
Abstract
The application of CRISPR/Cas9 has opened a new era in gene therapy, making it possible to correct mutated genomes in vivo. Exon replacement can correct many mutations and has potential clinical value. In this study, we used a lentivirus-delivered transgene to obtain transgenic mice in which Cas9 and green fluorescent protein (GFP) were driven by the hTBG promoter and were specifically expressed in the liver. In Cas9-positive mice, only ∼11.6% of hepatocytes were GFP positive. The newborn Cas9-positive F1 mice were injected via the temporal vein with rAAV carrying a modified homologous replacement sequence for exon 8 of Atp7b and a pair of single-strand guide RNAs targeting the introns surrounding exon 8. When the Cas9-positive hepatocytes were sorted and analyzed by PCR and next-generation deep sequencing with different labels, ∼16.34 ± 4.02% to 19.37 ± 6.50% of the analyzed copies of exon 8 were replaced by the donor template in the genome of GFP-positive hepatocytes, that is, 1.81 ± 0.29% to 2.09 ± 0.54% replacement occurred in all liver genomes. However, when rAAV carrying a modified homologous replacement sequence was injected into the adult spCas9 mice, a double-cut deletion ratio of up to 99%, only about 1.10–1.13% of the exon 8 replacement rate was detected in Cas9-positive hepatocytes. This study is the first to achieve exon replacement via CRISPR/Cas9, which will benefit research on CRISPR/Cas9 technology for gene therapy.
Introduction
The application of CRISPR/C
In recent years, CRISPR/Cas9 technology-mediated gene therapy has made major breakthroughs in a variety of hereditary diseases. In 2014, researchers used the CRISPR/Cas9 system to perform gene editing on the mouse model of Duchenne muscular dystrophy (DMD), corrected the mutant DMD gene, and successfully alleviated muscle atrophy in mice. 13 In 2016, researchers in the Wilson Laboratory used the dual AAV (adeno-associated virus) coinfection protocol to repair point mutations in the ornithine carbamoyltransferase gene that results in a rare metabolic urea cycle disorder, eventually achieving a cell repair rate of ∼10%. 14
The liver is an important metabolic organ in which many important metabolic processes occur. Therefore, there are many inherited liver diseases, such as hemochromatosis, Wilson's disease (WD), α1-antitrypsin deficiency, and citrin deficiency. 15 However, the liver is also a blood-rich organ, and most hepatocytes are immersed in plasma. The above characteristics make the liver a suitable target organ for blood-based delivery of drugs and therapeutic carriers. In humans, WD is a genetic disease caused by a mutation in the ATP7B gene, which encodes an important enzyme in the metabolism of copper ions. The mutation of ATP7B may cause ceruloplasmin synthesis disorder and biliary copper excretion disorder, thus resulting in excessive copper deposition in various organs, particularly in the liver. 16 In WD, there are at least 500 mutation types, although point mutations predominate, and there are regional and ethnic differences. 17 –20 In particular, exon 8 is the mutation hotspot in WD patients (Supplementary Fig. S1). Arg778Leu is the most common mutation in Asia, but the W779X and 2299insC, 18 Gly710Ala, 21 Gly710Ser, 22 and Gly710Arg 23 mutations have also been reported. Therefore, the replacement of exon 8 can correct various mutations and has clinical value.
In this study, we obtained transgenic mice that specifically express spCas9 and green fluorescent protein (GFP) in the liver by injecting packaged lentivirus into the perivitelline space of the fertilized egg. The newborn transgenic spCas9 mice were injected with rAAV carrying a homologous repair sequence and a pair of sgRNAs targeting the introns surrounding exon 8 of Atp7b. Through next-generation sequencing (NGS), we found that ∼16.34 ± 4.02% (4.49–28.31%) to 19.37 ± 6.50% (5.25–37.24%) of the analyzed copies of exon 8 were replaced by donor template in the genome of GFP-positive hepatocytes, that is, 1.81 ± 0.29% (0.86–2.29%) to 2.09 ± 0.54% (0.71–3.07%) replacement occurred in all liver genomes.
Methods
All experimental protocols, including animal care, the microinjection protocol, and embryo transfer, were performed according to the approved guidelines (IACUC No. A-2015-002) established by the Ethics Committee of the School of Medicine, Shanghai Jiao Tong University. All media and chemicals were from Sigma–Aldrich without special declaration. C57 mice were maintained in an AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care)-accredited facility.
Plasmids and construction of the lentiviral vector
Lenti-CRISPRv2GFP, psPAX2, and pMD2G were gifts from David Feldser (Plasmid No. 82416; Addgene). Lenti-CRISPRv2 was a gift from Zhang Feng (Plasmid No. 52961; Addgene). pX602-AAV-TBG and NLS-SaCas9-NLS-HA-OLLAS-bGHpA; U6:BsaI-sgRNA were gifts from Zhang Feng (Plasmid No. 61593; Addgene).
The Lenti-TBG-spCas9-GFP plasmid was constructed as follows. (1) The U6-sgRNA element in the Lenti-CRISPRv2GFP vector was digested with KpnI (No. 1068S; TaKaRa, Dalian, China) and EcoRI (No. 1040S; TaKaRa), and then, primers containing KpnI and EcoRI sticky ends (F: CCTCGAGAAGCTTCGGATCCTCTAGAG and R: AATTCTCTAGAGGATCCGAAGCTTCTCGAGGGTAC) were annealed and connected by T4 DNA Ligase (No. E140879; TaKaRa). (2) The EF-1a promoter in Lenti-CRISPRv2GFP was replaced with the hTBG promoter amplified from the pX602-AAV-TBG plasmid with the following primer pair: F: CGGGGTACCTGCATGTATAATTTCTACA and R: CTAGTCTAGATTATAGCATGTCCTGTAT. Then, the hTBG promoter was inserted into Lenti-CRISPRv2GFP at the KpnI and XbaI (No. 1093S; TaKaRa) sites. Lenti-CRISPRv2 carrying sgRNA1F or sgRNA2R was cloned using BsmbI (No. ER0451; Thermo Fisher Scientific) and T4 DNA Ligase according to the lenti-CRISPRv2 user manual.
Cell culture and transfection
AML12 cells (ATCC) were maintained in F12/DMEM (Gibco, Grand Island, NY) supplemented with 10% FBS (Gibco) at 37°C with 5% CO2. For in vitro target or donor template testing, plasmids were transfected into AML12 cells using Lipofectamine 2000 with Plus Reagent (Life Technology, Shanghai, China) according to the manufacturer's recommendations. Transfected cells were cultured in the presence of puromycin (2 μg/mL) for 4 days to enrich transfected cells.
sgRNA and donor DNA design and vector construction
Two sgRNAs (sgRNA1F and sgRNA2R) were devised using the CRISPR Design tool to target the sequence beside exon 8 of mouse Atp7b. The DNA sequences of sgRNA1F and sgRNA2R omitting NGG in the PAM were capped with a T7 promoter sequence and named P-sgRNA1 and P-sgRNA2, respectively (Supplementary Table S1).
The donor DNA contained the Atp7b exon 8 complete sequence with homologous arms (left 501 bp and right 268 bp). The GG site (8q22013858–22013859) in the PAM (AGG) of the sgRNA1F recognition site in the homologous arm of the donor DNA was replaced with CC. At the same time, the corresponding site, TGG, in the PAM region of the sgRNA2R recognition site in the donor DNA (8q22013268–22013270) was deleted to prevent donor DNA from being cut again (Fig. 1A).
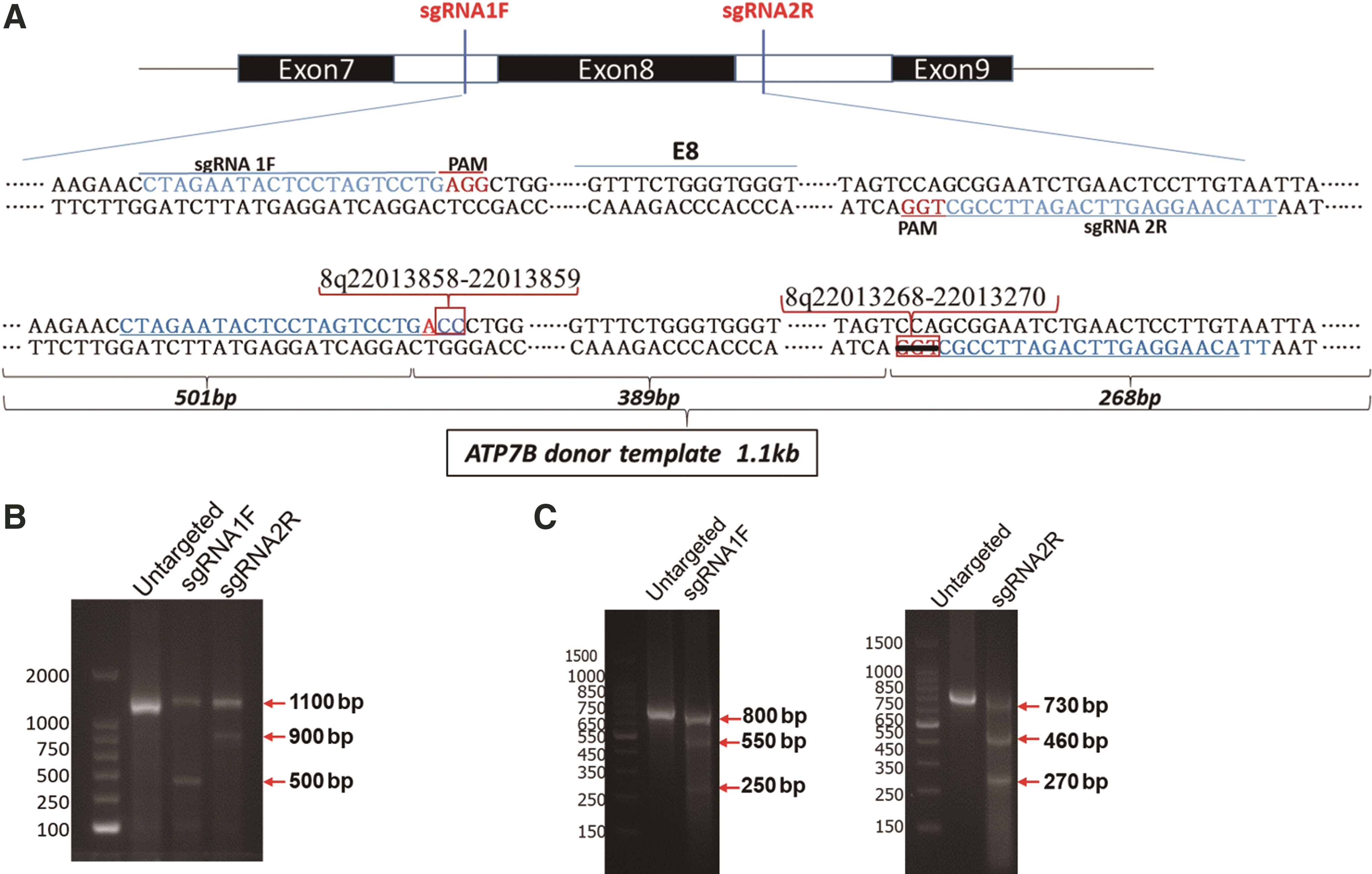
sgRNA design and cleavage efficiency.
The rAAV plasmid (AAV-U6sg-DDFE8; Fig. 2C) contained three PCR fragments (donor DNA, U6-sgRNA1F, and U6-sgRNA2R) that were fused into a single product by complementary fragments and cloned into the rAAV vector (pX602-AAV-TBG NLS-SaCas9) at the XhoI (No. 1094S; TaKaRa) and NotI (No. 1166S; TaKaRa) sites using T4 DNA Ligase.

The experimental schedule and plasmids constructed for Cas9 expression and exon replacement.
The three fragments (donor DNA, U6-sgRNA1F, and U6-sgRNA2R) were constructed as follows. (1) The donor DNA containing exon 8 of Atp7b with CC replaced with GG in the PAM (AGG) of the sgRNA1F recognition site was amplified from the wild-type (WT) Atp7b genomic sequence by three primer pairs (rep-1F and rep-1R; Rep-CC-2F and Rep-CC-2R; and rep-3F and rep-3R). These three PCR fragments were fused into a single product and named the donor DNA template. (2) The donor DNA template was amplified by the primers Fu-rep-F and Fu-rep-R. (3) The U6-sgRNA1F fragment was amplified from lenti-CRISPRv2 carrying sgRNA1F by the primers XhoI-U6-sgRNA1F-F and Fu-sgRNA1F-R. The U6-sgRNA2R fragment was amplified from lenti-CRISPRv2 carrying sgRNA2R by the primers rep-sgRNA2R-F and NotI-sgRNA2R-R. All the primers mentioned above are shown in Supplementary Fig. S2, and the sequences are shown in Supplementary Table S2.
RNA synthesis
The pT7-cas9 plasmid was digested with XbaI and purified by isopropanol precipitation. The linearized plasmid was transcribed to messenger RNA (mRNA) in vitro using the mMESSAGE mMACHINE T7 Ultra Transcription Kit (Ambion, Austin, TX). Cas9 mRNA was purified with lithium chloride precipitation according to the manufacturer's instructions. DNA templates for sgRNA1F and sgRNA2R were generated from sgF0 by PCR amplification using the forward primers P-sgRNA1F and P-sgRNA2R with the reverse primer T7R1. The above two templates were amplified with T7primer and T7R1. The products of the second PCR were purified and used for in vitro RNA synthesis by the MEGAshort T7 High Yield Transcription Kit (Invitrogen). The sgRNAs were purified with the MEGAclear-96 Kit in a 96-well format (Ambion). Cas9 mRNA and sgRNAs were dissolved in TE buffer (Ambion).
Detection of external cutting efficiency of the sgRNAs
The efficacy of cleavage by sgRNAs in vitro was detected by the Guide-it sgRNA In Vitro Transcription and Screening System Kit (Takara). The sgRNA-targeted fragments were amplified by PCR from WT mouse DNA with the primers m78JDFand m78JDR. The cleavage reaction containing 20 ng of the experimental sgRNA sample and 100 ng of the sgRNA-targeted fragments was incubated at 37°C for 1 h and terminated by incubation at 70°C for 10 min. The entire 10 μL reaction was analyzed on a 2% TAE agarose gel.
The TAE agarose gel result was analyzed by a custom image processing software package developed using the ImageJ (NIH) scripting platform to perform most of the data processing to confirm cleavage efficiencies. The MIT CRISPR design tool is available online.
In vitro validation of sgRNAs
AML12 cells were infected with lenti-CRISPRv2 carrying sgRNA1F or sgRNA2R, followed by 4 days of selection with 2 μg/mL puromycin. Genomic DNA from transfected AML12 cells was extracted using the QuickExtract DNA Extraction Solution (Epicentre Biotechnologies, Germany). The efficiency of each individual sgRNA was tested by the Guide-it™ Mutation Detection Kit (Takara).
Lentivirus and AAV vector production
The lentivirus and rAAV8 vector (AAV-U6sg-DDFE8) used in this study were produced by Genomeditech (Shanghai, China). The genome titer (genome copies/mL) of AAV vectors was determined by quantitative PCR (qPCR) by the manufacturer to be 1.09E + 12 VG/mL. The lentivirus titer was identified by gradient dilution to be ∼1 × 107 TU/mL.
Microinjection and embryo transfer
Mature female mice were superovulated by injection of 5 IU pregnant mare stimulation gonadotropin (PMSG; Ningbo Sansheng Second Hormone Factory, China), followed by 10 IU human chorionic gonadotropin (hCG; Ningbo Sansheng Second Hormone Factory) before they were mated. The fertilized eggs were flushed from the oviducts with prewarmed M2 medium and harvested in KSOM medium. The Lenti-TBG-spCas9-GFP virus was injected into the perivitelline space by a 3-μm-diameter pipette using a Piezo instrument (PMM-4G; Prime Tech, Japan). After microinjection, zygotes were cultured in KSOM medium in a humidified atmosphere containing 5% CO2 and 95% air at 37°C for 24 h.
Approximately 20 embryos at the 2- or 4-cell stage were surgically transferred to both oviducts of each recipient mouse. Offspring were born naturally 20 days after embryo transfer.
Genotyping
Genomic DNA was extracted from tail snips of newborn mice using a TIANgen Genomic DNA Kit (TIANGEN, Beijing, China). The fragments were amplified from genomic DNA by PCR with a pair of primers (spCas9-F: ATGGACAAGAAGTACAGCATCGGCC; spCas9-R: GCGGTAGCCTTGCCGATTT) and then analyzed on a 1.5% TAE agarose gel. Furthermore, some PCR products were sequenced by Lifetech Co. (Shanghai, China).
Off-target analysis
The potential off-target sites of the two sgRNAs were predicted using the CRISPR Design tool. The top three POTS of each sgRNA were selected according to the ranking scores. The potential off-target sites were analyzed by BLAST search of the predicted position in nucleotide sequences. Approximately 400–600 bp genomic fragments containing the off-target sites were amplified by PCR and sequenced.
AAV injection into the newborn mice and the 10-week-old mice
Mating cages were monitored daily for births. Newborn (postnatal day 2) spCas9 pups received a temporal vein injection of 3 μL rAAV8 vector (AAV-U6sg-DDFE8) diluted in 50 μL DMEM, as described previously. 24 Mice were euthanized 1 week after rAAV8 vector treatment, and liver samples were harvested for analyses. The DMEM-S0 group included three spCas9 pups injected with 50 μL DMEM and served as a control. Mice were genotyped at weaning or at the time of necropsy to confirm their genotype.
For the 10-week-old F1 mice, a tail vein injection of 150 μL rAAV8 vector or DMEM per mouse was given. After 1 week of AAV injection, liver single cell suspension preparation and GFP-based flow sorting were performed to extract positive cells for further analysis.
Hepatocyte separation and flow cytometry
One week after the AAV injection, spCas9-positive mice were anesthetized. The liver was perfused with 0.5 mM EGTA through the portal vein and then perfused and digested with 0.5 g/L type IV collagenase (C0130; Sigma) to obtain single dispersed hepatocytes for flow cytometry sorting to harvest GFP-positive cells.
Immunohistochemical analysis of liver tissue
The livers of some spCas9-positive F1 mice were fixed with 4% polyformaldehyde and then embedded in paraffin. Then, liver tissue sections were subjected to immunohistochemical staining with a GFP antibody (ab290; Abcam) containing AEC substrates. Liver tissues of spCas9-positive mice should be recognized by GFP antibodies and stained red.
NGS analysis
We designed different 6-nt labels upstream of the forward PCR primers and then mixed the PCR products from different templates for deep sequencing. Then, we used an R platform-based analysis based on label sequence to quantify total reads and CC tag-positive reads or CCA deletion tag reads in each subject to calculate the replacement ratio.
Quantitative PCR
We designed primers in the genomic region between sgRNAF and sgRNAR for the quantitation of WT and replaced sequences: F2: TATGCCTACTCCCTGGTCATC and R2: CAGGGCGATGAACACAAAGAG. Other primers were designed upstream of the donor DNA template for the quantitation of total genomic material: F3: TAGTTCCAGTGGTTCCTCATG and R3: TTCTTCTTAGTTCCCACATTT (Fig. 3B). Real-time qPCR was performed with a 7900 HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA) using FastStart Universal SYBR Green Master Mix (Roche), with WT genomes as the reference for samples.

AAV-mediated Atp7b exon 8 replacement in the newborn TBG-spCas9-GFP mice.
Data presentation and statistical analysis
All the results are presented as the mean ± standard error of the mean. Real-time qPCR data were analyzed with GraphPad Prism 5.0 software.
Results
Construction of the TBG-spCas9-GFP lentiviral vector
To facilitate the replacement of genomic exons in living liver tissue, mice expressing Cas9 in the liver were designed (Fig. 2A). To specifically express spCas9 in hepatocytes, the EF-1a promoter in the Lenti-CRISPRv2GFP plasmid was replaced with the TBG promoter amplified from the pX602-AAV-TBG plasmid, and the new plasmid was named Lenti-TBG-spCas9-GFP (Fig. 2B). The three plasmids, Lenti-TBG-spCas9-GFP, psPAX2, and pMD2G, were transfected into 293T cells, and the Lenti-TBG-spCas9-GFP virus particles were collected after 3 days.
When the AML12 hepatocyte line was infected with the virus solution (multiplicity of infection [MOI] = 100), 95.3% of the cells became GFP positive (fluorescence ratio) (Fig. 2D). The spCas9 sequence could be amplified from GFP-positive cells (Fig. 2E). This result proved that the constructed TBG-spCas9-GFP lentivirus can successfully carry the spCas9 gene into the AML12 cell genome.
Production of TBG-spCas9-GFP mice by microinjection of lentivirus
The constructed TBG-spCas9-GFP lentivirus was injected into the perivitelline space of fertilized eggs from C57BL/6N mice for transgenic mouse production. A total of 40 injected embryos that developed to the 2-cell stage were transferred to pseudopregnant recipients, and five offspring were born. We extracted the genome of the five mice and analyzed it by PCR. Three spCas9-positive offspring were found, including one female (number 2) and two males (numbers 3 and 4) (Fig. 4A). When spCas9-positive F0 mice were mated with WT C57 mice, seven F1 mice were first obtained. The PCR results (Fig. 4B) showed that five F1 mice carried spCas9, demonstrating that the spCas9 gene can be inherited by offspring of the F0 generation. The F1 generation mice were continuously produced and used for the next experiments.

Analysis of genomic DNA and Cas9 expression in TBG-spCas9-GFP mice.
spCas9 and GFP expression in TBG-spCas9-GFP mice
In the Lenti-TBG-spCas9-GFP transgenic mice, spCas9 and GFP are co-expressed under control of the TBG promoter, so GFP expression can reflect spCas9 expression. Hepatocytes were isolated from F1 spCas9-positive mice and analyzed by flow cytometry (Fig. 4C). The results showed that some hepatocytes expressed GFP protein, but no GFP-positive cells were detected in kidney tissue (Supplementary Fig. S3). The results indicated that spCas9 protein was specifically expressed in the liver of spCas9-positive mice; however, the entire liver did not express GFP, and only ∼11.6% of hepatocytes were GFP positive. To further confirm the tissue distribution of GFP-positive cells, liver tissue sections were subjected to immunohistochemical analysis using a GFP antibody (Fig. 4D). The results showed that compared with liver tissue in the immunoglobulin G control group, the liver tissue in the spCas9-positive group was recognized by the GFP antibody and stained red, which confirmed GFP protein expression and thus spCas9 protein expression. The rate of positive cells per section was similar to that determined by flow cytometry, and the positive cells are unevenly distributed. Only a small number of clusters of 5–10 cells expressed GFP protein, and the distribution of positive cells had no significant association with anatomy. These results indicated that only some cells in liver tissue expressed spCas9 protein.
sgRNA design for exon 8 of Atp7b
Serotype 8 AAV has been widely used for the introduction of gene editing vectors into the liver 25,26 and thus was selected to deliver sgRNA expression vectors and HR fragments in our study. To achieve the replacement of exon 8 in Atp7b, a pair of sgRNAs targeting the introns beside exon 8, sgRNA1F and sgRNA2R (Fig. 1A; Supplementary Tables S1, S3, and S4 for specific information), were devised in accordance with the design requirements of the CRISPR design website developed by Zhang Feng.
In vitro, both sgRNA1F and sgRNA2R guided spCas9 protein to efficiently cleave the target fragment into obviously detected bands at ∼500 and 900 bp, respectively (Fig. 1B), and the ImageJ gray analysis showed that the in vitro cleavage efficiencies of sgRNA1F and sgRNA2R were 85% and 67%, respectively, compared with the untargeted control.
Subsequently, we cloned sgRNA1F and sgRNA2R into the lentiviral vector lenti-CRISPRv2, which contains the spCas9 gene and puromycin gene driven by EF-1a. After the packaged lenti-CRISPRv2 lentivirus (MOI = 100) was transfected into AML12 cells, genomic material was extracted to detect the efficiency of sgRNA cleavage. The results demonstrated that both sgRNAs directed the spCas9 protein to cleave the cellular genome, and the ImageJ gray-scale analysis of the cleaved band (Fig. 1C) confirmed that the intracellular cleavage efficiencies of sgRNA1F and sgRNA2R were 34% and 63%, respectively.
In vitro double cutting and replacement of exon 8 of Atp7b in the AML12 cell line
A donor DNA fragment (1.1 kb) containing exon 8 with a modified PAM in the sgRNA1F and sgRNA2R sequences was produced by overlap PCR (Fig. 1A). The bases modification in the PAMs of sgRNAs could prevent the donor DNA fragment from being recognized and cut by spCas9. The GG bases in the PAM of sgRNA1F were replaced with CC and the CCA deletion in PAM of sgRNA2R, which can be also used as a marker for gene replacement. The probability of gene replacement can be calculated by detecting the probability of occurrence of the CC sequence or CCA deleted sequence. rAAV plasmids harboring sgRNA1F and sgRNA2R under control of the U6 promoter and the donor DNA fragment containing exon 8 were constructed (AAV-U6sg-DDFE8; Fig. 2C). To test whether the AAV vector can successfully cleave and replace exon 8 of the Atp7b gene in a cell line expressing spCas9, the hepatocyte-derived AML12 cell line was used for the following experiment.
First, the AML12 cell line was infected with the above-mentioned Lenti-TBG-spCas9-GFP lentivirus, and GFP-positive cells stably expressing spCas9 protein were sorted. Then, the constructed AAV-U6sg-DDFE8 plasmid was transiently transfected into these cells with Lipofectamine 2000. Subsequently, the cellular genome was extracted to detect the efficiency of Atp7b exon 8 replacement by PCR and DNA sequencing. The results of PCR showed that three bands were amplified in the transfected cells relative to the control cells (Fig. 5A). The largest band was ∼894 bp and included the replaced fragment. The smallest band suggested that genomic double cleavage was achieved by sgRNA1F- and sgRNA2R-directed Cas9 cleavage because it was ∼430 bp between the sgRNA1F and sgRNA2R recognition sites, and the remaining band was ∼465 bp (Fig. 5B). The band at ∼894 bp was sent for Sanger DNA sequencing, and the result is shown in Fig. 5C. Compared with the control genome sequence, the modified sequence showed a significant CC peak under the GG peak, indicating that exon 8 replacement occurred.
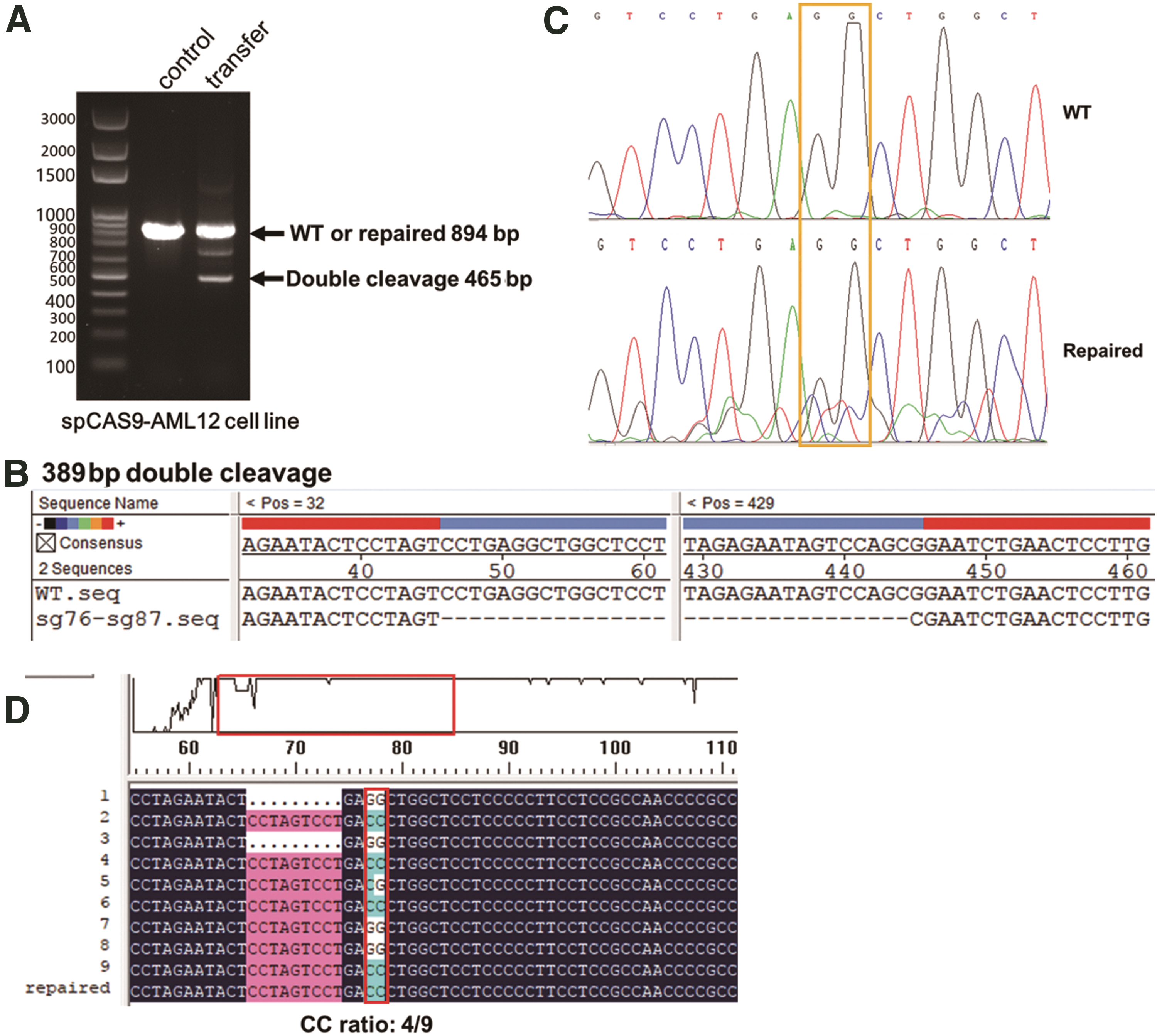
In vitro double cutting and replacement of exon 8 in Atp7b in the AML12 cell line.
To determine the replacement efficiency, we used the 894 bp band (Fig. 5A) as a template for PCR, which was performed using primers designed within 100 bp upstream and downstream of the CC tag site. Then, the PCR product was cloned into the pMD20 vector to construct a T-A clone for DNA sequencing. NGS was performed using the PCR product. The result in Fig. 5D shows that the CC ratio was 4/9, which means that the replacement efficiency was ∼44.4%. The NGS results showed that the CC replacement ratio was 48.17% in the PCR products (Table 1).
Analysis of replacement ratio on CC label by next-generation sequencing and quantitative PCR in newborn mice
NA, not analyzed; SEM, standard error of the mean; WT, wild type.
AAV-mediated Atp7b exon 8 replacement in the newborn spCas9 mice
Based on the results of the in vitro Atp7b exon 8 replacement experiments, we performed liver-specific replacement experiments in TBG-spCas9-GFP mice. We packaged the AAV-U6sg-DDFE8 plasmid into AAV8. For the delivery of packaged AAV-U6sg-DDFE8 virus, F1 mice received a temporal vein injection (1.09E + 12 VG/mL AAV, 3 μL per mouse) on the second day after birth. One week later, mouse liver tissues were isolated and digested to obtain individual cells, and GFP-positive cells were sorted by flow cytometry. The genome of GFP-positive cells was used for genetic analysis using a pair of primers spanning the donor DNA fragment, as was performed in the in vitro experiments. The PCR results showed that three bands were amplified from the genome of the five mice injected with the rAAV8 vector (AAV-U6sg-DDFE8); however, only one band was amplified from the DMEM-S0 mice (Fig. 3A), identical to the findings in the previous in vitro replacement experiments (Fig. 5A). Therefore, we used the band at ∼894 bp as a template for PCR amplification of the CC tag site and CCA deletion tag site (Supplementary Fig. S4A, B). The PCR product underwent NGS, and the results are shown in Tables 1 and 2, respectively. The CC-positive ratio of PCR products from the five rAAV8-injected mice was 21.4%, 8.41%, 25.8%, 18.02%, and 34.87%, with an average of 21.7 ± 4.36%; the CCA deletion-positive ratio of PCR products from the five rAAV8-injected mice was 21.64%, 9.62%, 39.65%, 9.22%, and 45.87%, with an average of 25.2 ± 7.57%. The result also suggests that the homologous DNA recombination (HDR) process in the method system is likely to be systematically performed, and there is no significant difference in HDR efficiency between the two ends.
Analysis of replacement ratio on CCA label by next-generation sequencing and quantitative PCR in newborn mice
Furthermore, the WT and replacement fragment ratios among all GFP-positive cells were detected via real-time qPCR by designing specific primers for WT and replaced sequences and a control for total genomic material. As shown in Fig. 3C, 80.20%, 54.56%, 78.62%, 62.96%, and 81.18% of all GFP-positive cells from different mice were WT and replaced. It is possible that the episomal AAV vector genome was extracted into the genomic sample, thereby interfering with the authenticity of the qPCR. A real-time qPCR experiment were set up to determine the content of AAV vector in each genomic sample. The result shows that the AAV genome has a very low percentage of residues in the sample, about 0.22% (Supplementary Fig. S5), and the effect on the WT and replaced ratio is extremely weak and negligible.
According to the proportion of WT and replaced sequences among all GFP-positive cells, the average exon 8 replacement ratio in GFP-positive cells was 16.34 ± 4.02% on label CC and 19.37 ± 6.50% on label CCA deletion (Tables 1 and 2; Fig. 3D). As only a small proportion of all liver cells were GFP positive, the in vivo replacement efficiency in the five rAAV8-injected mice was then converted to 2.28%, 0.86%, 2.21%, 1.40%, and 2.29% (average 1.81 ± 0.29%; Table 1) on label CC or to 2.32%, 0.99%, 3.40%, 0.71%, and 3.01% (average 2.09 ± 0.54; Table 2) on label CCA deletion in all liver genomes.
Based on the NGS results and the relative level of WT and replaced to WT reference genomes, we obtained depletion without replacement ratios of 19.80%, 45.44%, 21.38%, 37.04%, and 18.82%. Together with the depletion with replacement ratio, total depletion occurred at a rate of 36.96%, 50.02%, 41.66%, 48.39%, and 47.13% (Fig. 3D), with an average of 44.83 ± 2.42%.
AAV-mediated Atp7b exon 8 replacement in the adult spCas9 mice
When 150 μL rAAV8 vector was injected into the adult spCas9 mice, the same analysis was performed as in the treatment of newborn mice. The result in Fig. 6A and B demonstrated that in GFP-positive hepatocytes of adult mice, a significantly elevated sgRNA double-cut deletion ratio compared with neonatal mice occurs, but HDR is much lower than that of the neonatal mouse treatment group. To further quantify the exon 8 replacement rate, we still recovered the 894 bp band and performed NGS analysis of the positive rate of CC replacement tags and CCA deletion tags on PCR productions (Supplementary Fig. S6), analyzed the positive rate of HDR, and the total replacement of exon 8 in the liver of adult mice, as shown in Tables 3 and 4. The results showed that in spCas9 transgenic adult mice injected with AAV-U6sg-DDFE8, a double-cut deletion ratio of up to 99% was found in the spCas9-positive hepatocytes; however, the HDR efficiency was extremely low, and finally, only 1.10 ± 0.65% to 1.13 ± 0.66% in the spCas9-positive hepatocytes. In view of only a small proportion of all liver cells were GFP positive, the in vivo replacement efficiency was only 0.092–0.096% in all adult liver genomes.
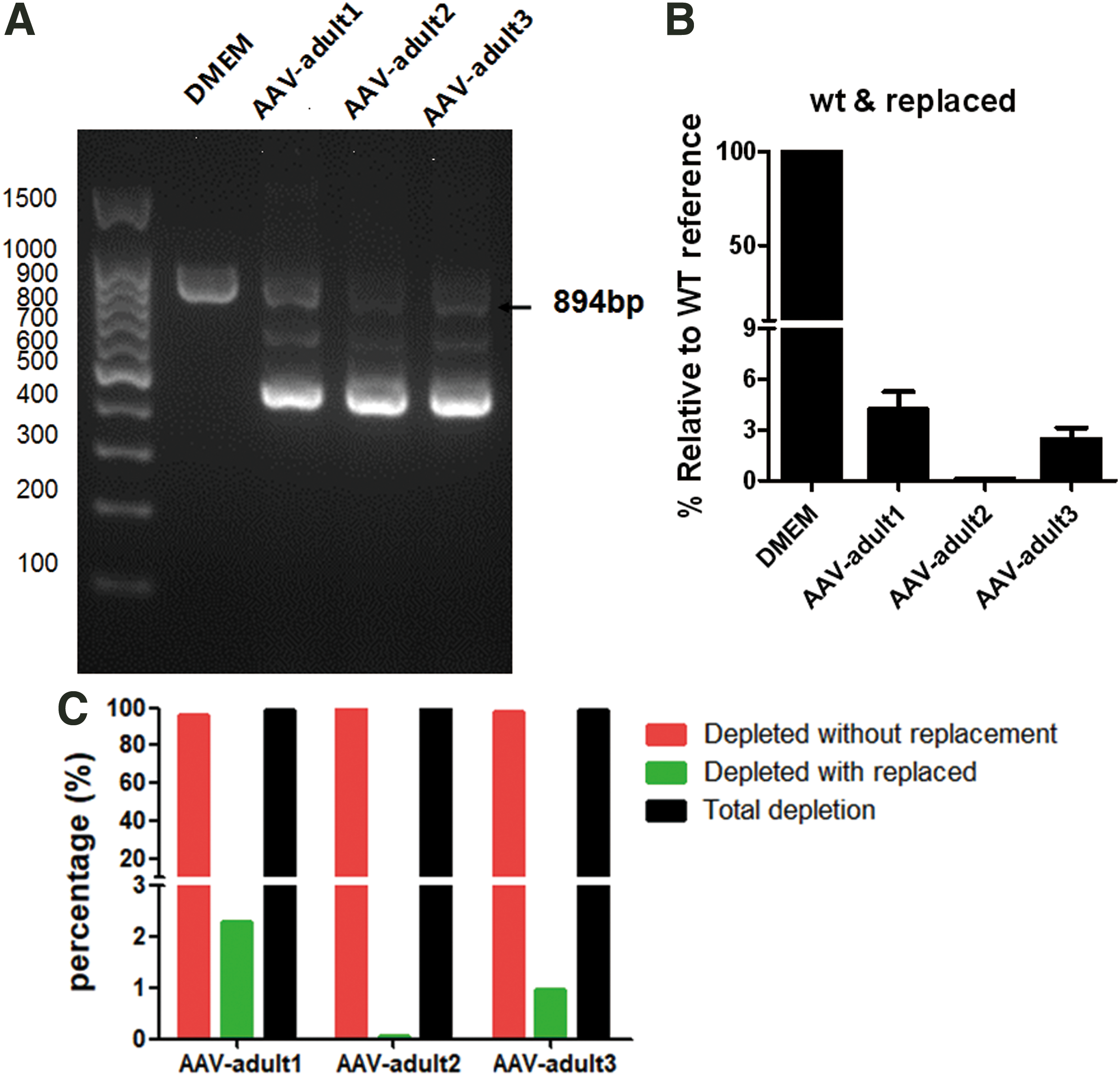
AAV-mediated Atp7b exon 8 replacement in the adult TBG-spCas9-GFP mice.
Analysis of replacement ratio on CC label by next-generation sequencing and quantitative PCR in adult mice
Analysis of replacement ratio on CCA label by next-generation sequencing and quantitative PCR in adult mice
Off-target effects of the sgRNAs in mouse hepatocytes
Off-target effects must be considered with the CRISPR/Cas9 system. To detect the off-target effects in the five newborn mice in this replacement experiment, the three potential off-target sites (POTS) with the highest scores (MIT CRISPR Design tool) were selected for further analysis (Supplementary Table S5). The sequence was amplified with primers ∼300 bp upstream and downstream of the target sequence, and Sanger sequencing was performed to detect any off-target activity. No off-target effects were detected in the mice (Supplementary Table S6).
Discussion
Mice that specifically expressed spCas9 protein in the liver were produced in this study. In the liver of newborn transgenic mice, gene cleavage and exon replacement were successfully achieved in some spCas9-positive cells using rAAV8 to deliver sgRNAs and Atp7b exon 8 replacement fragments. Approximately 16.34 ± 4.02% to 19.37 ± 6.50% of exon 8 replacement was achieved in spCas9-positive hepatocytes, and the replacement efficiency among all hepatocytes was ∼1.81 ± 0.29% to 2.09 ± 0.54%.
The traditional concept of gene therapy is to replace genes rendered nonfunctional by mutation with the correct gene. This method is commonly used in nonproliferating nerve cells 27 or visual cells to restore protein function and improve pathological conditions. 28
CRISPR/Cas9 technology has provided breakthroughs in gene repair, including the ability to correct frameshift mutations by gene fragment excision, 13 gene fragment replacement, 14 and repair of individual mutated genes by base editing. 29 Loss of gene function is generally caused by point mutations, and many genetic diseases are caused by multisite mutations or have multiple mutation hotspots. Gene fragment replacement, such as exon replacement, should be a suitable method to resolve multiple mutation hotspots in some exons. The first gene replacement using the CRISPR/Cas9 system was reported in human cells, 30 and then, AAV-mediated gene repair was reported in mice for different diseases. 14,31
In the obtained TBG-spCas9-GFP mice, only ∼10% of hepatocytes in F1 newborn mice were GFP positive, and the expression efficiency was lower than that in the transgenicAML12 cell line. The reason for this finding may be that the hTBG promoter is regulated differently in embryonic development than in the in vivo cultured AML12 cell line. The activity of the hTBG promoter is mainly regulated by HNF1 to achieve liver specificity. 32 The sequence of the core regulatory element of the rat and human TBG promoter regions is 80% homologous, but TBG production in adult rats is only 2%, equivalent to that in the human body, suggesting differential regulation of the hTBG promoter in the liver. 33 Using the Mouse ENCODE project data, we found that the expression of hTBG in mice was higher in the fetal liver during the embryonic period and was significantly decreased in the postnatal and adult liver, indicating that the promoter activity was significantly weakened. When the mouse liver was infected with a lentivirus carrying the hTBG driver unit, there was sustained low expression with no significant change in gene silencing. 34 Our immunohistochemical results show that the GFP-positive rate in F0 and F1 mice was low, and there was no significant difference between these two generations (Supplementary Fig. S3).
sgRNA1F- and sgRNA2R-induced double-strand cleavage beside exon 8 is the first step of replacement. The deletion efficiency reported in the article was 44.83% ± 2.42% in newborn mice. This deletion efficiency was determined by the cleavage efficiency of the two sgRNAs, which was comparable to the previously reported 38% depletion ratio but significantly lower than NHEJ-based deletion efficiency. 30 Moreover, the deletion efficiency is also related to the length of the targeted fragment. When the targeted fragment reaches 10 kb, the deletion efficiency in vivo is only 16–28%; when such large fragments are deleted in vivo, the efficiency is only 8.6%. 35 Therefore, the 37–50% in vivo cutting efficiency in newborn mice may benefit from the short deletion of 389 bp.
When the same rAAV8 vector was injected into the 10-week-old spCas9 mice, the sgRNA double-cut deletion ratio significantly elevated up to 99%. This may be result from high copy vectors that were injected. However, the HDR efficiency was extremely low, and finally, only 1.10% of the exon 8 replacement rate in spCas9-positive hepatocytes. Similar results have been reported in another study. 14 The specific mechanism is still unclear. Compared with data from neonatal mice, in which hepatocytes undergo extensive cell proliferation, possibly promoting HDR, the low replacement rate in adult mice may be caused by a much lower proliferation index. Therefore, age is a key factor in gene repair therapy based on Cas9 technology, but it has little effect on the gene knockout design that wants to delete a short sequence.
In humans, exon 8 of the ATP7B gene is a hotspot for multiple mutations associated with WD. This experiment attempted to replace exon 8 of Atp7b in mice. The strategy can also be used to replace exons of other genes. The vectors carrying sgRNA and HDR templates are easy to construct and deliver; however, the efficiency of delivery of spCas9 will play a decisive role in the efficacy of gene therapy. In addition, the growth rate of liver cells in young children is lower than that in neonatal mouse liver cells, and the proliferation rate of hepatocytes may affect the efficiency of exon replacement. Therefore, it is still necessary to perform gene exon replacement on animals with long growth cycles.
In conclusion, when sgRNAs and replacement fragments targeting exon 8 of Atp7b were introduced into spCas9-positive hepatocytes by AAV8, the introns beside exon 8 were cleaved successfully, and 16.34 ± 4.02% to 19.37 ± 6.50% of the analyzed copies of exon 8 were replaced by the donor sequence, and the replacement efficiency among all hepatocytes was ∼1.81 ± 0.29% to 2.09 ± 0.54%. This study is the first to achieve exon replacement via CRISPR/Cas9, which will benefit research on CRISPR/Cas9 technology for gene therapy.
Footnotes
Acknowledgments
This study was supported by grants from the National Key Research and Development Program of China (Grant No. 2016YFC1304805) and the Shanghai Natural Science Fund (Grant No. 17140900104).
Authors' Contributions
S.L., X.C., and Y.L. developed the methodology and wrote the article. L.L. conducted the experiments and performed data analysis. Q.C., F.X., G.Y., L.F., H.W., and Z.M. performed the micromanipulation experiments and participated in critical discussions and data analysis. All authors read and revised the article before submission.
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.