
Abstract
Preclinical studies showed that tumor necrosis factor
Graphical Abstract
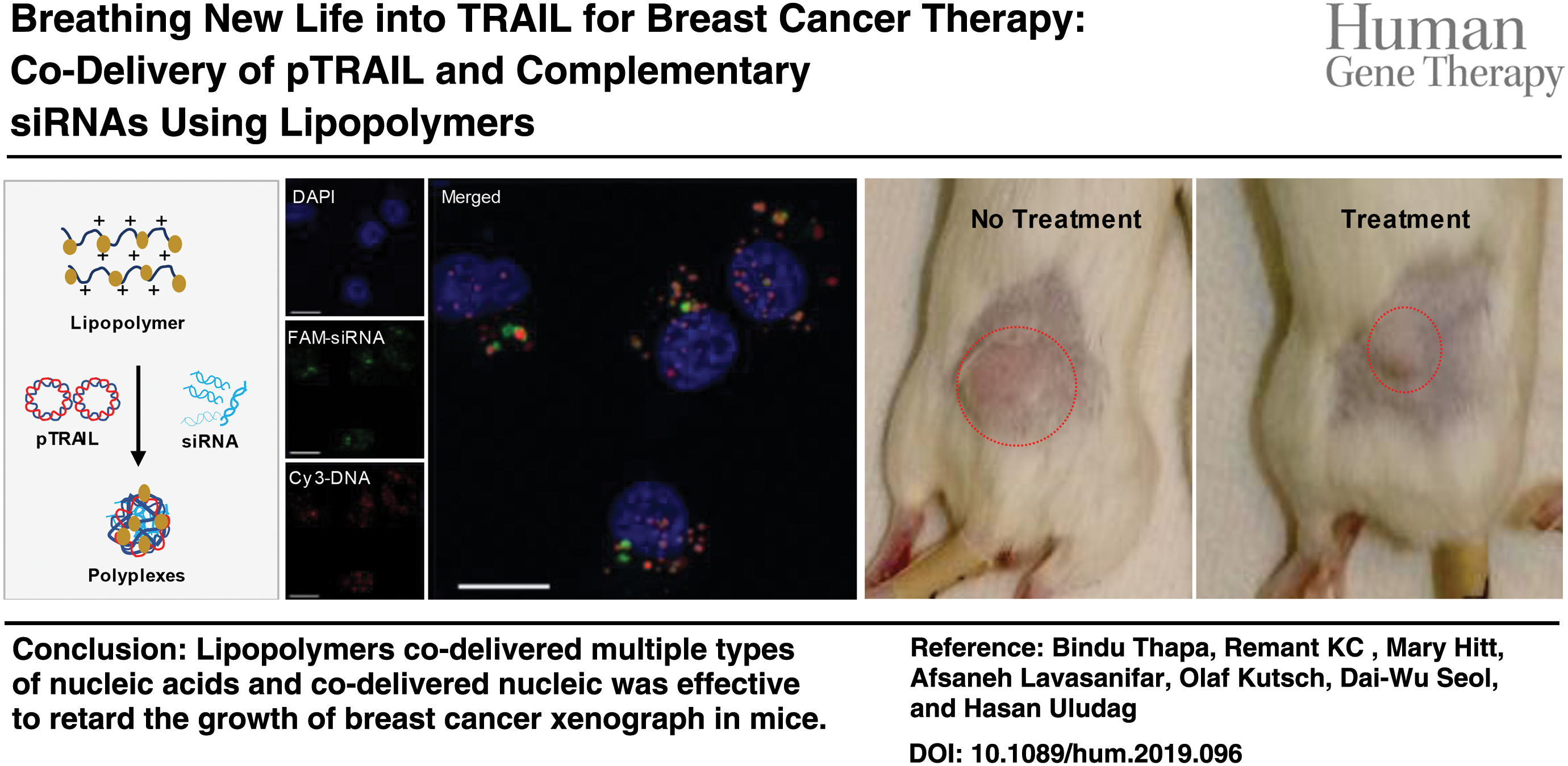
Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has emerged as a promising cancer therapeutic because of its ability to induce apoptosis in a variety of cancer cells while sparing normal cells. 1,2 TRAIL binds and induces trimerization of death receptors, which is a prerequisite for death-induced signaling complex formation and apoptosis induction. The p53 appears to be dispensable in apoptosis induction by death receptors, so that TRAIL therapy could be the most effective therapeutic approach for cancers with p53 mutations. TRAIL exerted potent tumor-suppressor activity after systemic administration in tumor-bearing mice without affecting normal tissues. 2 To translate these promising preclinical outcomes into the clinical realm, recombinant human TRAIL and TRAIL receptor agonist antibodies were explored. 3 The TRAIL therapies tested so far were safe and well tolerated in patients, but they failed to exert a robust anticancer activity. 3 –7 The ineffectiveness of these therapies was attributed to (1) development of intrinsic or acquired resistant to TRAIL therapy, 3,8 (2) poor pharmacokinetics profile of recombinant TRAIL proteins due to short half-lives (3–5 min in rodents and 23–31 min in nonhuman primates), 9 and (3) weak apoptosis induction by TRAIL receptor agonistic antibodies due to bivalent nature of antibodies, which cannot induce trimerization. 3 Therefore, improved pharmacokinetics as well as increased potency of TRAIL therapy are needed.
PEGylation 10 and fusion of TRAIL to polyhistidine (His), 11 Flag, 12 human serum albumin, 12 isoleucine 13 and leucine zipper, 2 and Fc portion of IgG 14 have been explored to increase stability and extend the half-life of the protein. 2,4,14,15 However, His-TRAIL and Flag-TRAIL were observed to induce hepatotoxicity, 11,13,16 and PEGylation decreased the TRAIL efficacy due to interference in TRAIL receptor binding. 17 To this end, TRAIL gene therapy can overcome the pharmacokinetics limitations by continuous in situ production of the protein, allowing sustained presence of TRAIL at the tumor microenvironment. 15 Successful studies on viral TRAIL gene therapy 18,19 were reported, but the safety issues related to viral vectors (e.g., unpredictable immunogenicity and toxicity) are always a concern for their clinical translation. 20,21 Alternatively, nonviral vectors (e.g., cationic lipopolymers, 22 dendrimers, 23 peptides, 24 and lipid nanoparticles 25 ) were explored for TRAIL gene delivery. These studies initially used a TRAIL gene encoding a membrane-bound full-length TRAIL. After the discovery of homotrimeric structure of receptor binding domain of TRAIL, 26,27 a modified trimeric form of TRAIL protein was found to be more potent. 2,28 Resistance to TRAIL action, however, is a significant concern that cannot be readily solved with improved pharmacokinetics. 3 To address this issue, we had previously showed that silencing Bcl2-like 12 (BCL2L12) and superoxide dismutase 1 (SOD1) using small interfering RNA (siRNA) sensitized breast cancer cells to TRAIL-induced apoptosis. 29 Therefore, co-delivery of a trimeric TRAIL expressing plasmid (pTRAIL) and sensitizing siRNAs (BCL2L12 and SOD1) was proposed in this study to improve the potency of TRAIL therapy.
Co-delivery of pDNA and siRNA allows simultaneous knockdown of undesirable proteins with siRNA and forced expression of desirable proteins with pDNA in same cells. 30 In addition, co-delivery allows exposure of both agents to the cells at a defined ratio to better control the pharmacological effects. Co-delivery of different therapeutics will be possible only with an appropriate carrier that can accommodate both agents. The structural differences between pDNA and siRNA make it difficult to identify a single carrier for co-delivery of both agents. Functional pDNAs are long (>3,000 base pairs) flexible molecules, whereas siRNAs are short (<30 base pairs) rigid molecules. 31 We have been tailoring cationic lipopolymers generated by grafting lipophilic ligands onto low–molecular weight (0.6–1.8 kDa) polyethylenimines (PEIs) for delivery of nucleic acids. The resultant amphiphilic lipopolymers are relatively nontoxic and possess high charge density suitable for polynucleotide interactions. Our past work identified carriers that were suitable for either pDNA or siRNA delivery to a variety of cells and preclinical tumor xenografts. 29,32 In this study, we report cationic lipopolymers prepared by grafting aliphatic lipids onto PEI via thioester and amide linkages 33 and demonstrate their promise for co-delivery of pDNA and siRNA for TRAIL therapy in in vitro and in vivo breast cancer models.
Materials and Methods
Materials
Branched 1.2 kDa PEI (PEI1.2) was obtained from Polysciences, Inc. (Warrington, PA) and used without further purification. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), human insulin, heparin, and organic solvents were obtained from Sigma–Aldrich (St. Louis, MO). SYBR Green I was purchased from Cambrex BioScience (Rockland, MD). The unlabeled scrambled siRNA (CsiRNA) and 5′-carboxyfluorescein (FAM)-labeled scrambled siRNA (FAM-siRNA), BCL2L12 siRNA, SOD1 siRNA, and all primers were obtained from Integrated DNA Technologies, Inc. (IDT; Coralville, IA). Cell culture medium, Dulbecco's modified Eagle's medium (DMEM)/F12, supplied with L-glutamine and 25 mM HEPES, and penicillin (10,000 U/mL)/streptomycin (10 mg/mL) were obtained from Invitrogen (Grand Island, NY). FB essence 100% U.S. origin serum was obtained from VWR Life Science Paradigm (Radnor, PA). Cholera toxin was obtained from List Biological Laboratories (Campbell, CA). Annexin V-FITC Apoptosis Kit I was purchased from BD Biosciences (San Jose, CA). The preparation and characterization of trimeric secretable TRAIL encoding plasmid was described before. 28 The gWIZ and gWIZ-GFP plasmids were obtained from Aldevron (Fargo, ND).
Cell culture
Human breast cancer cells MDA-MB-231, MCF-7, and MCF-10A were obtained from Dr. Judith Hugh (Department of Oncology, University of Alberta), and SUM-149 and MB-MB-436 were obtained from Dr. Raymond Lai (Department of Laboratory Medicine and Pathology, University of Alberta). GFP-positive MDA-MB-231 cells (MDA-MB-231-GFP+) were obtained by retroviral transformation as described before. 34 MDA-MB-231, MCF-7, SUM-149, and MDA-MB-436 were maintained in DMEM/F12 supplemented with 10% FB essence, 100 U/mL penicillin, and 100 μg/mL streptomycin. Human umbilical vein cells (HUVEC) were obtained from Dr. Janet Elliott (Department of Chemical and Materials Engineering, University of Alberta) and cultured in rat tail type I collagen-coated flasks with endothelial growth medium-2 with the manufacturer's growth factor BulletKit™ (Lonza), 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. MCF-10A cells were maintained in DMEM/F12 supplemented with 5% horse serum, 100 U/mL penicillin, 100 mg/mL streptomycin, hydrocortisone (500 ng/mL), epidermal growth factor, human (20 ng/mL), human insulin (0.01 mg/mL), and cholera toxin (100 ng/mL). Human bone marrow stromal cells (hBMSC) were obtained and maintained as described earlier. 35 All the cell lines were authenticated by STR DNA profiling analysis at the Genetic Analysis Facility, The Hospital for Sick Children (Toronto, ON).
Lipopolymer synthesis, complex preparation, and characterization
Hydrophobically modified PEI were synthesized via N-acylation using carboxyl end-capped aliphatic lipids, which were prepared by coupling α-linoleoyl chloride (αLA) with mercaptopropionic acid (MPA) through thioester (-S-CO-) bonding (PEI-modified αLA [tαLA]; Fig. 1), as reported previously. 33 As a control group, PEI1.2 modified with αLA (PEI-αLA), LA (PEI-LA), and tLA (PEI-tLA) were prepared according to previous protocols. 33 The lipopolymer complexes of pDNA, siRNA, or siRNA/pDNA combination were prepared at room temperature by incubating the lipopolymers with nucleic acids at a ratio of 5:1 (w/w) in nuclease-free water for 30 min. Size of complexes and surface charge were measured using Zeta Nano-ZS (Malvern, UK). Since all complexes were used within 30 min of preparation, long-term colloidal stability was not investigated for the purpose of this study. Dissociation of complexes was studied in the anionic environment of heparin by agarose gel retardation assay. 33

Synthesis of thioester-linked lipopolymer.
Uptake of lipopolymer/siRNA/pDNA complexes
The cellular uptake of complexes was assessed in MDA-MB-231 cells through flow cytometry and confocal microscopy using FAM-labeled siRNA and Cy3-labeled gWIZ (Cy3 labeling according to the manufacturer's protocol). MDA-MB-231 cells were seeded (105 cells/mL) in 24-well plates and allowed to attach overnight. The lipopolymer complexes with Cy3-gWIZ, FAM-siRNA, or FAM-siRNA/Cy3-gWIZ combination were prepared at room temperature by incubating the lipopolymers with nucleic acids at a ratio of 5:1 (w/w) in DMEM and then directly added to the cells. After 24 h of incubation, cells were harvested and analyzed by flow cytometry. 32 For confocal microscopy, attached MDA-MB-231 cells on cover glasses were treated with the complexes, processed, and observed under confocal microscope as described earlier. 32
pDNA transfection and effect of siRNA
Transfection efficiency of the lipopolymers was assessed in MDA-MB-231 cells through flow cytometry using the GFP expression plasmid gWIZ-GFP. Cells were seeded (105 cells/mL) in 24-well plates and grown overnight. Lipopolymer/gWIZ-GFP complexes (lipopolymer:nucleic acid ratio of 5:1, w/w) prepared in DMEM were added to the cells. To reveal the effect of siRNA on transfection efficiency, negative control scrambled siRNA (CsiRNA) was loaded into the complexes along with gWIZ-GFP plasmid. Briefly, gWIZ-GFP and CsiRNA were well mixed in separate tubes at different ratios and then added to lipopolymers solution (in DMEM) making the final lipopolymer:nucleic acid ratio of 5:1 (w/w). After 30 min of incubation, complexes were added directly to the cells and GFP expression was analyzed by flow cytometry after 48 h of transfection, as described earlier. 32
siRNA transfection and effects of pDNA
The siRNA transfection efficiency of the lipopolymers was assessed by measuring GFP silencing in MDA-MB-231-GFP+ cells using flow cytometry and a GFP-specific siRNA (GFP-siRNA). Cells were seeded (105 cells/mL) in 24-well plates and grown overnight. Lipopolymer complexes with GFP-siRNA or CsiRNA (lipopolymer:nucleic acid ratio of 5:1, w/w) prepared in serum-free DMEM were directly added to the cells. To reveal the effect of pDNA on transfection, gWIZ plasmid was loaded into the complexes along with GFP-siRNA. Briefly, gWIZ and GFP-siRNA were mixed at different ratios (w/w) in separate tubes and added to lipopolymers solution in DMEM (final lipopolymer:nucleic acid ratio of 5:1, w/w) and incubated for 30 min at room temperature. The complexes were added to the cells, and the cells were processed after 3 days with flow cytometry for GFP expression.
Cell viability
Anticancer efficacy of pTRAIL, and BCL212 and SOD1 siRNA co-delivery was evaluated in breast cancer cell lines MDA-MB-231, MCF-7, SUM-149, and MDA-MB-436 as well as in nonmalignant HUVEC, hBMSC, and MCF-10A cells by monitoring cell growth using the MTT assay. Cells were seeded (105 cells/mL) in 48-well plates and treated with lipopolymer/pDNA/siRNA complexes as described earlier. Custom-synthesized siRNAs targeting BCL2L12 (cat# HSS.RNAI.N001040668.12.2; IDT) and SOD1 (cat# HSC.RNAI.N000454.12.1; IDT), and pTRAIL were used for the formation of complexes. 29 The scrambled siRNA (CsiRNA) and gWIZ-GFP were used as negative controls in complex formation for siRNAs and pTRAIL, respectively. In parallel, separate complexes containing either pTRAIL or specific siRNAs were prepared, and their efficiency was compared with the complexes containing both the pTRAIL and siRNAs. After 72 h of incubation with complexes, cells were processed for the MTT assay. The MTT reagent in Hank's balanced salt solution (5 mg/mL) was added to each well to give a final concentration of 1 mg/mL. After 1 h incubation at 37°C, the medium was replaced with 200 μL of DMSO to dissolve the formazan crystals. Optical density of solution was measured at λ = 570 nm using a microplate reader (ELx808; Bio-Tech Instrument, Inc.). Cell viability was expressed as percentage of nontreated cells.
Gene silencing by real-time PCR
Knockdown of BCL2L12 and SOD1 genes as well as TRAIL expression at the mRNA level in MDA-MB-231 and MCF-7 cells transfected with lipopolymer/nucleic acid complexes was assessed by real-time PCR (qPCR), as described before. 29 Briefly, total RNA was isolated using TRIzol (Invitrogen) and converted into cDNA using SensiFAST cDNA Synthesis Kit (Bioline) according to the manufacturer's instruction. The qPCR was performed using StepOnePlus Real-Time PCR Systems (Applied Biosystems, Foster City, CA) with SensiFAST SYBR Hi-ROX Kit (Bioline). Reaction mixture (10 μL) containing SensiFAST SYBR Hi-ROX Kit, primers, and cDNA template was added in triplicate to the MicroAmp Fast Optical 96-Well Reaction Plate. The reaction mixture was then heated at 95°C for 10 min and 40 cycles of denaturation step 95°C for 15 s and annealing/elongation step 65°C for 1 min were run. Specific primers against human endogenous housekeeping gene β-actin (forward: 5′-GCG AGA AGA TGA CCC AGA T-3′ and reverse: 5′-CCA GTG GTA CGG CCA GA-3′), BCL2L12 (forward: 5′-CCC GCC CCT ATG CCC TTT TT-3′ and reverse: 5′-ATA ACC GGC CCA GCG TAG AA-3′), SOD1 (forward: 5′-GTG TGA CTT TTT CAG AGT TGC T-3′ and reverse: 5′-AAG TCT GGC AAA ATA CAG GTC A-3′), and TRAIL (forward: 5′-ATT GTC TTC TCC AAA CTC CAA G-3′ and reverse: 5′-TGC TCA GGA ATG AAT GCC CAC-3′) were used for amplification. Levels of mRNA for each gene were determined using the comparative threshold cycle method 36 and presented as fold-change relative to β-actin.
Analysis of apoptotic cell population and caspase-3 activity
The MDA-MB-231 cells were seeded in 12-well plates and treated with the complexes, as described above. After 72 h, induction of apoptosis was assessed using FITC-Annexin V and propidium iodide staining according to the manufacturer's protocol. To elucidate apoptosis mechanism, caspase-3 activity in cells was assayed after 48 h of transfection, as described before. 29 The caspase-3 activity was expressed as an increase in relative fluorescence units per hour and normalized with respect to the total protein content in the samples (from BCA assay).
TRAIL secretion by enzyme-linked immunosorbent assay
The secretion of TRAIL protein from MDA-MB-231 and MCF-7 cells after treatment with the indicated lipopolymer/nucleic acid complexes was assayed with a validated enzyme-linked immunosorbent assay (ELISA). The cells were seeded in 24-well plates (105 cells/mL) and transfected as described above. After 48 h of transfection, secreted TRAIL protein in the supernatants was determined by human TRAIL Duo Set ELISA kit (R&D Systems; Minneapolis, MN) according to the manufacturer's instructions.
Animal studies
All experiments were conducted in accordance with the pre-approved procedure by the Health Sciences Laboratory Animal Services (HSLAS), University of Alberta. To create breast cancer xenografts, 7- to 8-week-old male ICR-Prkdcscid mice (Taconic Farms, Seattle, WA) were kept in a biocontainment unit. Mice were anesthetized using isoflurane, shaved left flank (about 2 × 2 cm), and ∼3 million MDA-MB-231 cells in Matrigel and PBS (1:1) were injected subcutaneously into shaved area. Tumor growth was monitored every 72–96 h, and tumor volume was measured using a digital caliper. Once the tumor volume reached ≥50 mm3 (length × width2 × 0.5), 40 μL of pDNA/siRNA/lipopolymer complexes (lipopolymer:nucleic acid ratio of 5:1, w/w) was injected subcutaneously into the tumor vicinity. Four injections were performed with 72–96 h apart with simultaneous measurements of tumor volume. BCL2L12 siRNA was chosen for co-delivery with pTRAIL. The injected dose of siRNA was 6 μg per mouse (∼0.2 mg/kg), whereas the dose of pTRAIL (3 μg per mouse; ∼0.1 mg/kg) was fairly lower than the typical 10 μg per mouse intratumoral doses used in other studies. 37,38 Co-delivery of gWIZ-GFP/CsiRNA (3/6 μg per mouse) was used as control to evaluate the nonspecific toxicity, if any, related to the delivery system and the nucleic acid. Co-delivery of pTRAIL/CsiRNA (3/6 μg) was performed to evaluate the effect of pTRAIL only. After 72 h of the last injection, mice were euthanized, tumors were collected, weighted, and stored in RNAlater in −20°C until qPCR analysis. Mice with large tumor volumes (>1,500 mm3), necrotic spot(s), or excessive weight loss (>20%) were euthanized for humane considerations.
Statistical analysis
The data are presented as mean ± standard deviation (SD) of three replicates for all results except the in vivo data (see Figure legends). For comparison between two group means, the results were analyzed using Student's t-test. In vivo experiment represents mean ± SD for groups with six to eight replicates, and statistical significance was analyzed by one-way analysis of variance followed by Tukey's post hoc analysis for comparison of between-group means. A value of p < 0.05 was considered significant throughout the study.
Results
Lipopolymer synthesis, characterization, and transfection efficiency
We prepared a series of lipopolymers by grafting unsaturated lipids α-LA and LA onto PEI1.2 via thioester bonding (Fig. 1A) as described before 33 and explored their co-delivery efficiency into breast cancer cells. Modification of PEI with α-LA and LA was confirmed with 1 H-NMR (Supplementary Fig. S1). We were able to graft as much as 2.2 mol of lipid/mol PEI1.2. As a control carrier, we chose the best performing lipopolymers for pDNA and siRNA delivery from our previous studies, namely PEI-LA (LA/PEI = 2.5 mol/mol), PEI-tLA (tLA/PEI = 2.7 mol/mol), and PEI-αLA (αLA/PEI = 2.4 mol/mol), and explored their dual loading capacity (Fig. 1B).
These lipopolymers showed a good capability to condense siRNA, pDNA, or siRNA/pDNA mixtures into nanosized complexes, without any differences in size and charge density (Supplementary Fig. S2). We monitored dual loading of nucleic acids into single complexes through flow cytometry. When the complexes were prepared separately with either Cy3-pDNA or FAM-siRNA, clear populations of corresponding fluorescent particles were confined to expected quadrant of the histogram in flow cytometry analysis. The ternary complexes with Cy3-pDNA and FAM-siRNA were localized in the quadrant specific for double-labeling, indicating co-loading of both pDNA and siRNA into the complexes (Supplementary Fig. S3). Dual (pDNA+siRNA) loading capability of the lipopolymers was further confirmed by cellular uptake study in MDA-MB-231 cells (Fig. 2). Flow cytometry showed that cells treated with individual nucleic acid (pDNA or siRNA) complexes were localized in the quadrant of the corresponding fluorescence, whereas cells treated with the dual pDNA/siRNA complexes were localized in the quadrant associated with both fluorescent labels, indicating efficient uptake of the dual complexes. Broadly effective l-PEI40 was able to deliver pDNA into MDA-MB-231 cells, but not siRNA, unlike the prepared lipopolymers. Higher siRNA uptake was achieved by the lipopolymers with amide bonding (PEI1.2-LA and PEI1.2-αLA) than the lipopolymers with thioester bonding (PEI1.2-tLA and PEI1.2-tαLA) (Fig. 2A). Approximately 80% of cells were FAM-siRNA positive with amide-linked lipopolymers, whereas ∼60% of cells were FAM-siRNA positive with thioester-linked lipopolymers. In case of Cy3-pDNA uptake, ∼90% of cells were Cy3-pDNA positive with all lipopolymers. Interestingly, ∼90% of cells were pDNA and siRNA positive with dual-loaded complexes, indicating uniform uptake among the cell population (Fig. 2A). Confocal micrographs of MDA-MB-231 cells after co-delivery of pDNA and siRNA (Fig. 2C) showed distinct particles that are mostly orange indicating the entrapment of both siRNA and pDNA in the same complexes. Unlike co-delivery, separate delivery of pDNA and siRNA complexes led to the distribution of distinct green (FAM-siRNA) and red (Cy3-pDNA) particles inside the cells (Fig. 2B).
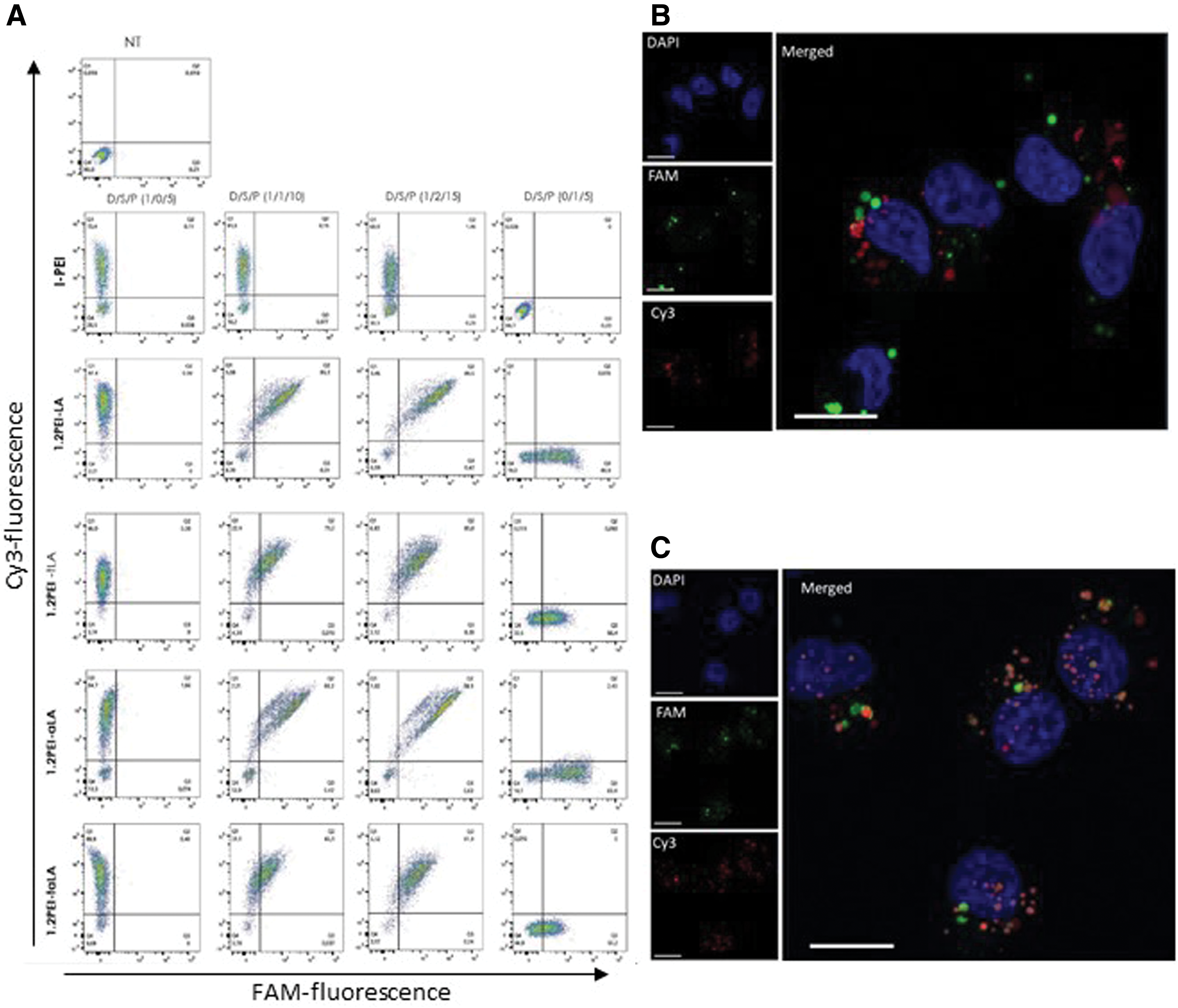
Cy3-pDNA and FAM-siRNA uptake in MDA-MB-231 cells by flow cytometry (
To measure the transfection efficiency of lipopolymers, gWIZ-GFP complexes were formed with and without CsiRNA. The thioester-linked lipopolymers (PEI1.2-tLA and PEI1.2-tαLA) displayed higher transfection efficiency with and without siRNA than the amide-linked lipopolymers (PEI1.2-LA and PEI1.2-αLA) (Fig. 3A). Uptake of pDNA with and without siRNA by amide-linked lipopolymers was higher than by thioester-linked lipopolymers (Supplementary Fig. S4). Therefore, the higher transfection efficiency of thioester-linked lipopolymers was attributed to better dissociation of complexes to release the nucleic acid cargo (Fig. 3B) rather than the delivery efficiency. The transfection efficiency of thioester lipopolymers was increased with the addition of siRNA (Fig. 3A). Compared with complexes without siRNA (D/S/P = 1/0/5), complexes with siRNA (D/S/P = 1/0.5/7.5, 1/1/10, and 1/2/15) resulted in higher transfection (polymer:nucleic acid ratios were 5 in all complexes). Despite the significant increase in transfection efficacy in the presence of siRNA, the effect of siRNA on the uptake of PEI1.2-taLA/pDNA was minimal (Supplementary Fig. S4). Instead, siRNA enhanced the dissociation of complexes since the PEI1.2-taLA/pDNA complexes showed higher release of pDNA when siRNA was added upon heparin challenge (Fig. 3D). The transfection efficiency of thioester lipopolymers was increased with the addition of siRNA (Fig. 3A); as an example, compared with pDNA/siRNA/lipopolymer weight ratios of 1/0/5 (i.e., no siRNA), higher transfection was achieved in the presence of siRNA with weight ratios of 1/0.5/7.5, 1/1/10, and 1/2/15 (where the lipopolymer:nucleic acid ratios were maintained at 5).
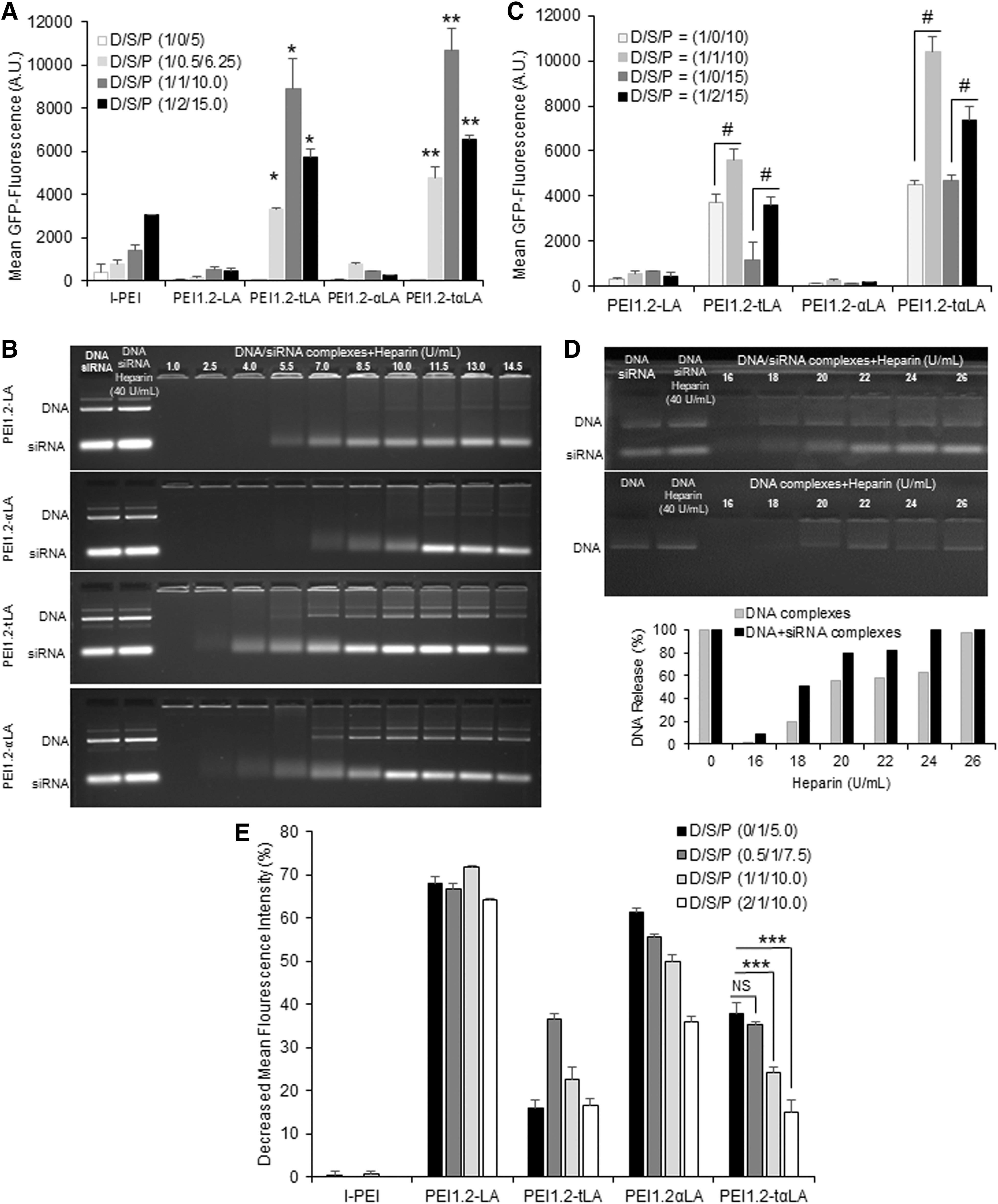
Transfection efficiency of polymers and dissociation of complexes.
Transfection efficiency of lipopolymers was generally increased with increasing lipopolymer amount. To evaluate the net effect of siRNA addition, we also tested complexes with fixed ratio of lipopolymer/pDNA (10 or 15) and with or without siRNA. The effect of siRNA on pDNA transfection (i.e., GFP expression) was clear where the complexes with siRNA gave higher transfection than the complexes without siRNA (Fig. 3C). Therefore, the co-delivery of pDNA and siRNA may not only improve therapeutic responses derived from either agent but can also enhance transgene expression. In addition, transfection efficiency of PEI1.2-tαLA and PEI1.2tLA lipopolymer was higher than other lipopolymers and commercial lipid transfection reagent, lipofectamine (Supplementary Fig. S5).
We further evaluated the complexes with similar composition for siRNA silencing efficiency. Analogous to the use of scrambled siRNA in previous studies, blank gWIZ plasmid was used to assess the effect of pDNA on siRNA silencing efficiency. The silencing efficiency was expressed as percentage of mean GFP fluorescence in MDA-MB-231-GFP+ cells by using GFP-specific siRNA. Unlike pDNA transfection, amide-linked lipopolymers gave higher GFP silencing than thioester-linked lipopolymers (Fig. 3E). The addition of pDNA did not increase the siRNA silencing efficiency. The siRNA silencing efficiency by polyplexes with siRNA (D/S/P = 0.5/1/7.5) and without siRNA (D/S/P = 0/1/5) was similar when using PEI1.2-tαLA. However, increasing pDNA amount in complexes that is, D/S/P = 1/1/10 and 2/1/15 was detrimental to siRNA silencing efficiency. Since, PEI1.2-tαLA lipopolymer facilitated high transgene expression and siRNA silencing efficiency with optimal pDNA to siRNA ratios, this lipopolymer was further explored for co-delivery of pTRAIL and its complementary siRNAs.
Co-delivery of pTRAIL and siRNAs for induced cell death
The effects of pTRAIL treatment in combination with BCL2L12 and SOD1 siRNAs on growth inhibition of different breast cancer cells (MDA-MB-231, MCF-7, MDA-MB-436, and SUM-149) are summarized in Fig. 4A. The pTRAIL on its own (within indicated doses) was not effective in breast cancer cells, but when pTRAIL was co-delivered with CsiRNA, significant cell death was observed. The co-delivery of “inactive” gWIZ-GFP/CsiRNA remained nontoxic, which indicated that the enhanced cell death was due to the secretion of TRAIL. When CsiRNA was replaced with BCL2L12 and SOD1 siRNAs, significantly enhanced cell death was observed in all cells compared with pTRAIL/CsiRNA treatment. Responses to separate delivery of pTRAIL combination with CsiRNA or BCL2L12 or SOD1 siRNAs varied with cell lines. Separate delivery of pTRAIL and BCL2L12 siRNA resulted higher cell death in MCF-7 cells than other cells. More importantly, the co-delivery of pTRAIL and BCL2L12 or SOD1 siRNAs within the same complex displayed significantly higher cell death than separate delivery of the same dose of pTRAIL and siRNAs complexes (Fig. 4A), giving synergistic activity for therapeutic agents only in the co-delivered formulations. All pTRAIL and siRNAs combinations, either co-delivered or separately delivered, had minimal effect on nonmalignant hBMSC, HUVEC, and MCF-10A cells compared with the effect achieved in breast cancer cells under similar conditions (Fig. 4B).
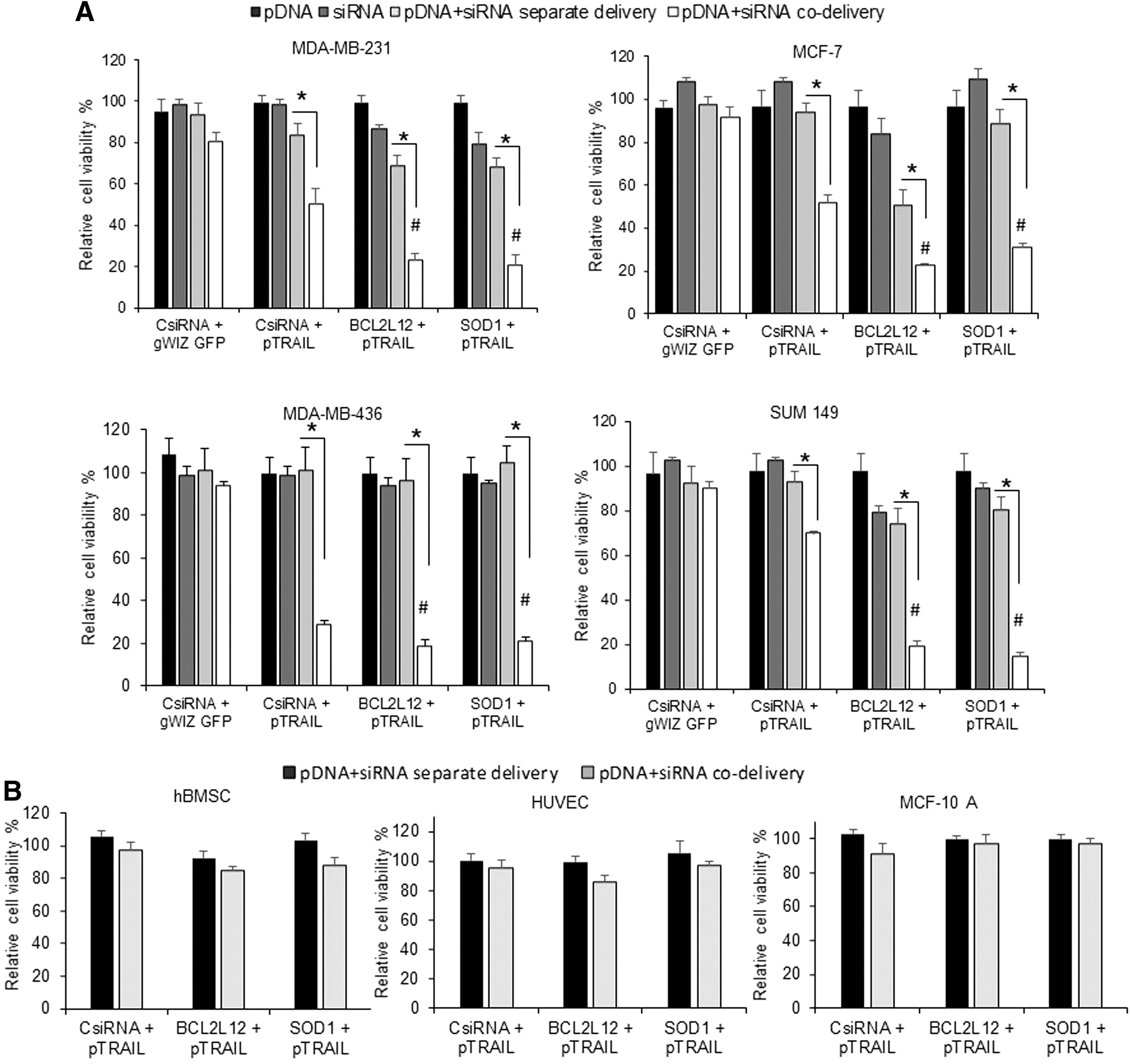
Effect of complexes on growth inhibition of breast cancer cells and effect on normal cells.
Secretion of TRAIL protein and siRNA silencing in breast cancer cells
To confirm the underlying basis of cell death following pTRAIL/siRNA delivery, TRAIL protein secretion and target silencing by BCL2L12 and SOD1 siRNAs were investigated. Co-delivery of pTRAIL and the siRNAs resulted in higher TRAIL secretion than the separate delivery in both MDA-MB-231 and MCF-7 cells (Fig. 5), similar to the GFP expression results above. We observed higher TRAIL secretion in MCF-7 cells (∼2,500 pg/mL) compared with MDA-MB-231 cells (∼200 pg/mL), in line with higher induction of TRAIL mRNA in the MCF-7 cells (Supplementary Fig. S6). The relative quantity of SOD1 mRNA after specific siRNA treatment either alone or in combination with pTRAIL (i.e., separate delivery vs. co-delivery) was significantly less than the CsiRNA treatment in both cell lines. SOD1 mRNA silencing after co-delivery of pTRAIL/SOD1 siRNA was not as effective as delivery of SOD1 siRNA alone in MDA-MB-231 (∼17% vs. 30% silencing, respectively, vs. nontreatment) and MCF-7 (∼21% vs. 47% silencing, respectively, vs. nontreatment). However, the BCL2L12 mRNA levels displayed a more complex behavior; BCL2L12 mRNA expression was increased upon pTRAIL/siRNA co-delivery (with CsiRNA or SOD1 siRNA), but the increase was attenuated with BCL2L12 siRNA inclusion in the complexes. The BCL2L12 silencing after pTRAIL/BCL2L12 siRNA co-delivery was found to be equivalent to CsiRNA treatment group, but the effect of BCL2L12 siRNA was manifested by preventing its increase above the levels found in nontreated cells.
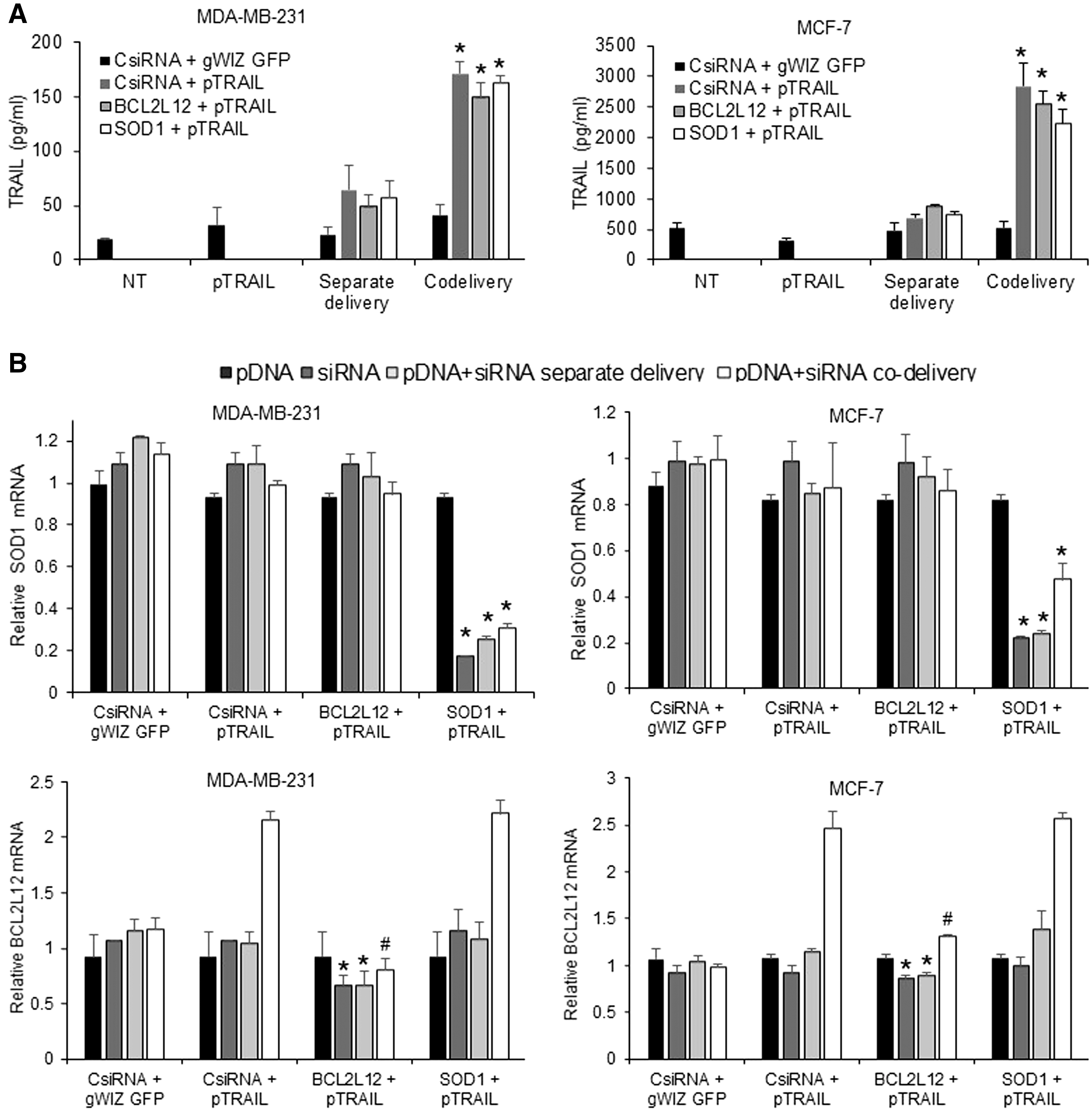
Analysis of TRAIL secretion and siRNA-mediated silencing.
Apoptosis and caspase-3 induction by pTRAIL and siRNA co-delivery
We further explored the effect of separate and co-delivery of pTRAIL and specific siRNAs on apoptosis induction (Fig. 6A). No significant increase in apoptosis was observed with pTRAIL alone nor by the BCL2L12 and SOD1 siRNAs compared with CsiRNA at the chosen doses. Most of the cells in these groups were Annexin-FITC and PI negative (Supplementary Fig. S7). In line with Fig. 4, co-delivery of pTRAIL/siRNAs (CsiRNA, BCL2L12, or SOD1) induced more apoptosis than the separate delivery. Co-delivery of pTRAIL and BCL2L12 or SOD1 siRNAs resulted in significantly higher levels of apoptosis (64.7% and 65.3%, respectively) in MDA-MB-231 cells compared with co-delivery of pTRAIL/CsiRNA (44.6%
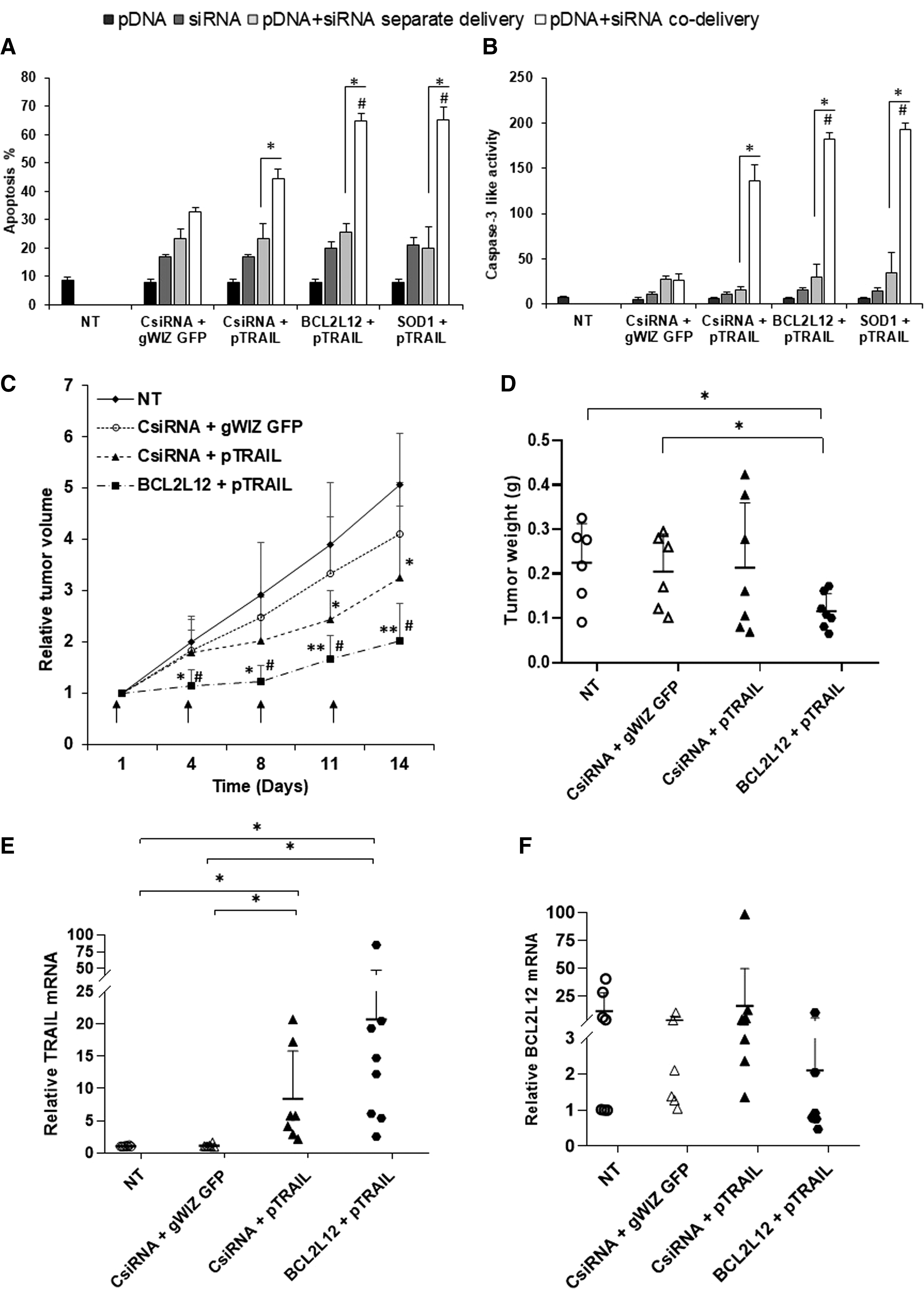
Apoptosis and caspase activation in MDA-MB-231 cells. Cells were treated with combination of pTRAIL and siRNAs (CsiRNA, BCL2L12, or SOD1 siRNAs) as a separate or co-delivery of complexes. Apoptosis
Antitumor activity of pTRAIL/BCL2L12 siRNA delivery in vivo
MDA-MB-231 xenografts were established in mice and treated with combinations of gWIZ-GFP/CsiRNA (as negative control), pTRAIL/CsiRNA (TRAIL therapy alone), and pTRAIL/BCL2L12 siRNA (TRAIL and specific siRNA therapy) at indicated time points (Fig. 6C). Co-delivery of gWIZ-GFP/CsiRNA did not affect the tumor growth (not significantly different from nontreated tumors). Co-delivery of pTRAIL/CsiRNA was effective in significantly reducing tumor volume only after day 11. Co-delivery of pTRAIL/BCL2L12 siRNA showed the most potent response, giving significant tumor retardation compared with nontreatment group after day 4 and to gWIZ-GFP/CsiRNA co-delivery group after day 8 (Fig. 6C). The tumor weights recovered at the end of the study were in line with measured changes in tumor volume, where the lowest tumor weights were observed with the pTRAIL/BCL2L12 siRNA treatment (Fig. 6D). We also assessed TRAIL expression by quantification of mRNA through qPCR analysis; high TRAIL mRNA expression was evident in pTRAIL treatment group, which was significantly higher than the other groups (Fig. 6E). Despite similar level of TRAIL mRNA, pTRAIL/CsiRNA was not as effective as pTRAIL/BCL2L12 siRNA in reducing the tumor volume, which further confirmed the role of BCL2L12 siRNA in sensitizing TRAIL. Although we were not able to observe a significant reduction of BCL2L12 mRNA in the pTRAIL/BCL2L12-treated group compared with untreated group (due to high variations among samples in the study groups), we observed low levels (<1) of BCL2L12 mRNA in most of the pTRAIL/BCL2L12-treated tumors (Fig. 6F).
Discussion
To facilitate TRAIL therapy in a clinical setting, we explored the possibility of co-delivering pTRAIL to enhance in situ availability of the protein and specific siRNAs that sensitize malignant cells to TRAIL both in vitro and in vivo models. Co-delivery of pDNA and siRNA could be a milestone for the treatment of drug-resistant cancers since it can trigger synergistic effects via complementary pathways, which are greater than the sum of the constituent components. Many aggressive heterogeneous cancers where targeting individual signaling pathway have failed to block abnormal proliferation will benefit from this strategy. 30 Identification of functional carriers that can accommodate such nucleic acid combinations is always challenging due to variations in ionic charge density, size, and stiffness of the constituent nucleic acids. Here, we designed specific cationic lipopolymers that are tailored by grafting aliphatic lipids via thioester linkages for this particular purpose. One can independently vary the polymeric backbone, the nature of hydrophobe, and the linkage chemistry of these lipopolymers to make them suitable for co-delivery of nucleic acids. These lipopolymers efficiently accommodated both pDNA (pTRAIL) and siRNAs (BCL2L12 and SOD1 siRNAs) into single complexes. After co-delivery, the complexes resulted in TRAIL expression and TRAIL protein secretion as well as silencing of BCL2L12 and SOD1 at the mRNA level (reductions in protein levels of BCL2L12 and SOD1 remain to be evaluated). The lipid modification of low–molecular weight PEI was implemented with cholesteryl chloroformate initially, 39 after which a broad range of hydrophobic moieties were added to low–molecular weight PEIs to transform it into effective nucleic acid carriers. 39 –42 This is the first study where co-delivery of pDNA and siRNA was reported using a lipid-modified low–molecular weight PEI.
In our previous study, using high-throughput screening, we identified siRNAs against BCL2L12 and SOD1 that sensitized breast cancer cells to TRAIL protein-induced apoptosis. 29 In this study, we assessed the therapeutic potency of pTRAIL and specific siRNAs (targeting BCL2L12 and SOD1) to inhibit breast cancer growth. We were able to enhance cell death in breast cancer cells via simultaneous induction of apoptosis with pTRAIL and inhibition of antiapoptotic proteins with specific siRNAs. More importantly, the co-delivery of pTRAIL and BCL2L12 or SOD1 siRNAs elicited potent anticancer response than the separate delivery at similar doses. Better therapeutic outcome after co-delivery of these nucleic acid was because of (1) improved TRAIL expression due to additive actions of the supplemented siRNAs and (2) sensitization TRAIL action by the co-delivered siRNAs targeting BCL2L12 and SOD1. We previously showed that polyanion additives (e.g., hyaluronic acid) enhanced the release of payload in complexes and resulted in higher transfection. 33 The siRNA in the current complexes also acted in the same way and improved the dissociation of the complexes (Fig. 3D). In line with our findings, pDNA transfection was increased with siRNA when co-delivered with arginine-based PEI 43 and poly(L-lysine) with or without oligomeric sulfadiazine. 44 However, other nonviral delivery systems used to co-deliver pDNA and siRNA such as lipopolymer-coated gold nanoparticles 45 and PEI-modified poly-(lactide-co-glycolic acid) nanoparticles 46 were unable to show beneficial effect of siRNA additive on transgene expression. To co-deliver pDNA and siRNA by lipopolymer-coated gold nanoparticles and PEI-modified poly-(lactide-co-glycolic acid) nanoparticles, layers of pDNA and siRNA were added on the surface of pre-made nanoparticles without affecting compactness of nanoparticles, so that the presence of the second polyanion had no effect on transgene expression.
In addition to enhanced transgene expression, the BCL2L12 and SOD1 siRNAs sensitized the TRAIL activity by facilitating apoptosis induction. The BCL2L12 protein is known to inhibit the caspase-3/7, thereby impeding apoptosis, whereas SOD1 maintains the levels of reactive oxygen species (ROS) under a critical threshold to protect cells from ROS damage and apoptosis induction. 47 Therefore, the co-delivery of pTRAIL and BCL2L12/SOD1 siRNAs generated higher apoptosis via increased caspase-3 activity in MDA-MB-231 cells, which was in line with our previous results where the delivery of BCL2L12 and SOD1 siRNAs with recombinant TRAIL protein substantially induced apoptosis. 29 Despite the absence of caspase-3, MCF-7 can undergo apoptosis via activation of caspase-6 (i.e., independent of caspase-3/7). 48 It was not surprising to observe increased BCL2L12 expression (mRNA) upon pTRAIL treatment since it could facilitate survival of malignant cells in the face of TRAIL threat, 49 but the co-delivery of pTRAIL and BCL2L12 siRNA prevented the increased BCL2L12 mRNA. Upregulation of the prosurvival transcription factor nuclear factor-kappaB by TRAIL in different pancreatic cell lines is another example of TRAIL-induced survival response. 50 Although TRAIL protein has minimal effect on nonmalignant cells, strategies to increase the TRAIL potency may increase the toxicity of TRAIL therapy. This was not the case in this study. Co-delivery of pTRAIL and BCL2L12 or SOD1 siRNAs did not induce any obvious toxicity in nontransformed cells in vitro. While SOD1 is an important regulator of cell damage in all cells, the higher levels of SOD1 in transformed cells 51,52 are likely to enhance the activity of SOD1-specific siRNAs on malignant cells.
Co-delivery of pTRAIL and the specific siRNAs not only enhanced the TRAIL potency in TRAIL sensitive cells but also reversed the TRAIL resistance. Among the cells lines used, MCF-7 was resistant to TRAIL therapy, which was attributed to the higher expression of decoy receptor TRAIL-R4 53 and the defects in apoptosis pathway, such as the lack of caspase-3, minimal caspase-8 expression, and expression of higher antiapoptotic proteins relative to proapoptotic protein. 54 Despite the resistance to TRAIL, the co-delivery of pTRAIL and BCL2L12 or SOD1 siRNA to MCF-7 cells turned out to be as effective as TRAIL treatment in sensitive cells. TRAIL expression (based on mRNA and protein secretion levels) was ∼16-fold higher in MCF-7 cells compared with MDA-MB-231 cells, which could have caused the reversal of TRAIL resistance. The separate delivery of pTRAIL and BCL2L12 siRNA also resulted in significant death in MCF-7 cells, which was increased with the co-delivery of the nucleic acids. Therefore, silencing BCL2L12 adds another mechanism to reverse the TRAIL resistance in MCF-7 cells.
In vivo results further confirmed the potency of pTRAIL and BCL2L12 siRNA co-delivery in retarding tumor growth in a breast cancer xenograft model. We are aware of only two studies that evaluated nonviral delivery systems for co-delivery of pDNA and siRNA in vivo. 46,55 One study explored poly (lactic-co-glycolic acid) nanoparticles to co-deliver SOX9-expressing pDNA and anti-Cbfa-1 siRNA to stimulate chondrogenesis in human mesenchymal stem cells (MSCs) in vivo. 46 In that study, human MSCs were transfected in vitro and transfected cells were administered to mice, which did not rely on the efficacy of delivery systems under physiological conditions. Another study utilized surface-functionalized lipopolymer microparticles for dual delivery of IL-10 siRNA and pDNA vaccines to dendritic cells to modulate T cell responses. 55 Since siRNA was encapsulated and pDNA was coated on the surface of microparticles, siRNA did not affect the transfection efficiency of pDNA. To the best of our knowledge, this is the first in vivo study demonstrating the co-delivery of pTRAIL and BCL2L12 siRNA in breast cancer xenografts, reflecting the limited success of co-delivering pDNA and siRNA with a single carrier in the past. Most importantly, co-delivery enabled us to achieve tumor growth inhibition with low dose of pTRAIL (3 μg per mouse) compared with typical 10 μg per mouse intratumoral dose used in other studies. 37,38 Reducing the dose will be especially beneficial to attenuate the side effects when the TRAIL gene therapy is employed in clinics. Future studies to co-deliver the pTRAIL and BCL2L12 siRNA systemically will expand the scope of the proposed TRAIL therapy.
Conclusions
We describe ternary complexes of lipopolymer/pDNA/siRNA that were more efficient than the binary complexes (lipopolymer/pDNA and lipopolymer/siRNA). Upon co-delivery of pTRAIL and BCL2L12 siRNA in an in vivo model, the growth of breast cancer tumor was significantly retarded, which was the consequences of (1) increased in situ TRAIL protein secretion due to polyanionic siRNA and (2) sensitization of cells to TRAIL action by BCL2L12 silencing. The convergent action of these two mechanisms ultimately induced a stronger apoptotic effect and inhibited the antiapoptotic response, resulting in a promising therapeutic activity in both in vitro and in vivo models. Hence, the dual mode of nucleic acid delivery with a single polymeric carrier may provide a novel framework for nucleic acid combination therapy to potentiate the anticancer activity of TRAIL.
Footnotes
Acknowledgments
The authors are grateful for the technical assistance of Mr. Cezary Kucharksi and Dr. Juliana Valencia Serna (Department of Chemical and Materials Engineering) with the animal studies.
Author Disclosure
R.K.C. and H.U. are founders of a company (RJH Biosciences, Inc., Edmonton, Alberta) intended to commercialize the lipopolymers for transfection.
Funding Information
B.T. is supported by Alberta Innovates Graduate Studentship. This study was supported by an Operating Grant from the Canadian Institutes of Health Research (CIHR) and a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). Equipment support was provided by the Edmonton Civic Employees Charitable Assistance Fund.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.