
Abstract
Metabolic liver diseases are attractive gene therapy targets that necessitate reconstitution of enzymatic activity in functionally complex biochemical pathways. The levels of enzyme activity required in individual hepatocytes and the proportion of the hepatic cell mass that must be gene corrected for therapeutic benefit vary in a disease-dependent manner that is difficult to predict. While empirical evaluation is inevitably required, useful insights can nevertheless be gained from knowledge of disease pathophysiology and theoretical approaches such as mathematical modeling. Urea cycle defects provide an excellent example. Building on a previously described one-compartment model of the urea cycle, we have constructed a two-compartment model that can simulate liver-targeted gene therapy interventions using the computational program Mathematica. The model predicts that therapeutically effective reconstitution of ureagenesis will correlate most strongly with the proportion of the hepatic cell mass transduced rather than the level of enzyme-encoding transgene expression achieved in individual hepatocytes. Importantly, these predictions are supported by experimental data in mice and human genotype/phenotype correlations. The most notable example of the latter is ornithine transcarbamylase deficiency (X-linked) where impairment of ureagenesis in male and female patients is closely simulated by the one- and two-compartment models, respectively. Collectively, these observations support the practical value of mathematical modeling in evaluation of the disease-specific gene transfer challenges posed by complex metabolic phenotypes.
Introduction
Preclinical gene therapy research relies heavily on the use of animal models, most commonly mice, to address the disease-specific challenges of achieving therapeutically effective and safe gene transfer.
Important ethical considerations in the use of animals for research are the principles of replacement, reduction, and refinement. 1 While the complete replacement of animals is not plausible, other adjunctive approaches have the potential to reduce and refine animal usage. These include disease modeling in ex vivo cellular and organoid systems, and theoretical strategies such as mathematical simulation. 2,3 The latter has particular merit in modeling intricate systems such as biochemical pathways, where the combined complexities of enzyme kinetics, rate-limiting reactions, substrate concentrations, and metabolite fluxes can be simulated and dissected.
Disorders of the urea cycle provide an excellent paradigm for the development of gene therapy for metabolic liver disease and are ideally suited to mathematical modeling. Many important questions of direct relevance to the challenges of liver-targeted gene transfer are potentially tractable using this approach. These include the relative therapeutic value of achieving efficient transduction in a maximal number of hepatocytes versus achieving physiological or even supraphysiological expression in a smaller number of hepatocytes. For example, might it be possible to increase the ureagenic capacity of individual hepatocytes to supraphysiological levels such that therapeutic benefit could be achieved at reduced transduction efficiencies?
In the context of the most promising gene delivery system for liver-targeted gene transfer, recombinant adeno-associated virus (AAV), 4 –6 the answers to these questions have the potential to influence vector capsid selection and whether or not it is important to optimize the vector expression cassette such that physiological or even supraphysiological levels of enzyme-encoding transgene expression are achieved. This in turn could have implications for vector safety as the risk of genotoxicity associated with vector integration events increases with the use of stronger enhancer/promoter elements that may be necessary to achieve higher levels of transgene expression. 7
With these considerations in mind, we set out to explore the use of mathematical modeling to simulate the challenge of liver-targeted gene therapy for urea cycle defects (UCDs), particularly deficiency of the proximal enzymes ornithine transcarbamylase (OTC) and argininosuccinate synthetase (ASS), both of which we have explored in preclinical mouse models. 8 –12
A one-compartment model of the urea cycle, previously developed by Kuchel et al., 13,14 formed the starting point for the work described. By simulating urea cycle enzyme deficiency, the resultant increases in plasma ammonia concentrations can be inferred from corresponding decreases in ureagenesis, although the strength of this inference declines for the more distal UCDs. An essential requirement was to develop a two-compartment model, with one-compartment representing the proportion of the hepatic cell mass successfully transduced and the other representing the remaining urea cycle-deficient population of hepatocytes.
The original model was also extended to include an output simulating the accumulation of carbamoyl phosphate as a surrogate marker for orotic aciduria, a feature of the biochemical phenotype of the more proximal UCDs lying beyond carbamoyl phosphate synthetase (CPS1) deficiency. 15 This enhanced two-compartment model allowed simulation of the potential therapeutic impact of differing gene transfer efficiencies, differing levels of enzyme-encoding transgene expression in individual hepatocytes, and the influence of residual levels of endogenous enzyme expression in patient cells.
Materials and Methods
Addition of CPS1 to the one-compartment model of the urea cycle
All simulations were performed using the computational package Mathematica (Wolfram Research) and the graphical output was further manipulated in GraphPad Prism. Kuchel et al. constructed the first one-compartment model using enzyme kinetic equations (nonlinear differential equations) to describe the rate of reactions for OTC, ASS, argininosuccinate lyase (ASL), and Arginase. 13
To extend this basic one-compartment model, the equations describing the interaction of CPS1 with the relevant urea cycle substrates and products were generated using the substrate binding and product release mechanism described by Elliot 16 (Supplementary Fig. S1). The steady-state rate equations were then generated using the “RateEquationDeriver” Package that is written in Mathematica. 17
The rate equations for CPS1, OTC, ASS, ASL, and Arginase were then combined in an array. The rate equations describing influx and efflux of related cosubstrates and products were defined by simple first-order kinetics. The pool concentrations of ATP, bicarbonate, ammonium, and aspartate were assumed to be held constant by external processes/reactions. Differential equations were then formulated to describe this extended one-compartment model and solved using the numerical algorithm for stiff differential equations in “NDSolve” in Mathematica.
To simulate OTC deficiency using the one-compartment model, the enzyme concentration (eotc1) was reduced from 100% to 1% of the normal value. Time courses of urea and carbamoyl phosphate synthesis were graphed over 600 s. Analogous simulations were performed for ASL deficiency, ASS deficiency, CPS1 deficiency, and Arginase deficiency.
Formulating a two-compartment model to simulate gene therapy
The one-compartment model was simply duplicated to obtain two separate sets of equations to represent two discrete compartments (Fig. 1). Exchange between the cells in each compartment was added by including rate equations for the transport of metabolites from one-compartment to another. Designated rate constants were set to simulate rapid and free exchange (nonconcentrative) between the two compartments.

Schematic showing how the two-compartment model used for gene therapy simulations was generated. A normal compartment with 100% OTC activity (blue) and an OTC-deficient compartment (red). Lack of OTC causes a deficiency of urea cycle intermediates in the deficient cells. Arrows represent exchange of solutes between the two cellular compartments. OTC, ornithine transcarbamylase.
Once the system of equations was defined, it was solved using the “NDSolve” function in Mathematica, as noted above. Changes in substrate and metabolite concentrations were thus expressed for the two different compartments. The volume fraction of each compartment was specified to gain insights into cross-correction of metabolic outputs by the overall cell population.
To simulate gene therapy for OTC deficiency, cell population 1 was designated as the native cells, which expressed no OTC, while cell population 2 expressed OTC levels at the normal value. The amount of urea produced after 600 s was graphed against the proportion of gene-corrected cells increasing from 0% to 100%. Analogous simulations were also performed for the remaining urea cycle enzyme defects.
The Mathematica program is given in the Supplementary Data.
Results
Simulation of UCDs and effects of gene therapy on ureagenesis
For each urea cycle enzyme deficiency, simulations were performed sequentially using the one- and two-compartment models (Fig. 1). The one-compartment model was used to simulate the untreated state, while the two-compartment model was used to simulate the effects of genetic correction of variable proportions of the hepatic cell mass on ureagenesis. In the latter, the two compartments represent populations of untreated and treated hepatocytes.
Simulation of OTC deficiency, the most common UCD, was undertaken first with added interest conferred by the fact that carrier females provide a naturally occurring model of the two-compartment state as a consequence of random X chromosome inactivation. 18 As an initial step, the one-compartment model was used to simulate the ureagenic capacity of the male liver (all hepatocytes affected) for a series of residual OTC activities ranging from 1% to 10%, and the data compared with wild-type activity (100%).
The predicted rate of ureagenesis, an adequate surrogate of ammonia clearance for this proximal UCD, did not decrease substantially until the residual OTC activity fell below 3% (Fig. 2A), while perturbation of the rates of production of urea cycle amino acid intermediates (ornithine, citrulline, argininosuccinate, and arginine) did not become evident until OTC activity was reduced to 1% (Fig. 2B).
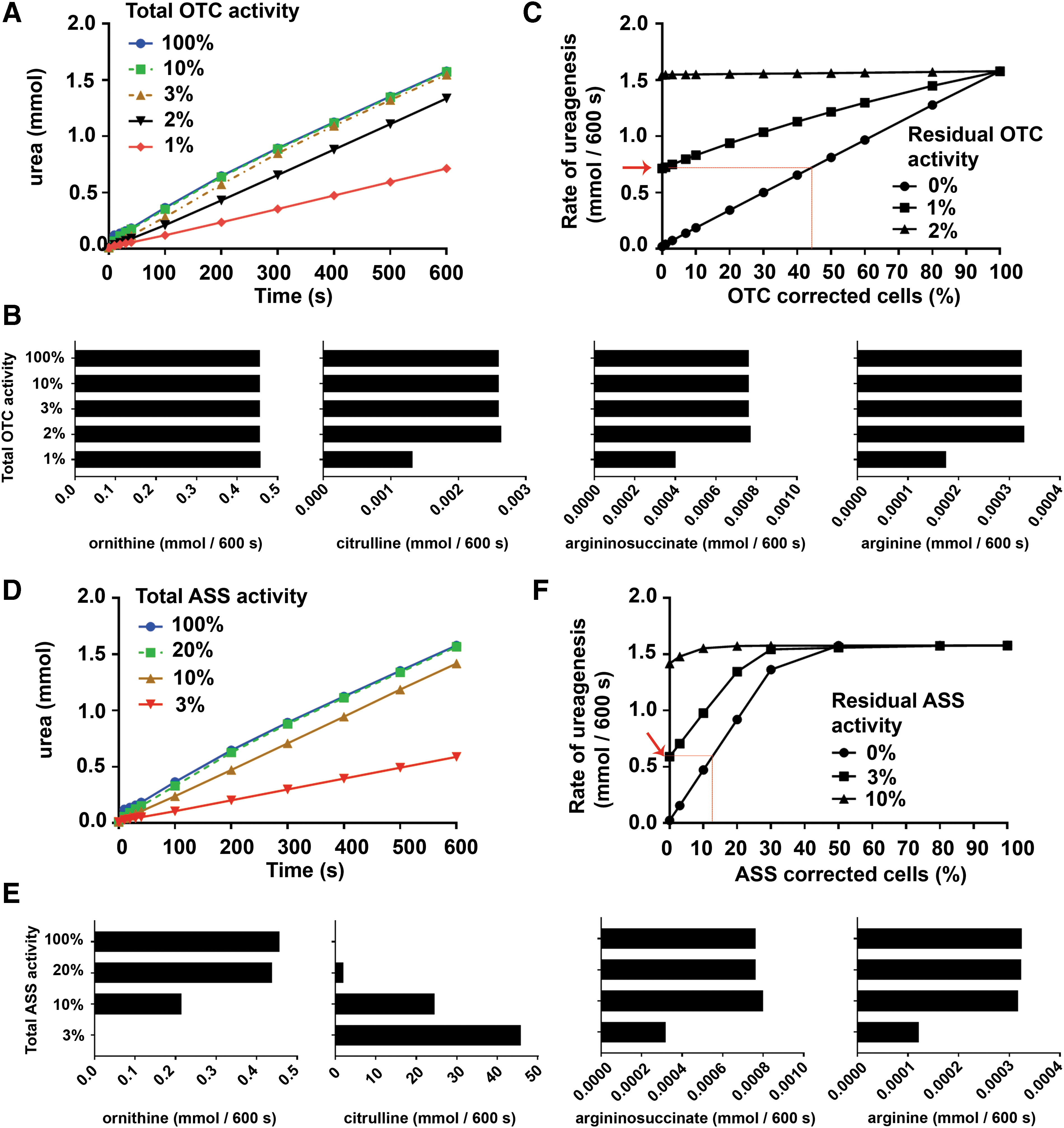
Simulation of OTC deficiency and citrullinemia and the benefit obtained from subsequent gene therapy.
Next, gene therapy was simulated by using the two-compartment model, which predicts the rate of ureagenesis in a mixed population of enzyme-deficient and gene-corrected hepatocytes. The ratio of deficient to corrected hepatocytes was varied, for cells with 0% to 3% residual OTC activity, to predict the rate of ureagenesis as the proportion of treated cells was increased. As expected, higher percentages of gene-corrected cells corresponded with an increase in the rate of ureagenesis (Fig. 2C).
Notably, however, correction of more than 40% of hepatocytes with no residual endogenous activity was required to achieve a rate of urea production that was equivalent to untreated hepatocytes each with 1% OTC activity (Fig. 2A, C). Interestingly, simulation of overexpression of OTC activity up to 10-fold supraphysiological levels in a given population of treated hepatocytes did not further increase the rate of ureagenesis (data not shown).
Similarly, ASS deficiency (citrullinemia) was initially simulated using the one-compartment model for a series of residual ASS enzymatic activities ranging from 3% to 20%, and the data compared with wild-type activity (100%) (Fig. 2D). Urea production was more sensitive to changes in ASS enzymatic activity compared with OTC, as ureagenesis declined after ASS was decreased below 20%. The model predicted that citrulline accumulation was sensitive to reduced ASS activity, with a marked progressive increase in citrulline levels between 20% and 3% residual activity.
Reduction in the rate of production of ornithine was observed from 20% residual ASS activity, while reduction in the rates of argininosuccinate and arginine production was only observed at 3% residual activity (Fig. 2E). In the gene therapy scenario, simulated by using the two-compartment model, correction of more than 40% of hepatocytes in an ASS-null background was required to restore the rate of ureagenesis to normal (Fig. 2D, F).
In addition, correction of more than 10% of cells in an ASS-null hepatocyte population resulted in urea production at equivalent levels to untreated hepatocytes each with 3% residual ASS activity. Since ASS is the rate-limiting enzyme in the urea cycle, overexpression of ASS at 200% physiological levels was simulated in concert with correction of increasing proportions of OTC-null hepatocytes by using the two-compartment model. No increase in ureagenesis was predicted above that achieved by correction of OTC-deficient cells alone (data not shown).
Further simulations were performed for deficiency of the remaining urea cycle enzymes, ASL, Arginase, and CPS1. Simulation of ASL deficiency by using the one-compartment model predicted a similar phenotype to OTC deficiency, with urea production decreasing when ASL activity was reduced below 3% (Fig. 3A). The urea cycle intermediates ornithine and argininosuccinate were most sensitive to changes in ASL activity, with the rates of production decreasing and increasing, respectively, as residual ASL activity was reduced below 10% (Fig. 3B).
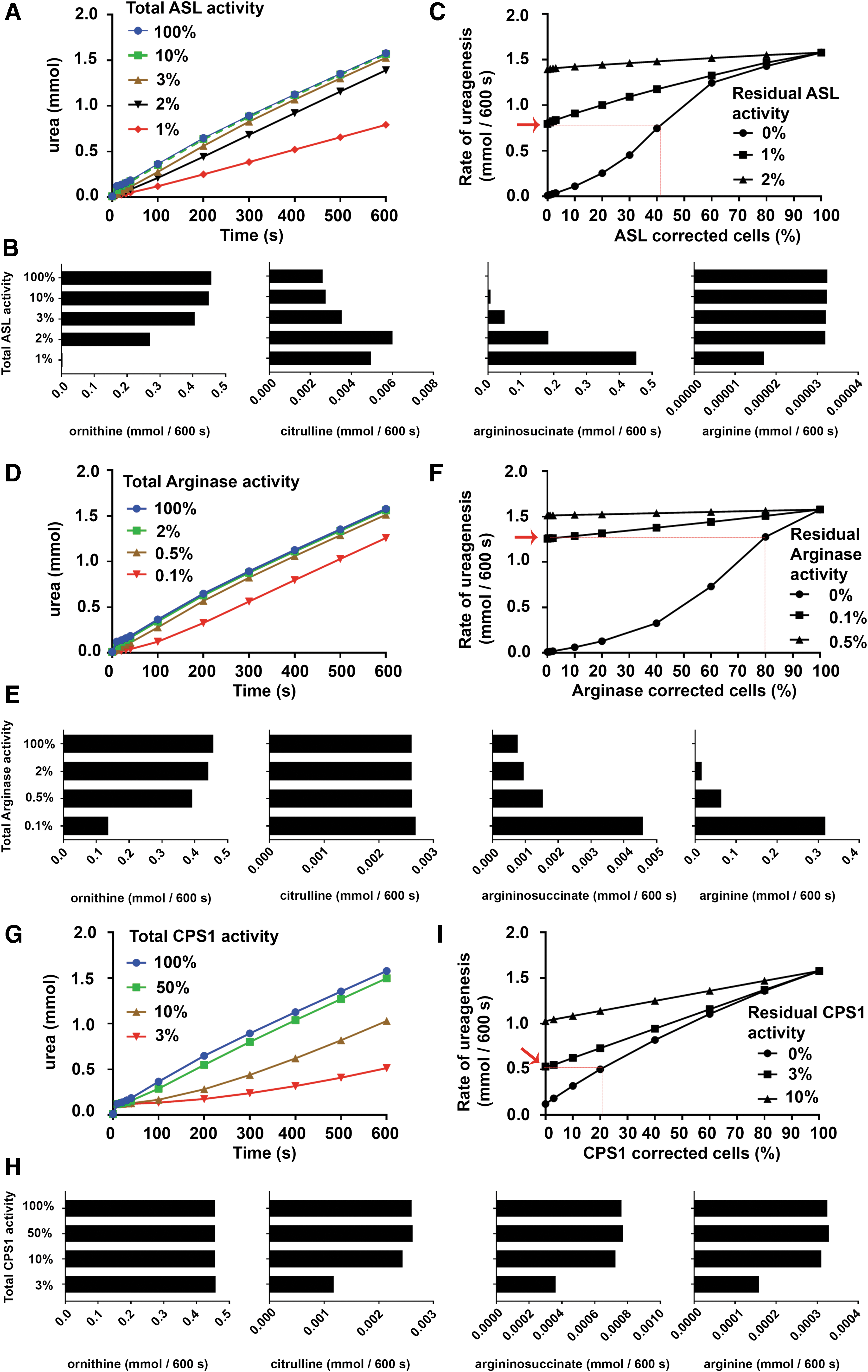
Simulation of enzyme deficiency for ASL, Arginase, and CPS1 using the one-compartment model, and the subsequent gene therapy using the two-compartment model. Change in urea synthesis is shown using the one-compartment model as enzyme activity in all cells was reduced for
The two-compartment model predicted that correction of ∼40% of hepatocytes would be required to achieve a rate of ureagenesis equivalent to that obtained from untreated hepatocytes each with 1% residual ASL activity (Fig. 3A, C).
Consistent with the clinical phenotype, simulation of Arginase deficiency by using the one-compartment model predicted preservation of ureagenic capacity even when residual Arginase activity was reduced to as little as 0.1% (Fig. 3D). This was associated with a marked increase in the rate of arginine production, and a more modest increase and decrease in the rate of argininosuccinate and ornithine production, respectively (Fig. 3E). The two-compartment model predicted that correction of ∼75% of hepatocytes would be required to achieve a rate of ureagenesis equivalent to that obtained from untreated hepatocytes each with 0.1% residual Arginase activity (Fig. 3D, F).
Finally, simulation of CPS1 deficiency by using the one-compartment model predicted a progressive decline in the rate of ureagenesis when liver-wide CPS1 activity was reduced below 50% (Fig. 3G). As CPS1 activity fell below 10%, there was no change in the rate of ornithine production, although there was a decrease in the rate of citrulline, argininosuccinate, and arginine production (Fig. 3H). The two-compartment model predicted that correction of ∼20% of hepatocytes would be required to achieve a rate of ureagenesis equivalent to that obtained from untreated hepatocytes each with 3% residual CPS1 activity (Fig. 3G, I).
In addition to allowing simulation of CPS1 deficiency and treatment by gene therapy, extension of the original mathematical model of the urea cycle to include CPS1 enzyme activity enabled simulation of the impact of individual UCDs on carbamoyl phosphate production, which serves as a surrogate measure for orotic aciduria. As expected, the predicted rate of carbamoyl phosphate production was sensitive to progressive reduction in OTC enzymatic activity, with log-fold increases observed (Fig. 4A).
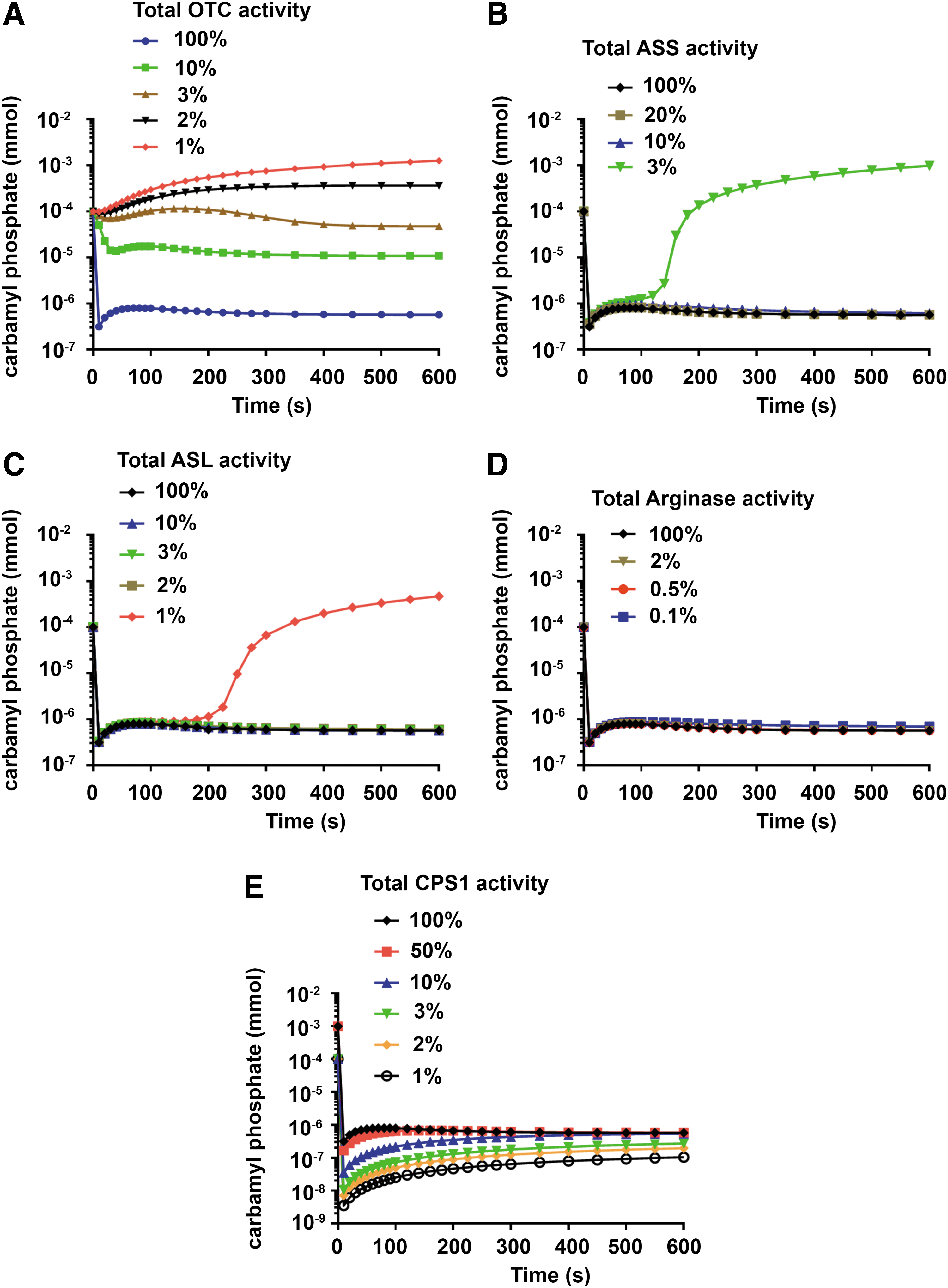
Steady-state levels of carbamoyl phosphate in urea cycle defects. Levels of carbamoyl phosphate were predicted as the amount of enzyme in all cells was reduced in
In contrast, the rate of carbamoyl phosphate production in ASS and ASL deficiency was not predicted to rise until enzyme activities were reduced to 3% and 1%, respectively (Fig. 4B, C). In Arginase deficiency, no change in the rate of carbamoyl phosphate production was predicted, even when enzyme activity was reduced to 1% of physiological levels (Fig. 4D). Finally, in CPS1 deficiency, the rate of carbamoyl phosphate production was sensitive to progressive reduction in enzyme activity with a marked decline becoming evident at <10% of residual activity (Fig. 4E).
Discussion
The human liver is an immensely promising target for gene therapy interventions. 4,10,19 A prerequisite for success, however, is the selection of disease phenotypes where the threshold for therapeutic benefit is within reach of contemporary gene transfer technology/genome editing. This threshold varies in a disease-dependent manner and is a function of both the number of target cells that must be gene modified and the level of transgene expression required in individual cells. This information is often difficult to deduce based on theoretical considerations and is most commonly addressed empirically in preclinical studies using animal models.
In the present study, we sought to explore the value of mathematical modeling for predicting the gene transfer/expression thresholds required for gene therapy to be of therapeutic benefit in UCDs. The study was facilitated by reconfiguring a previously published one-compartment model of the urea cycle, developed by Kuchel et al., 13,14 as a two-compartment model representing treated and untreated populations of urea cycle-deficient hepatocytes. We also extended the model to encompass CPS1 deficiency, with the added utility of being able to use carbamoyl phosphate accumulation as a surrogate for orotic aciduria.
The metabolic disturbances in ureagenesis, urea cycle intermediate, and carbamoyl phosphate concentrations associated with each of the five urea cycle enzyme deficiencies, simulated using the modified one-compartment model, were consistent with clinically recognized patterns, thereby supporting the general veracity of the model. Notably, the model predicts that disturbances of citrulline, argininosuccinate, and arginine are sensitive biomarkers of the underlying enzymatic defects in citrullinemia, argininosuccinic aciduria, and Arginase deficiency, respectively.
For the four proximal enzymes, CPS1, OTC, ASS, and ASL, reduction in ureagenic capacity did not become notable until simulated enzyme activities were reduced to 10%, 2%, 10%, and 2%, respectively, while reduction in Arginase down to 1% residual activity had no effect on ureagenic capacity. Using impairment of ureagenesis as a surrogate for hyperammonemia, these predictions align well with observed clinical patterns.
Notably, patients with deficiency of one of the four proximal enzymes tend to present with episodic hyperammonemia beyond the neonatal period until residual enzyme activities fall well below 10% of normal physiological values, 20 –29 while even the most severely affected patients with Arginase deficiency tend to present late and with hyperammonemia as a less prominent feature of the disease phenotype. Similarly, using rates of carbamoyl phosphate production as a surrogate for orotic aciduria, the model accurately predicts that orotic aciduria has the greatest sensitivity in the diagnosis of OTC deficiency and progressively lower sensitivity for detection of deficiencies of the more distal enzymes, ASS, ASL, and Arginase.
The model also accurately predicted disease-specific disturbances in urea cycle intermediates, most notably the sensitivity of citrulline levels to reduction in ASS activity, argininosuccinate levels to reduction in ASL activity, and arginine levels to reduction in Arginase activity, the three disturbed analytes from which deficiency of each of these enzymes earn their names, citrullinemia, argininosuccinic aciduria, and argininemia respectively. 30 –32
Simulation of liver-targeted gene therapy using the two-compartment model predicted that the number of hepatocytes undergoing genetic repair is the major determinant of therapeutic efficacy as measured by reconstitution of liver-wide ureagenic activity, rather than the absolute level of expression of the deficient urea cycle enzyme achieved in individual hepatocytes. This prediction was consistent for each of the five UCDs simulated and aligns qualitatively with published preclinical data generated in mouse models of OTC and ASS deficiency, 9,10,12 and also with clinical observations made in symptomatic OTC-deficient females, whose livers provide a natural model of the two-compartment state. 18,33
In a later context, the model predicts that even in the absence of skewing of X-inactivation (i.e., 50% mosaicism), the rate of ureagenesis is significantly impaired and that only modest skewing toward preferential inactivation of the X-chromosome carrying the healthy allele pushes ureagenesis down to critical levels known to be associated with hyperammonemia in males (one-compartment models Fig. 2A).
It follows that similarly modest levels of gene transfer to the population of hepatocytes carrying the inactivated healthy allele would be predicted to have a clinical benefit. From a vector development perspective, the implications for UCD gene therapy are significant in that they provide a strong rationale for focusing on achieving maximally efficient hepatocyte transduction rather than physiological or even supraphysiological transgene expression in individual hepatocytes. Indeed, the model predicts that markedly subphysiological levels of transgene expression would be therapeutic provided an adequate number of hepatocytes are transduced.
In the case of AAV vectors, which are at present the leading system for liver-targeted gene transfer, this translates to a research focus on the identification of capsids with enhanced tropism in primary human hepatocytes 34,35 rather than optimization of expression cassettes to achieve higher levels of transgene expression. This in turn could have important safety implications as the use of less powerful enhancer-promoter elements in vectors designed for UCD gene therapy would reduce the probability of insertional mutagenesis through enhancer-mediated gene dysregulation. 7,36
The model also predicted that among the five urea cycle enzyme deficiencies examined, there are likely to be differing gene transfer thresholds for therapeutic efficiency, and that low residual levels of enzymatic activity are likely to markedly reduce the gene transfer threshold required for clinical benefit. Modeling of gene therapy for deficiency of ASS, the rate-limiting enzyme in the urea cycle, was particularly interesting with the model predicting this UCD to have the lowest gene transfer threshold for therapeutic efficacy.
Conversely, Arginase deficiency was predicted to have the highest gene transfer threshold required for normalization of ureagenesis. It should be noted, however, that for this distal enzyme, reduced rates of ureagenesis would not be expected to provide a useful surrogate measure for hyperammonemia. Further studies in mice and ultimately human clinical trials will be required to address the strength of these predictions.
Predictions of the two-compartment model can also be extended to hepatocyte transplantation, an approach that is being explored as bridging therapy for pediatric patients awaiting liver transplantation. 37,38 A major limitation of the approach is the relatively small number of donor hepatocytes that can be safely infused, in the order of 1% of the hepatic mass, without the risk of portal hypertension and systemic embolization. 39
Based on the two-compartment model, in this instance represented by urea cycle-competent donor hepatocytes and urea cycle-deficient hepatocyte in the host liver, infusion of an equivalent of only 1% of donor hepatocytes would be predicted to exert a negligible effect on host ureagenic capacity and deliver minimal clinical benefit. Again, this is consistent with clinical experience in hepatocyte transplantation where multiple independent infusions are required to achieve modest and frequently transient clinical effects. 39 –41
While the one- and two-compartment models have clear predictive utility, they do not fully recapitulate the complexity of urea cycle function, and have the potential for further refinement. The present model does not directly predict ammonia levels, which are at present inferred by the effects of urea cycle enzyme deficiency on the rates of ureagenesis. This inference becomes progressively less robust for the more distal urea cycle enzymes, but nevertheless remains useful. The same limitation prevents modeling of the effects of increased ammonia load on steady-state ammonia levels as would commonly occur under conditions of catabolic stress or protein loading.
Other complexities that are not captured include metabolic zonation of urea cycle enzyme activity across the hepatic lobule and the interplay between hepatic and extrahepatic urea cycle activity in controlling urea cycle intermediate levels such as the amino acids arginine and citrulline. Both introduce minor caveats when considering gene therapy predictions. In relation to metabolic zonation, the model would therefore fail to capture the therapeutic consequence of differential transduction across the hepatic lobule.
Similarly, in relation to systemic citrulline/arginine levels, the model would not predict the ongoing need for urea cycle intermediate supplementation even after successful liver-targeted gene therapy. These subtleties do not substantially detract from the utility of the model, but do serve to emphasize that the success of gene therapy interventions requires detailed knowledge of the pathophysiology of target disease and of the gene transfer technology being used.
In summary, the one- and two-compartment models of the urea cycle described make predictions that align well with metabolic disturbance observed in patients with UCDs and also the effect of liver-targeted gene therapy in preclinical murine models. This outcome, combined with evidence from an earlier study exploring the use of mathematical modeling to inform strategies for genetic intervention in sickle cell disease, 3 supports the view that this approach can be a useful adjunct to empirical studies in the gene therapy field. Such modeling has the potential to deepen understanding of the disease-specific challenges of gene therapy, enhance preclinical experimental design, and optimize the use of preclinical animal models of human disease.
Footnotes
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council (NHMRC) project grants 423400 and 1008021 to IEA; and an Australian Research Council Discovery Project Grant DP140102596 to PWK. CYK was the recipient of a Children's Medical Research Institute postgraduate scholarship.
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Data
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.