
Abstract
Exogenous delivery of messenger RNA (mRNA) is emerging as a new class of medicine with broad applicability including the potential to treat rare monogenic disorders. Recent advances in mRNA technology, including modifications to the mRNA itself along with improvements to the delivery vehicle, have transformed the utility of mRNA as a potential therapy to restore or replace different types of therapeutic proteins. Preclinical proof-of-concept has been demonstrated for mRNA therapy for three different rare metabolic disorders: methylmalonic acidemia, acute intermittent porphyria, and Fabry disease. Herein, we review those preclinical efficacy and safety studies in multiple animal models. For all three disorders, mRNA therapy restored functional protein to therapeutically relevant levels in target organs, led to sustained and reproducible pharmacology following each dose administration of mRNA, and was well tolerated as supported by liver function tests evaluated in animal models including nonhuman primates. These data provide compelling support for the clinical development of mRNA therapy as a treatment for various rare metabolic disorders.
Introduction
A new era of genomic medicine is on the horizon with the potential to treat a myriad of disorders including rare monogenic diseases with nucleic acid therapies. Messenger RNA (mRNA) therapy is emerging as a new class of medicine that has broad applicability across a range of therapeutic areas, such as oncology/immune-oncology, 1,2 cardiovascular diseases, 3 infectious diseases, 4 and more recently, rare metabolic diseases as supported by preclinical proof-of-concept studies in animal models. Multiple clinical trials are ongoing to evaluate mRNA-based vaccines and cancer immunotherapies, 4 and first-in-human clinical trials of mRNA therapy are forthcoming in various rare metabolic disorders with high unmet medical needs.
As a potential therapy for various monogenic disorders, exogenously delivered mRNA is attractive due to its ability to encode any type of therapeutic protein, 5 including cytosolic, intramitochondrial, transmembrane, and secreted proteins (Fig. 1). By using the cell's endogenous translational machinery, mRNA allows for the natural production of such therapeutic proteins with the proper post-translational modification. Once the mRNA-encoded therapeutic protein is produced, its subcellular localization is determined by signal peptides, which can direct the protein to targeted compartments within the cell. Alternatively, the mRNA-encoded therapeutic protein can be secreted to have an effect on distal organs. Thus, mRNA could be administered to restore deficient or defective intracellular proteins or to use an organ, such as the liver, as a protein-production depot to produce secreted therapeutic proteins. 6,7
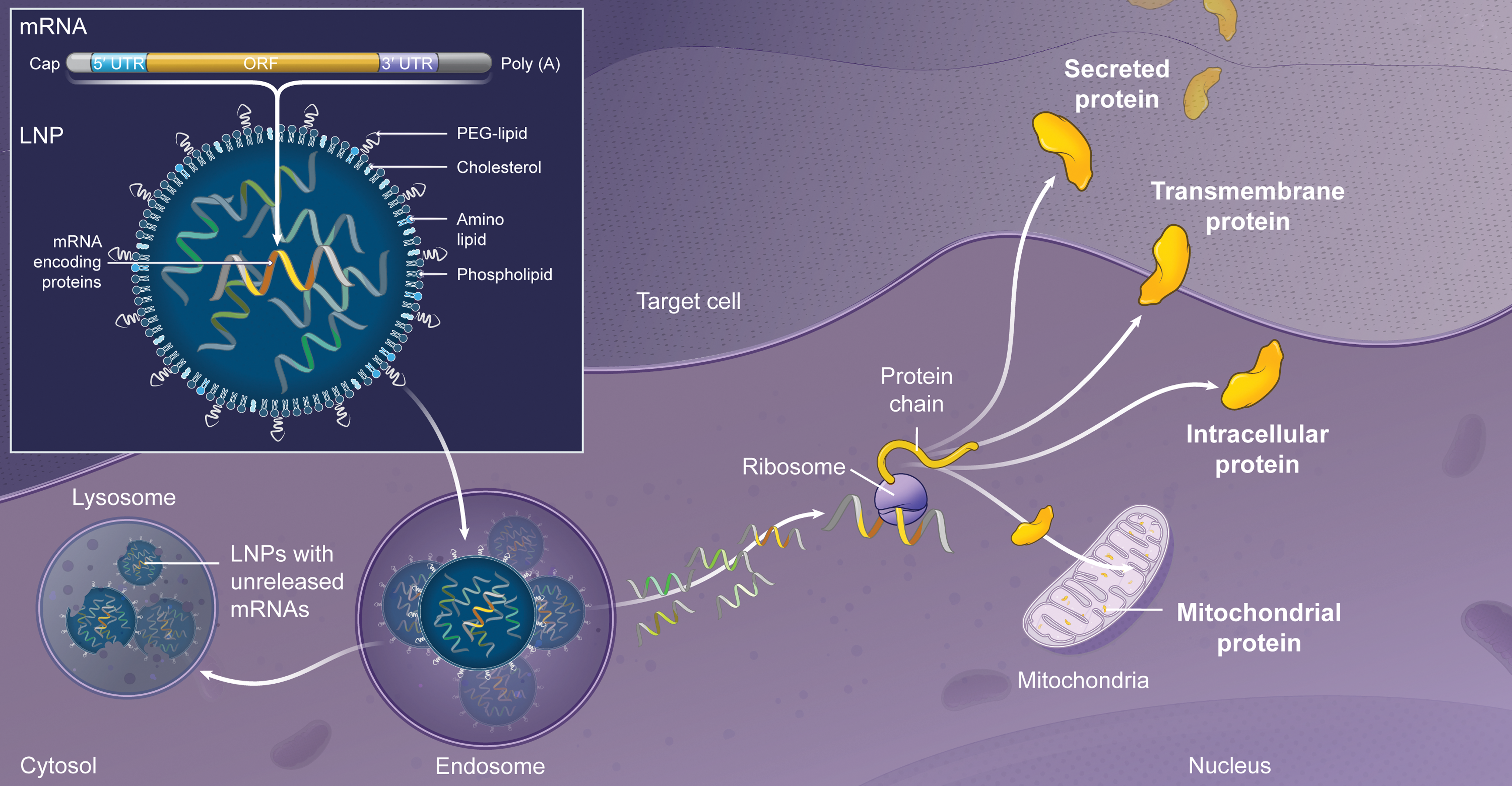
Overview of mRNA encapsulated in LNPs. mRNA structure includes the classical elements required for ribosome translation: cap, 5′ UTRs, ORF, 3′ UTRs, and poly(A) tail. Encapsulated in LNPs, mRNA is delivered to targeted cells via endosome escape and translated into protein following endosomal escape. Proteins can be translocated to the designated organelles or secreted based on their natural signal peptides. LNPs, lipid nanoparticles; mRNA, messenger RNA; ORF, open reading frame; UTRs, untranslated regions.
To realize the potential of mRNA therapy for monogenic disorders, mRNA technology has had to overcome a series of hurdles. Historically unstable and easily degradable, significant efforts have been made to improve both the stability and translation of mRNA with modifications to the 5′ cap, 5′ and 3′ untranslated regions, and poly(A) tail. 8,9 Codon optimization of the open reading frame 10 can design an mRNA with high affinity to engage ribosomes and improve translation. Furthermore, chemical modification of the nucleoside uridine 11,12 (e.g., pseudouridine) attenuates the indiscriminate recognition of mRNA by pathogen-associated molecular receptors such as toll-like receptors, minimizing the inflammatory response. A number of pattern recognition receptors including toll-like receptors and RNA sensors RIG-I (retinoic-acid inducible gene I) recognize mRNAs and subsequently trigger immune responses and/or inactivation of the mRNAs. By modifying the chemical structure of uridine to a naturally occurring modified nucleoside such as pseudouridine, 2-thiouridine, and 5-methoxyuridine, the innate immune response toward the therapeutic mRNA is dramatically attenuated without compromising mRNA translation via the ribosomal machinery. 4,13
Exogenous mRNA is highly susceptible to rapid degradation from extracellular ribonucleases, and thus, the success of mRNA therapy is largely dependent on the availability of a safe and efficient delivery vehicle. mRNA formulated in lipid nanoparticles (LNPs) provides a necessary physical protective barrier to deliver mRNA into target cells. The cellular internalization mechanism of certain classes of LNPs begins with opsonization of LNPs by apolipoprotein E (ApoE) followed by low-density lipoprotein receptor-mediated endocytosis. 14 The mRNA then escapes from the endosome/lysosome compartments into the cytosol of the targeted cell and is translated into functional protein 14,15 (Fig. 1). Advances in LNP as a delivery vehicle have focused on identifying LNPs with improved endosomal escape, biodegradability, and safety and tolerability profiles in animal models including nonhuman primates (NHPs). 16 Additionally, as mRNA expresses protein transiently, the ability to repeatedly dose LNP-encapsulated mRNA with sustained and consistent pharmacology is required.
Gene therapy and gene editing are immensely attractive due to their “one and done” approach, that is, administer a single dose of treatment that will be effective for a lifetime or several years. Similar to mRNA, gene therapy and gene editing can be used to produce or edit any type of protein. Multiple early- and late-stage clinical trials are already underway for various rare monogenic disorders, potentially yielding a wave of additional gene therapy/editing drug approvals following the success of voretigene neparvovec (Luxturna™). The “one shot on goal” gene therapy/editing approach, however, may have potential risks such as drug-induced safety concerns that cannot be “turned off” by removal of drug (as can be performed for other nonpermanent drug modalities). Additionally, if the single administered dose was not efficacious or if the treatment effect wanes over time (e.g., due to dilution of gene of the transgene in cells like hepatocytes), it is difficult to administer subsequent effective doses due to the development of neutralizing antibodies against the delivery vector. For these reasons, novel gene therapy combined with immunotherapy approaches to enable repeat dose administration are under development. Pre-existing antibodies to vectors present additional challenges to widespread implementation of gene therapy and gene editing.
Contrary to gene therapy and gene editing, mRNA does not require nuclear localization and therefore has minimal to no risk of insertional mutagenesis. However, as the half-life of exogenous mRNA is relatively short, and the mRNA-encoded protein is transiently expressed, a key disadvantage of mRNA therapy is the requirement to chronically and repeatedly dose mRNA with frequent dosing intervals (e.g., every few weeks). However, the transient nature of mRNA therapy can also be viewed as an advantage. Due to the nonpermanent nature of mRNA therapy, one has the ability to titrate the dose within an individual to identify an efficacious dose level and the ability to stop dosing in the event of an unexpected safety concern. Additional limitations of mRNA therapy include the liver being the main target organ following systemic administration of LNP-encapsulated mRNA, restricting the spectrum of the treatable diseases, and the route of administration for liver targets (i.e., intravenous [IV] infusion). Additional research is ongoing to improve mRNA stability and translation, improve LNP delivery to extrahepatic tissues, and to enable alternative routes of administration (e.g., subcutaneous injections). While further studies are needed to demonstrate the translatability into the clinic, mRNA therapy seems poised to be part of the next generation of promising therapeutics for the treatment of monogenic disorders. An overview of current preclinical and clinical stage mRNA programs for rare monogenic disorders is shown in Table 1. Additional mRNA development programs, both preclinical and clinical stages, are ongoing in other therapeutic areas including oncology, hematology, infectious disease, and cardiovascular disease.
List of preclinical and clinical stage messenger RNA therapy programs for rare monogenic disorders
This list excludes preclinical and clinical stage mRNA therapy programs for therapeutic areas other than rare monogenic disorders.
α-Gal A, alpha-galactosidase A; CFTR, cystic fibrosis transmembrane conductance regulator; G6PC, glucose-6-phosphatase; GDE, glycogen debranching enzyme; IND, investigational new drug application; IV, intravenous; mRNA, messenger RNA; MUT, methylmalonyl-coenzyme A mutase; OTC, ornithine transcarbamylase; PAH, phenylalanine hydroxylase; PCCA, propionyl coenzyme A carboxylase alpha subunit; PCCB, propionyl coenzyme A carboxylase beta subunit.
mRNA as a Potential Therapeutic for Monogenic Disorders
Three different peer-reviewed publications from our laboratory and in collaboration with different academic institutions evaluated systemic mRNA therapy for three different monogenic disorders in relevant animal models. The mechanism of action of mRNA therapy for these monogenic disorders was to restore the deficient or defective enzyme/protein with mRNA-encoded functional protein in target organs. Using modified mRNA (uridines modified to 1-pseudouridine or 5-methoxyuridine) and novel LNPs, preclinical proof-of-concept for mRNA therapy was demonstrated for methylmalonic acidemia 17 (MMA), acute intermittent porphyria 18 (AIP), and Fabry 19 disease.
Methylmalonic Acidemia
Isolated MMA is an ultra-rare, devastating organic acidemia with no approved therapies and significant morbidity and mortality. 20,21 MMA comprises a group of genetically distinct subtypes characterized by impaired metabolism of propionate derived from certain proteins and fats. 22 The disorder is autosomal recessive and most frequently caused by a deficiency of methylmalonyl-coenzyme A (CoA) mutase (MUT), a vitamin B12-dependent mitochondrial enzyme that catalyzes the isomerization of methylmalonyl-CoA to the Krebs cycle intermediate, succinyl-CoA. 20 MMA due to MUT deficiency (MIM: 251000) can be further classified into defects without (mut0) or with residual (mut−) enzyme activity, correlating with severe and milder disease courses, respectively. 23 The disorder is biochemically characterized by marked elevations of methylmalonic acid in body fluids and tissues.
Metabolic instability is the hallmark of the disorder, as manifested by life-threatening acute metabolic decompensations that tend to occur more frequently within the first few years of life. 20,24 Mortality is significant and typically occurs during severe metabolic decompensations. Longer term sequelae include severe neurological complications and chronic renal failure. 23 Current management of the disorder is limited to strict dietary restrictions, carnitine supplementation, antibiotics, and other supportive measures. 22,25 Elective liver transplantation (LT) is an approach to increase enzyme activity for severely affected individuals. Patients with renal insufficiency can also consider combined liver–kidney transplantation (LKT). LT and LKT have led to significant reductions in circulating methylmalonic acid concentrations and near elimination of metabolic decompensations. 26 –28 The effectiveness of LT demonstrates the importance of the liver as a major metabolic organ for this disorder.
Systemic mRNA therapy could be envisioned as an alternative treatment option to restore functional MUT enzyme in the liver. In collaboration with Dr. Charles Venditti and his laboratory at the National Institutes of Health (NIH), we evaluated mRNA encoding for human wild-type MUT in human fibroblasts obtained from MMA mut0 patients and two different murine models of MMA representing the spectrum of MUT deficiency (mut0 and mut−). In vitro studies evaluating MUT mRNA 16 showed that the mRNA-encoded protein was enzymatically active and had the correct subcellular localization in mitochondria in mut0 patient fibroblasts. Similarly, an in vivo study in wild-type mice demonstrated production of therapeutically relevant concentrations of MUT enzyme as early as 2 h, which peaked at 16 h, and remained detectable 7 days after a single IV bolus administration of MUT mRNA formulated in LNPs (0.5 mg/kg).
A rapid onset of pharmacology was observed in both murine models of MMA, with a lowering of plasma methylmalonic acid occurring as early as 6 h following a single IV bolus administration of human MUT (hMUT) mRNA encapsulated in LNPs (0.2–0.5 mg/kg) (Fig. 2A). In a single-dose study in mut0 mice, a dose-dependent reduction in plasma methylmalonic acid concentrations was observed, with a rebound to pretreatment concentrations occurring 2 weeks after a single IV injection (Fig. 2B).

MUT mRNA ameliorates biochemical abnormalities and improves survival in a severe mouse model of MMA. Adapted from An et al.
17
Data are shown as mean ± SD.
When fed a regular chow diet, MMA mut0 mice display significant mortality, growth retardation, and significant metabolic disturbances similar to severe affected mut0 patients who survive the neonatal period. Weekly IV bolus administration of MUT mRNA (0.2 mg/kg) rescued these mut0 mice in a 6-week study, in contrast to control mice that all perished with the exception of a single control mouse (Fig. 2C). Body weight gain was ∼40% higher than the sole surviving control mouse, and plasma methylmalonic acid levels were reduced 60–90% from pretreatment concentrations in MUT mRNA-treated mice. The reduction of plasma methylmalonic acid concentrations was similar to that observed in liver transplanted patients. 27,29
Moreover, a safety study conducted in the mut− hypomorphic mouse model showed no changes in liver function tests nor increase in inflammatory markers following 3 or 5 weekly IV bolus doses of 0.2 mg/kg MUT mRNA, suggesting that LNP-encapsulated MUT mRNA was well tolerated after repeat dosing. Furthermore, no elevation of anti-MUT antibodies was observed after five consecutive doses in these hypomorphic mice. These data altogether suggest that LNP-encapsulated MUT mRNA is efficacious, as demonstrated by the dramatic improvement in survival and reductions in disease biomarkers at doses as low as 0.2 mg/kg, and was well tolerated in murine models of MMA.
These preclinical proof-of-concept data support the clinical development of MUT mRNA for this serious inherited metabolic disorder. A Phase 1/2 study in patients with MMA MUT deficiency will evaluate the safety, pharmacokinetics, and pharmacodynamics of MUT mRNA encapsulated in LNPs (
Acute Intermittent Porphyria
AIP (MIM: 176000) is a rare, autosomal dominant metabolic disorder caused by hepatic deficiency of porphobilinogen deaminase (PBGD), the third enzyme in the heme biosynthesis pathway that resides in the cytosol. 18,30 Heme is a key cofactor for critical cellular hepatic functions related to energy homeostasis, cell metabolism, synthesis of steroid hormones, and bile acid synthesis. PBGD deficiency and subsequent heme-mediated upregulation of the first enzyme of the heme biosynthesis pathway, δ-aminolevulinate synthase 1 (ALAS1), cause marked accumulation of the porphyrin precusors, δ-aminolevulinic acid (ALA) and porphobilinogen (PBG). Accumulation and renal excretion of ALA and PBG are associated with acute neurovisceral attacks, the predominant clinical feature of AIP. These acute attacks significantly alter the quality of life for these patients and their families. Longer term sequelae include hypertension, renal damage, and increased incidence of hepatocellular carcinoma. 31 –33
The current treatment for AIP is prophylactic hemin administration, which replaces the deficient heme pool and results in negative feedback inhibition of ALAS1. 34 Hemin replacement therapy is typically effective after at least three to four consecutive daily infusions, 35,36 although chronic administration can often lead to iron overload or thromboembolism. LT or LKT is a potential treatment option for severe patients, leading to normalization of ALA and PBG levels and elimination of porphyric attacks. 37,38 The liver transplant experience combined with the domino LT experience, in which non-porphyric LT recipients transplanted with the livers from AIP patients develop acute porphyric attacks, 38 demonstrate that the liver is the major organ of pathology for this disorder.
In collaboration with Dr. Antonio Fontanellas and his laboratory at the University of Navarra in Pamplona, Spain, PBGD replacement in animal models of AIP using LNP-encapsulated mRNA encoding for human wild-type PBGD is described as a potential new alternative therapeutic in Jiang et al. 18 IV bolus administration of two different dose levels of PBGD mRNA (0.2 and 0.5 mg/kg) in a mouse model of AIP showed high PBGD protein expression and enzymatic activity in the liver that was detected as early as 2 h post-injection. At day 10 post-injection, human PBGD protein concentrations were still detectable indicating a long tissue residence time of mRNA-encoded PBGD protein. A dose-dependent biomarker response following mRNA treatment was observed in AIP mice, with 0.2 and 0.5 mg/kg dose levels eliciting pronounced reductions in urinary excretion of ALA and PBG (Fig. 3A, B). Furthermore, at these efficacious dose levels, urinary levels of ALA and PBG were normalized (∼90% decreased) 24 h post-treatment. Repeat IV bolus administration of PBGD mRNA every other week for 6 weeks showed sustained and reproducible reduction of porphyrin precursors following each dose and was well tolerated in AIP mice. Additionally, mRNA treatment protected against mitochondrial dysfunction, pain, motor impairment, and hypertension in AIP mice, which are all clinically relevant endpoints in patients.
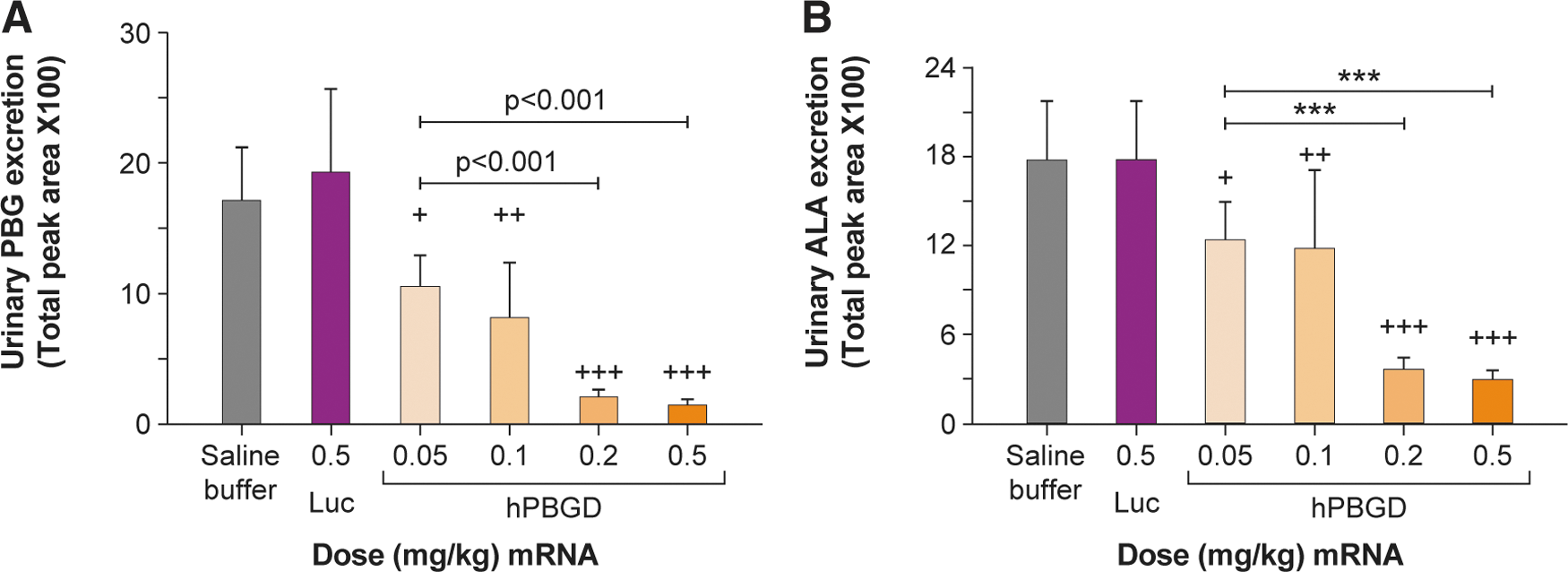
Normalization of urinary porphyrin precursors in a mouse model of AIP due to a single IV dose of PBGD mRNA. Adapted from Jiang et al.
18
Data are shown as mean ± SD. AIP mice received daily intraperitoneal injections of phenobarbital, a porphyrinogenic agent, to induce porphyric attacks in these mice. Two hours before the second phenobarbital injection, mice were treated with a single IV bolus injection of different hPBGD mRNA doses (0.05 mg/kg [n = 5], 0.1 mg/kg [n = 4], 0.2 mg/kg [n = 5], and 0.5 mg/kg [n = 11]), luciferase control mRNA (0.5 mg/kg, n = 10), or saline buffer (n = 4).
The effectiveness of PBGD mRNA was confirmed in two larger species: chemically induced porphyric rabbits and wild-type NHPs. LNP-encapsulated PBGD mRNA led to normalized urinary levels of ALA and PBG after single and repeat IV dose administrations (0.5 mg/kg) in porphyric rabbits. In wild-type NHPs, hepatic PBGD enzyme activity was increased ∼80% one day following single and repeat IV doses of 0.5 mg/kg PBGD mRNA formulated in LNPs. Importantly, anti-PBGD antibodies were not detected in both large species and liver function tests remained within normal range in NHPs following single and repeat dosing.
These data demonstrate preclinical proof-of-concept in small and large animal models of AIP and proof-of-bioactivity in wild-type NHPs. mRNA therapy was well tolerated after single and repeat IV dose administrations in all three species. These data altogether provide comprehensive support for the clinical development of PBGD mRNA as a treatment for AIP.
Other emerging technologies are also in development for AIP. The most advanced development program is Givosiran, a small interfering RNA targeted ALAS1. A phase 1 trial of givosiran in patients with AIP showed significant reductions in porphyrin precursors and porphyric attacks. 39 Recently, topline data from the ENVISION phase 3 trial of givosiran in acute hepatic porphyrias showed significant reductions in clinical outcomes and biomarkers. 40 Although mRNA therapy has not entered clinical testing for AIP, it is conceivable that mRNA therapy could provide an alternative or complementary treatment option to hemin and givosiran therapy. Liver-directed gene therapy is another alternative potential therapy for AIP. However, a phase 1 trial evaluating a recombinant AAV expressing PBGD (rAAV2/5-PBGD) in patients with AIP showed no change in porphyrin precursor levels, 41 and thus, further research is warranted for gene therapy as a treatment for AIP.
Fabry Disease
Fabry disease (MIM: 301500) is a rare, X-linked lysosomal storage disorder caused by mutations in GLA, the gene encoding the lysosomal enzyme alpha-galactosidase A (α-Gal A). Deficiency of α-Gal A enzyme results in accumulation of glycosphingolipids, specifically globotriaosylceramide (Gb3) and the deacylated corresponding analog globotriaosylsphingosine (lyso-Gb3), in most tissues and organs including the kidney, heart, and skin. 42,43 Progressive deposition of these glycosphingolipids in the vascular endothelium and tissues leads to development of angiokeratoma, acroparesthesias, hypohidrosis, corneal and lenticular opacities, and proteinuria. 44 As patients age into their third, fourth, and fifth decades of life, end-stage renal disease, cardiac, and cerebrovascular disease can develop.
Currently, there are several approved therapies for Fabry depending on the geographic region, specifically enzyme replacement therapy (ERT) (agalsidase alfa and agalsidase beta) and chaperone therapy (CT) (migalastat). Other therapeutic approaches are in development for this disorder, including gene therapy, gene editing, substrate reduction therapy (SRT), ERT, and combinatorial approaches using both ERT and SRT or CT. While ERT and CT are the current standard of care for patients with Fabry, declining renal and cardiac function in treated patients represents an unmet medical need to date. 45 –47 ERT is currently administered every other week and is burdensome on the quality of life of these patients; therefore, a less frequently administered therapy could be attractive to patients and their families.
Zhu et al. 19 reported preclinical proof-of-concept data for mRNA encoding α-Gal A enzyme in a mouse model of Fabry disease and wild-type NHPs. Unlike the liver-focused therapeutic approach for MMA and AIP, the target organs of pathology for Fabry include the liver, spleen, heart, and kidney. Thus, the mRNA therapy approach for Fabry is to use the liver as a protein-production depot. A dose-dependent restoration of α-Gal A enzyme was demonstrated in key disease organs (liver, spleen, heart, and kidney) with a concomitant reduction in glycosphingolipids, Gb3 and lyso-Gb3, in plasma and all key disease tissues 3 days following a single IV bolus administration of α-Gal A mRNA encapsulated in LNPs (0.05–0.5 mg/kg) in Fabry GLA-knockout mice. Enzyme restoration levels were correlated with substrate reductions in key disease tissues, including the heart and kidney. A long duration of effect was observed after a single dose of mRNA in Fabry mice as demonstrated by the kinetics of glycosphingolipids reductions in disease organs. Specifically, all tissue concentrations of Gb3 and lyso-Gb3 remained low 5–6 weeks after a single IV bolus dose of α-Gal A mRNA (0.5 mg/kg) and did not rebound to control levels until 12 weeks post-treatment.
In a 3-month repeat-dose study in which 0.2 or 0.5 mg/kg of α-Gal A mRNA formulated in LNPs was IV bolus injected in Fabry mice either on a biweekly (similar to ERT) or a monthly regimen, a dose-dependent reduction of glycosphingolipids occurred in similar manner between the two dose regimens, suggesting that mRNA therapy has the potential to be efficacious with less frequent dose intervals. Anti-α-Gal A antibodies were measured, as exposure to mRNA-encoded α-Gal A secreted into the circulation might trigger an immune response in Fabry GLA-knockout mice. No evidence of anti-α-Gal A antibodies was detected after multiple doses of α-Gal A mRNA contrary to ERT where an antibody response against recombinant α-Gal A can be triggered as early after two to three dose administrations in Fabry mice. 48 These data may suggest improved immune tolerability of an mRNA-encoded therapeutic protein produced in the liver with endogenous post-translational modifications compared with traditional ERT.
Finally, in a 6-week repeat-dose study of 0.5 mg/kg α-Gal A mRNA formulated in LNPs administered every other week in wild-type NHPs, reproducible kinetics of mRNA-encoded α-Gal A enzyme activity in plasma was observed following all four IV infusions (Fig. 4A). The α-Gal A enzyme activity in key disease tissues (heart, kidney, liver, and spleen) was well above background levels (Fig. 4B) correlating with immunohistochemistry staining of α-Gal A protein in these tissues following the last mRNA infusion. α-Gal A mRNA encapsulated in LNPs (0.5 mg/kg) was well tolerated in NHPs following four consecutive IV infusions every other week as supported by liver function tests and the lack of anti-α-Gal A antibodies.

Increased α-Gal A enzyme activity in plasma and key target tissues after every other week IV infusions of hα-Gal A mRNA in wild-type NHPs. Adapted from Zhu et al.
19
Data are shown as mean ± SD.
These data, similar to MMA and AIP, support the clinical development of α-Gal A mRNA as a treatment for Fabry. Based on the prolonged duration of biomarker response in Fabry mice, mRNA therapy could potentially translate into a more convenient dose regimen. Also, as mRNA therapy resulted in higher exposure of therapeutic enzyme in key disease tissues (namely, the kidney and heart) compared with traditional ERT in Fabry mice, mRNA therapy may result in improved efficacy as well.
Conclusion
In the three independent peer-reviewed studies evaluating LNP-encapsulated mRNA for the treatment of three different monogenic disorders, mRNA therapy was efficacious and well tolerated after single and repeat IV dose administrations in relevant small or large animal models. While the preclinical data in animal models are encouraging, translation into the clinic in rare monogenic disorders has yet to be evaluated. Clinical trials of mRNA therapy for rare monogenic disorders are on the horizon, and the next several years will reveal whether mRNA therapy is safe and efficacious in patients affected by these disorders. Key challenges faced by upcoming clinical trials of mRNA therapy will be to evaluate the translatability of preclinical mRNA efficacy and safety data, specifically with respect to the therapeutic index, potency, and optimal dosing interval in patients. The biodistribution of the mRNA and encoded therapeutic protein in humans will be particularly important for intracellular targets like MUT and PBGD for MMA and AIP disorders, respectively, where the aim is to restore functional enzyme in the liver. If mRNA therapy is successful in clinical trials, it could become a viable treatment option for patients in addition to other nucleic acid therapies, such as gene therapy or gene editing.
Footnotes
Acknowledgments
We thank the Moderna Rare Diseases and Platform group for their contribution to this review article.
Author Disclosure
All authors are employees and shareholders of Moderna, Inc. The authors have been named inventors on a patent application related to this work.