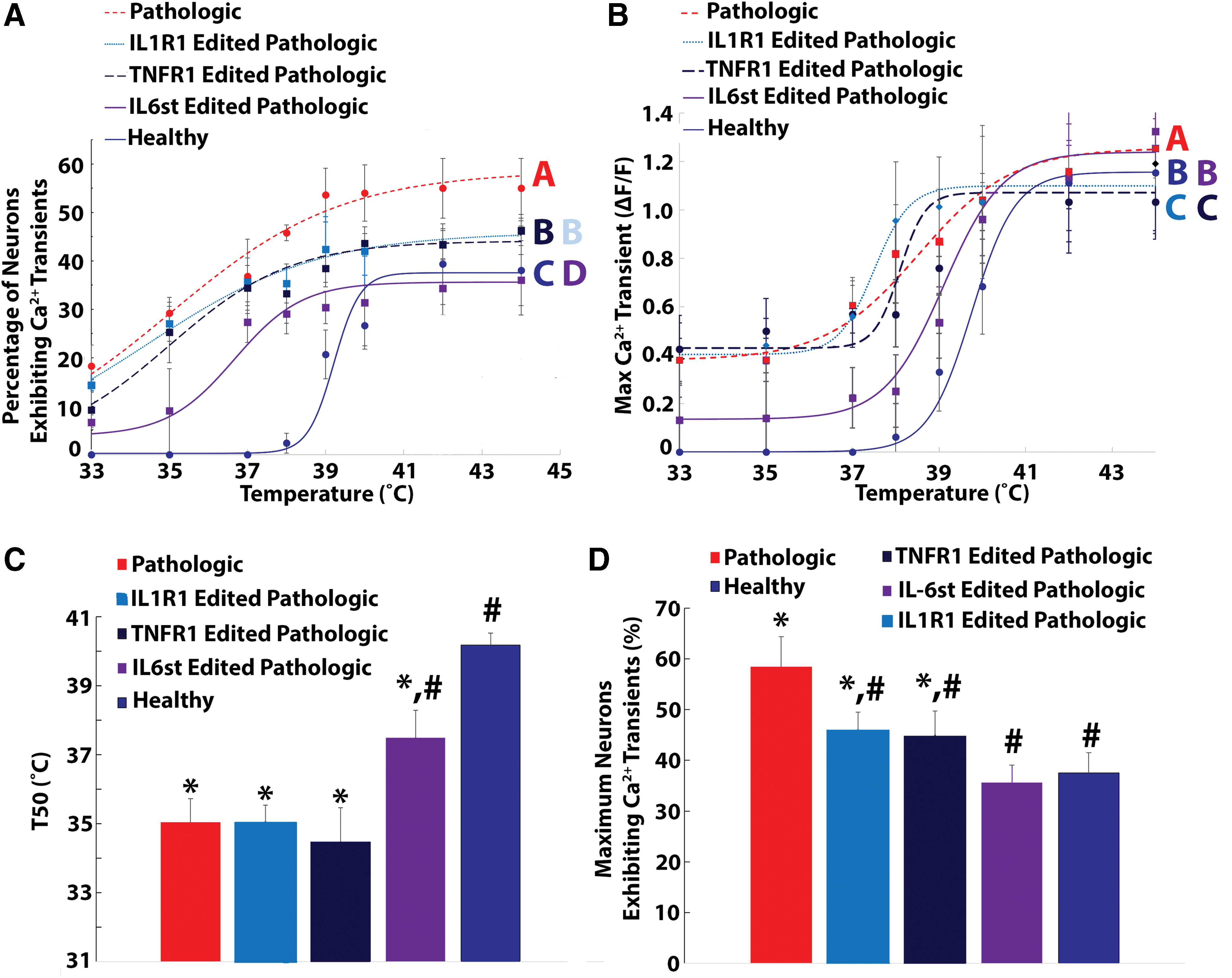
Singleplex epigenome editing of IL-1R1, TNFR1, and IL-6st reduced pathologic AF tissue induced neuron sensitization to thermal stimuli.
From the curve fitting of the percentage of DRG neurons exhibiting heat-induced calcium transients as a function of temperature, the T50 values of naïve neurons (35.03 ± 0.78°C) and IL-1R1 (35.0 ± 0.53°C), TNFR1 (34.47 ± 0.57°C), and IL-6st epigenome-edited (37.56 ± 0.75°C) epigenome-edited neurons seeded onto pathologic AF tissue were significantly less (p < 0.05) than the T50 of naïve neurons seeded onto control AF tissue (40.18 ± 0.34°C; Fig. 3C). However, the T50 (37.56 ± 0.75°C) of IL-6st epigenome-edited epigenome neurons seeded onto pathologic AF tissue is significantly elevated compared to the T50 values of naïve neurons seeded onto degenerative AF tissue (35.03 ± 0.78°C; Fig. 3C). In addition, the Tmax values of IL-1R1 (45.86 ± 3.7%) and TNFR1 (44.06 ± 4.6%) epigenome-edited neurons are significantly decreased compared to naïve neurons seeded onto pathologic AF tissue (58.45 ± 7.5%) while remaining significantly elevated (p < 0.05) over the Tmax values of naïve neurons seeded onto healthy AF tissue (37.5 ± 3.5%; Fig. 3D). In contrast, the Tmax of IL-6st epigenome-edited neurons seeded onto degenerative AF tissue (35.26 ± 4.5%) returned to baseline levels (Fig. 3D). Moreover, nontarget epigenome editing of DRG neurons had no effect on the T50 (Supplementary Fig. S2C) and Tmax (Supplementary Fig. S2D) of neurons seeded onto healthy or pathologic AF tissue. Together, these results demonstrate that while IL-6, IL-1β, and TNF-α each contribute to the elevated neuron activation observed, the complete elevated response cannot be attributed to any single cytokine signaling pathway.
Duplex epigenome editing of IL-6st, TNFR1, and IL-1R1 in DRG neurons reduces degenerative AF tissue-induced thermal sensitivity but does not eliminate it
Multiplex transduction of DRG neurons with CRISPR epigenome editing vectors targeting IL-6st, TNFR1, and IL-1R1 (Fig. 4A) simultaneously demonstrated robust downregulation of target gene expression at the RNA level when compared to DRG neurons transduced with nontarget lentiviral vectors (<25% of nontarget RNA expression; p < 0.05; n = 3 independent transductions) for all genes tested. In addition, multiplex epigenome-edited DRG neurons exhibited robust expression of epigenome editing vectors (Fig. 4B), with 54.9 ± 5.6% of transduced neurons showing expression of all three targeting vectors IL-6st, TNFR1, and IL-1R1.
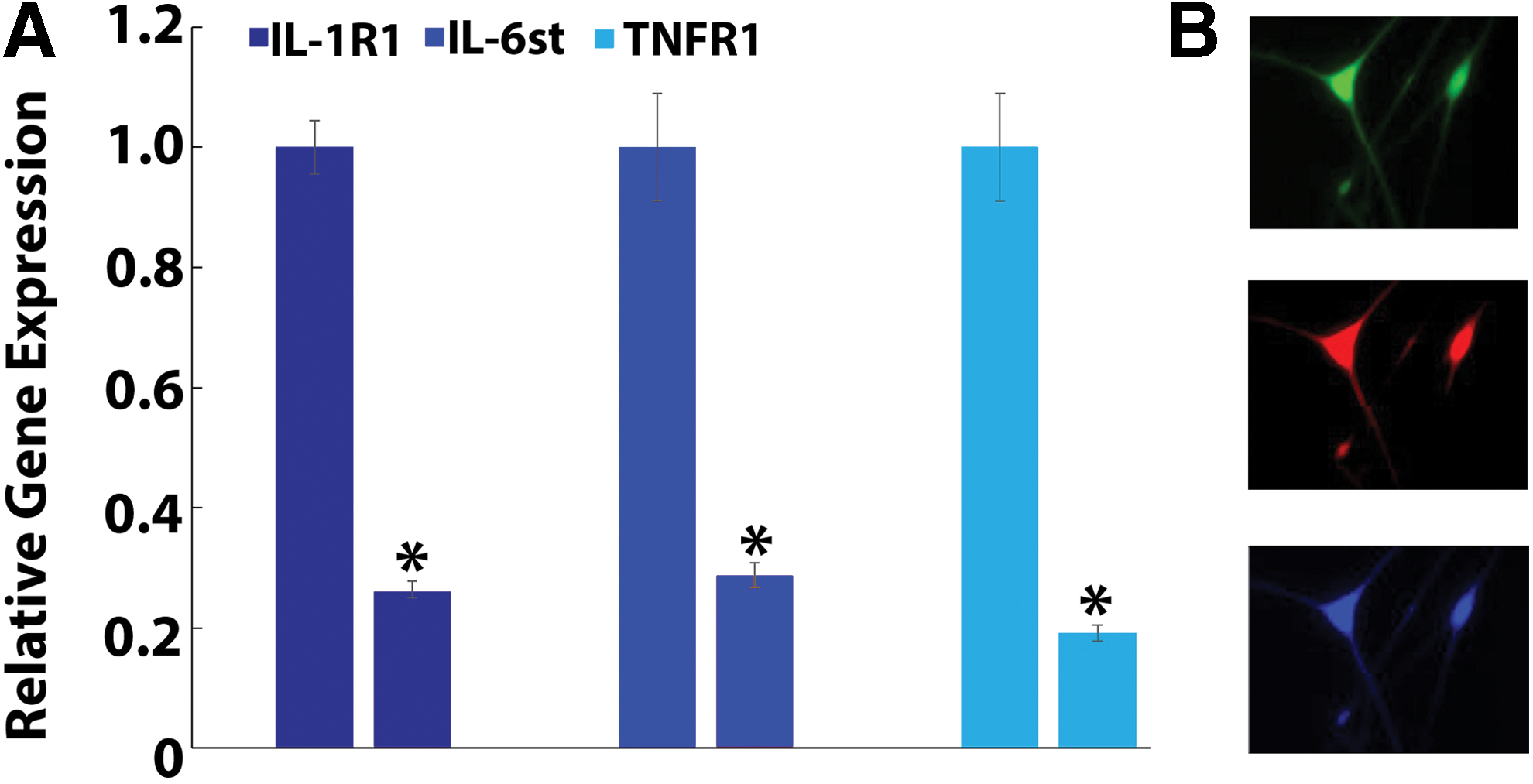
Multiplex epigenome editing of DRG neurons regulates IL-6st, IL-1R1, and TNFR1 gene expression.
The percentage of neurons exhibiting calcium transients was significantly elevated in naïve (non-transduced) neurons seeded onto pathologic AF tissue when compared to naïve neurons seeded onto healthy AF tissue (p < 0.05; Fig. 5A). In addition, the percentages of neurons exhibiting calcium transients in duplex epigenome-edited neurons (IL-1R1 + TNFR1 edited, IL-6st + IL-1R1 edited, and IL-6st + TNFR1 edited neurons) were significantly reduced (p < 0.05) when compared to naïve neurons seeded onto pathologic AF tissue (Fig. 5A). However, neuronal activity in duplex epigenome-edited neurons remained significantly elevated over baseline healthy levels (Fig. 5A).
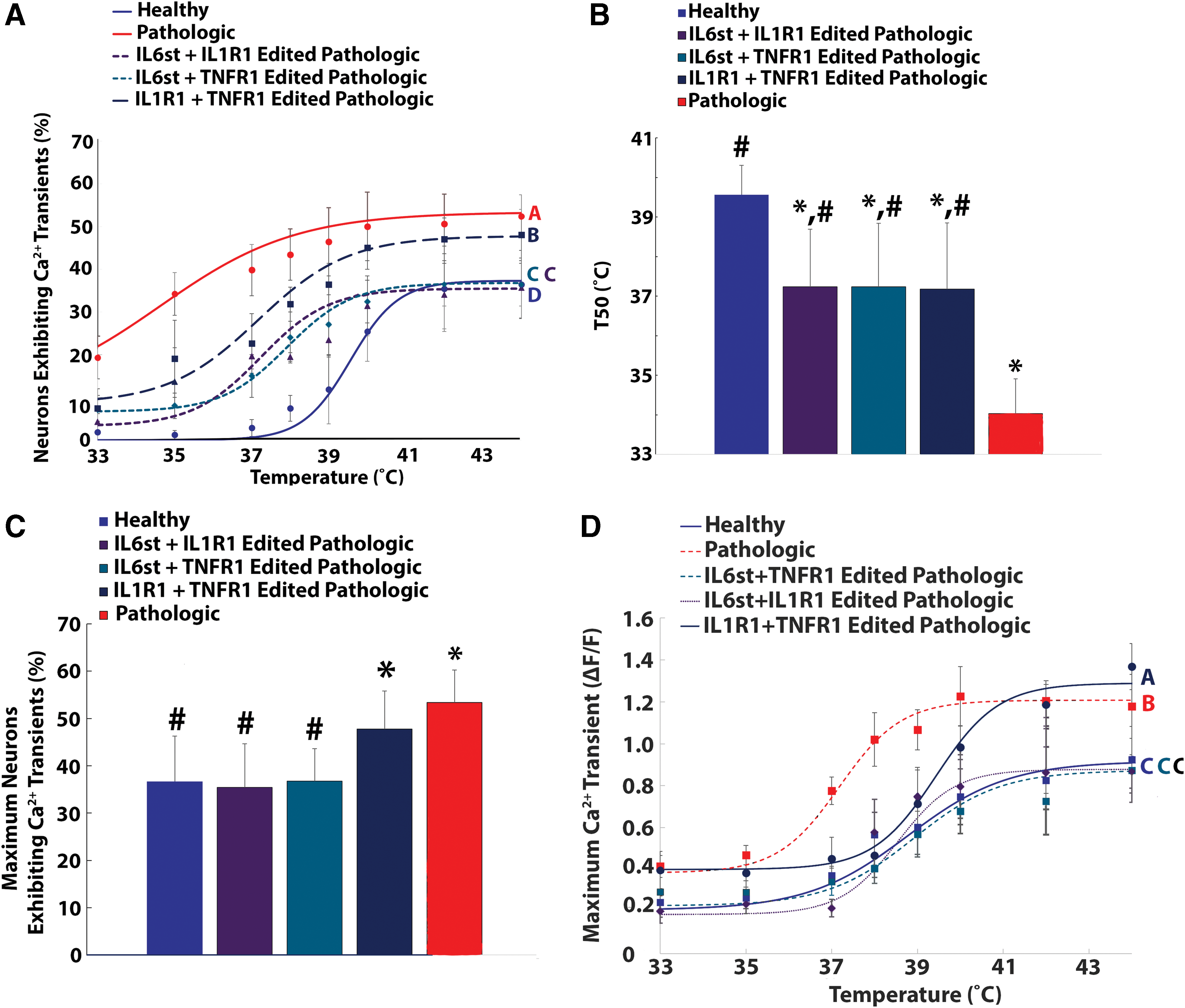
Duplex epigenome editing of DRG neurons reduced pathologic AF tissue induced enhanced thermal nociception.
From the curve fitting of the percentage of DRG neurons exhibiting heat-induced calcium transients as a function of temperature, the T50s of duplex-edited neurons were significantly reduced (p < 0.05) compared to the T50s (39.6 ± 0.74°C) of naïve neurons seeded onto control AF tissue (Fig. 5B). Nonetheless, the T50s of duplex epigenome-edited neurons remained significantly elevated over the T50 (34.03 ± 0.87°C) of naïve neurons seeded onto pathologic AF tissue (Fig. 5B). Additionally, IL1-R1 + TNFR1 epigenome editing of neurons had no effect on the Tmax of neurons seeded onto pathologic AF tissue (Fig. 5C). Contrastingly, the Tmax values of IL6st + IL1R1 and IL6st + TNFR1 duplex epigenome-edited neurons seeded onto pathologic AF tissue returned to baseline levels (Fig. 5C).
For ΔF/F, duplex epigenome editing of IL-6st + TNFR1 or IL-6st + IL-1R1 in DRG neurons seeded onto pathologic AF tissue returned the maximum calcium transients to baseline levels (Fig. 5D). However, duplex epigenome editing of TNFR1 + IL-1R1 significantly reduced the maximum calcium transients in DRG neurons seeded onto degenerative AF tissue, but they did not return to baseline levels (Fig. 5D).
Triplex epigenome editing of IL-6st, TNFR1, and IL-1R1 in DRG neurons eliminates degenerative AF tissue-induced thermal sensitivity
Again, it was demonstrated that neurons seeded onto degenerative AF tissue showed a significantly elevated percentage of neurons exhibiting calcium transients compared to neurons seeded onto healthy AF tissue (p < 0.05; Fig. 6A). However, triplex epigenome editing (IL-1R1 + TNFR1 + IL-6st) of neurons seeded onto pathologic AF tissue returned the percentage of neurons exhibiting calcium transients to baseline (healthy) levels (p = 0.28; Fig. 6A).
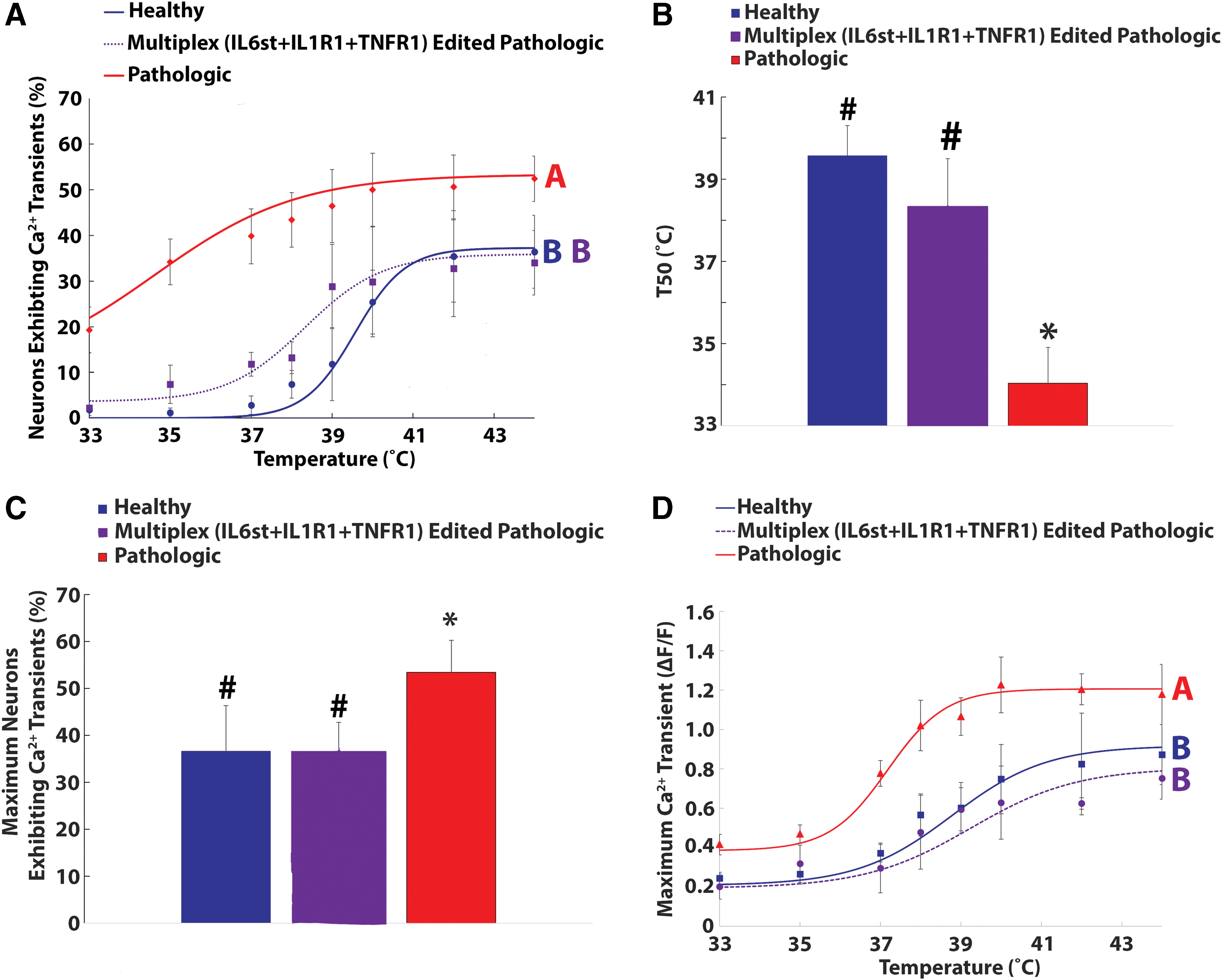
Triplex (IL-1R1 + TNFR1 + IL-6st) epigenome editing of DRG neurons abolished pathologic AF tissue-induced increased thermal nociception.
From the curve fitting, the T50 (34.03 ± 0.87°C) and Tmax (53.38 ± 6.8%) of neurons seeded onto pathologic AF tissue were significantly different (p < 0.05) from the T50 (39.6 ± 0.74°C; Fig. 6B) and Tmax (36.6 ± 9.7%; Fig. 6C) of neurons seeded onto healthy AF tissue. However, the T50 (38.32 ± 1.1°C) and Tmax (35.9 ± 5.8%) values of triplex (IL-1R1 + TNFR1 + IL-6st) epigenome-edited neurons returned to baseline (healthy) levels (Fig. 6B and C). Furthermore, for ΔF/F, the maximum calcium transients of triplex-edited (IL-6st + TNFR1 + IL-1R1) neurons seeded onto pathologic AF tissue returned to baseline levels (Fig. 6D). Together, these results demonstrate that triplex epigenome editing of IL-6st, TNFR1, and IL-1R1 expression in sensory neurons exposed to pathologic IVD tissue is required to return the activity of the neurons to baseline (healthy) levels.
Discussion
This study modeled and investigated the interactions between pathologic IVD tissue and sensory neurons to determine the ability of the degenerative IVD to alter sensory neuron activity in response to nociceptive stimuli present in the IVD. Utilizing this in vitro model, the key signaling pathways in the degenerative IVD and their relationship to neuronal activation were elucidated, and multiplex CRISPR epigenome editing was investigated for its ability to alter that response. Previously, a degenerative IVD supernatant model 54 was utilized to demonstrate that soluble factors (IL-6 and IL-6st) released from degenerative IVDs sensitize sensory neurons to nociceptive stimuli. In addition to soluble factors, the degenerative IVD contains other inflammatory cytokines that may sensitize nociceptive neurons. Furthermore, the degenerative IVD undergoes pathologic changes in ECM composition, such as the loss of hyaluronic acid (HA), which have been demonstrated to alter neuron function. 55
To determine the ability of these factors to alter neuronal activation, an in vitro model was developed in which nociceptive neuron activation, in response to nociceptive stimuli, was measured in sensory neurons seeded directly onto degenerative IVD tissue. This model allowed us the underlying mechanisms by which degenerative IVDs alter neuron activity to be identified and novel therapeutics to be screened for their ability to regulate these responses.
One potential limitation of this model was the usage of rat DRG neurons seeded onto human degenerative IVD tissue. This choice was made due to the scarcity of human DRG neurons available for research and is justifiable for this model. First, the conservation of neuron receptor profiles across species, as well as the ability of inflammatory cytokines to induce cross-species signaling has been well established in the literature. In addition, a previous study utilizing a similar model demonstrated IL-6/IL-6 soluble receptor-driven sensitization of sensory neurons to thermal stimuli—results consistent with in vivo and in vitro animal models of inflammation-induced pain. Furthermore, by utilizing human tissue, the model may provide a system to identify therapeutic targets and to test therapeutic efficacy in a way that better translates to the clinic than the use of animal models alone.
The data demonstrate that the pathologic IVD contains multiple factors that directly induce elevated neuron activity in response to thermal stimuli, suggesting the degenerative IVD enhances nociception in sensory nerves. This enhanced neuronal activation resulted in increased percentages of neurons exhibiting calcium transients in response to thermal stimuli, increased maximum calcium transients, and a T50 activation threshold (34.03 ± 0.87°C) below normal core body temperature ranges that were not observed in neurons seeded onto healthy IVD tissue. Increased nociception was observed in neurons exposed to pathologic IVD tissue from all patients tested in this study (Supplementary Table S1). The patient population was comprised of patients of both sexes, exhibiting varying degrees of disc degeneration at the time of surgical treatment (Supplementary Table S1). In addition, most patients had comorbidities ranking among the most prevalent comorbidities in the low back pain patient population. While the current study did not control for the contributions of these factors (i.e., sex differences) to pain, the consistent changes in sensory neuron activity triggered by degenerative IVDs from all patients tested in this study suggests similar mechanisms may exist in subpopulations of low back pain patients, and this merits future investigation.
Next, CRISPR epigenome editing of the cytokine receptors of sensory neurons was utilized to identify the primary cytokines mediating enhanced neuron activity induced by direct degenerative IVD tissue exposure. The data demonstrate that singleplex CRISPR epigenome editing-based cytokine receptor repression of IL-6st, IL-1R1, or TNFR1 independently reduced but did not abolish elevated neuron activity triggered by pathologic IVD tissue exposure. In addition, triplex epigenome editing of IL-6st, IL-1R1, and TNF1 expression simultaneously in DRG neurons abolished enhanced neuron activation induced by degenerative IVD tissue exposure. Together, these results implicate the combined activation of IL-6, TNF-α, and IL-1β signaling pathways as the primary mechanism of pathologic IVD-induced elevated neuron activity elucidate these redundant signaling pathways as potential mechanisms of discogenic pain, and suggest successful treatment strategies of discogenic pain will require targeting of multiple inflammatory signaling pathways in contrast to attempts to treat with singleplex inflammatory cytokine targeting. 56 –58 Multiple clinical trials have focused on treating discogenic back pain via intradisc injection of monoclonal blocking antibodies targeting IL-6, 55 TNF-α, 57 or IL-1β 56 signaling pathways individually. These treatments were found to provide short-term reduction in pain. 55,57 However, they were ineffective at providing long-term pain relief. 55 –57 One possible explanation is that redundant signaling through activation of a combination of IL-6, TNF-α, and IL-1β signaling pathways, as demonstrated in this study, contribute to discogenic pain, and successful treatment strategies for discogenic low back pain will require multiple inflammatory cytokine sensitization mechanisms to be targeted.
This study demonstrated that pathologic IVD tissue-driven activation of IL-6, TNF-α, and IL-1β signaling pathways in sensory neurons enhances sensory neuron activity in response to nociceptive stimuli, particularly thermal stimuli, present in the degenerative IVD. Previous studies have established the presence of pathologically elevated levels of the pro-inflammatory cytokines IL-6, TNF-α, and IL-1β in painful, 12 –16 degenerative IVDs. In addition, IL-6, TNF-α, and IL-1β have been implicated in animal models of disc degeneration and have been demonstrated to sensitize rodents to thermal and mechanical stimuli in the hind paw in models of radiculopathy and peripheral neuropathy. 59 Furthermore, it has previously been demonstrated that IL-6 released from painful, degenerative IVDs directly enhanced sensory neuron activity in response to thermal stimuli. 49 This study demonstrated the ability of degenerative, but not healthy, IVD tissue to trigger enhanced nociception directly. Together, these results suggest IL-6, TNF-α, and IL-1β present in the degenerative IVD are bound by their respective receptors (IL-6st, TNFR1, and IL-1R1) in sensory neurons, activating pro-inflammatory signaling pathways that lead to neuron sensitization to thermal stimuli.
To test this hypothesis, CRISPR epigenome editing was utilized to downregulate the expression of IL-6st, TNFR1, and IL-1R1 (the receptors for IL-6, TNF-α, and IL-1β, respectively) in sensory neurons seeded directly onto degenerative IVD tissue. Utilizing this strategy allowed the potential roles of the activation of the IL-6, TNF-α, and IL-1β signaling pathways to be simultaneously identified in degenerative IVD-induced enhanced thermal nociception in sensory neurons and CRISPR epigenome editing of IL-6st, TNFR1, and IL-1R1 expression in sensory neurons to be tested as a potential neuromodulation strategy for the treatment of discogenic pain. This study demonstrated that the ability of CRISPR epigenome editing of the gene promoter region of IL-6st, TNFR1, and IL-1R1 significantly downregulated their expression in sensory neurons. In addition, it demonstrated that epigenome editing of IL-6st, TNFR1, and IL-1R1 expression (the receptors for IL-6, TNF-α, and IL-1β, respectively) in DRG neurons significantly reduced degenerative IVD-driven enhanced nociception. These results suggest that degenerative IVDs drive enhanced thermal nociception in sensory neurons via the combined activation of the IL-6, TNF-α, and IL-1β signaling pathways, not solely the activation of the IL-6 signaling pathway, as indicated in the supernatant model utilized in a previous study. 49 Together, these findings support the hypothesis that inflammation-driven sensitization of sensory neurons in the degenerative IVD contributes to discogenic back pain via sensitization of nociceptive neurons to stimuli that would not be painful in healthy patients, identifies the simultaneous activation of IL-6, TNF-α, and IL-1β signaling pathways as the primary mediators of this sensitization, and establishes CRISPR-based neuromodulation of sensory neurons as a potential treatment strategy for discogenic pain. Additionally, these three cytokines appear to be responsible for all the sensitization observed.
In this study, neurons exposed to pathologic IVD tissue exhibited increased maximum calcium transient values, and a reduction of heat-induced calcium transient thresholds, indicating pathologic IVD directly sensitized sensory neurons to noxious stimuli at temperatures as low as 33°C. Under pathologic IVD conditions, the T50 value (34.03°C) of neurons occurs below the normal core body temperature region (36.1–37.8°C), and the Tmax is observed just above this range (38°C). A previous study demonstrated the lowering of the TRPV1 firing threshold by IL-6 released from degenerative IVDs. 54 Most neurons innervating the degenerative IVD are small, nociceptive neurons expressing TRPV1, indicating the lowering of TRPV1 thermal activation threshold to core body temperatures by IL-6, TNF-α, and IL-1β present in the pathologic IVD provides a mechanism for elevated nociception in the degenerative IVD.
In addition, the data suggest that degenerative IVD activation of IL-6, TNF-α, and IL-1β signaling pathways may drive enhanced thermal nociception in sensory neurons via multiple underlying mechanisms. This study demonstrated that duplex epigenome editing of all combinations of IL-6st, TNFR1, and IL-1R1 expression in sensory neurons increased the T50 value of neurons exposed to degenerative IVD tissue. However, triplex epigenome editing was required to return the T50 value to baseline (healthy) levels. Furthermore, duplex epigenome editing of TNFR1 + IL-1R1 had no effect on Tmax. In contrast, duplex epigenome editing of all IL-6st combinations (IL-6st + TNFR1 and IL-6st + IL-1R1), as well as triplex epigenome editing of sensory neurons, returned the Tmax to baseline (healthy) levels. Together, these results suggest that TNF-α and IL-1β play a larger role in reducing the thermal firing threshold of sensory neurons exposed to pathologic IVD, while IL-6 signaling appears to play a larger role in increasing the percentage of thermally responsive neurons. Further investigation is required to parse the underlying mechanisms by which IL-6, TNF-α, and IL-1β contribute to degenerative IVD-induced enhanced thermal nociception.
CRISPR epigenome editing systems have tremendous potential to modulate cell function. This study demonstrated their ability to modulate the cytokine receptor expression of DRG neurons in multiplex and alter the DRG neuron response to degenerative IVD tissue. The data demonstrate that sensory neurons with multiplex epigenome modifications of the TNFR1, IL-1R1, and IL-6st promoter regions maintained normal activation under pathologic conditions that sensitize non-edited neurons to noxious stimuli. These results demonstrate that CRISPR multiplex epigenome editing of pain-related genes in nociceptive neurons regulates degenerative IVD modulated sensitization and establishes epigenome modification of sensory neurons as a potential therapeutic strategy for the treatment of back pain.
This study demonstrated that TNF-α, IL-1β, and IL-6 present in the degenerative IVD induced elevated neuron activation at sub-37°C temperatures through the activation of TNFR1-, IL1R1-, and IL6st-dependent signaling pathways. These results demonstrate potential underlying mechanisms for the activation of nociceptive neurons in the degenerative IVD. Additionally, this study demonstrated that multiplexepigenome editing of the TNFRI, IL1R1, and IL6st gene promoter regions of sensory neurons abolished degenerative IVD mediated sensitization of sensory neurons to noxious (i.e. thermal) stimuli. These results elucidate an underlying mechanism of discogenic pain, identify therapeutic targets for the treatment of back pain, and establish multiplex epigenome editing of nociceptive neurons as a treatment strategy for discogenic back pain.
Footnotes
Acknowledgments
The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R03AR068777. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.