
Abstract
Phenylketonuria (PKU) is considered to be a paradigm for a monogenic metabolic disorder but was never thought to be a primary application for human gene therapy due to established alternative treatment. However, somewhat unanticipated improvement in neuropsychiatric outcome upon long-term treatment of adults with PKU with enzyme substitution therapy might slowly change this assumption. In parallel, PKU was for a long time considered to be an excellent test system for experimental gene therapy of a Mendelian autosomal recessive defect of the liver due to an outstanding mouse model and the easy to analyze and well-defined therapeutic end point, that is, blood
Introduction
Hyperphenylalaninemia (HPA) also termed phenylketonuria (PKU; OMIM 261600) is an inborn error of metabolism defined as blood
Current treatments for PKU
HPA caused by PAH deficiency can be treated by a low
Although lowering
An enzyme substitution therapy, pegvaliase (PALYNZIQ™; BioMarin, San Rafael, CA), has been approved by U.S. Food and Drug Administration (FDA) in May, 2018, for use in adults with PKU whose blood
Pegvaliase is a PEGylated recombinant Anabaena variabilis phenylalanine ammonia lyase (PAL) administrated by continuous subcutaneous injections that lowers blood
Several other groups have investigated in experimental setups alternatives for delivery of PAL. The group from Leuzzi and coworkers has shown to successfully normalize the blood
Pahenu2/2 mice: a model of human PKU
To explore the potential of novel therapies for PKU, an appropriate animal model that accurately recapitulates the human disease is necessary. The most commonly used mouse model for PKU is the Pahenu2/2
mouse that was first described by McDonald almost 30 years ago.
22
It was generated by random chemical mutagenesis in a Black and Tan Brachyury-mouse strain background using the alkylating agent N-ethyl-N-nitrososurea, which resulted in a missense mutation in exon 7 (p.Phe263Ser), a region that encodes the active site of the PAH enzyme.
23
Although protein levels of mutant PAH are reduced compared with wild type, it can be detected by Western blot in liver and kidney from Pahenu2/2
mice. PAH enzyme activity, nevertheless, is completely abolished.
24
Homozygous Pahenu2/2
animals exhibit blood
Experimental restoration of PAH enzyme activity by gene addition or correction in the homozygous Pahenu2/2 “PKU” mouse model has been the goal of many investigations. Several reviews have previously summarized different strategies for gene therapy in the PKU mouse. 33 –38 In this review, we compiled all the approaches with a focus on the more recent achievements using recombinant adeno-associated viral (rAAV) vectors as well as nonviral naked DNA vectors, genome editing by base editors, and integration of promoter-less PAH–mRNA into the Pah locus (for an overview of all successful approaches thus far see Table 1). Finally, the plans for clinical gene therapy trials in humans are discussed.
Overview of experimental gene therapy for phenylketonuria using the Pahenu2/2 mouse model
Note that all Pahenu2/2 mice were treated as adults.
hPAH, human Pah gene; mPah, murine Pah gene; mcoPah, murine, codon-optimized Pah gene; Gch1, guanosine triphosphate cyclohydrolase I; Pts, 6-pyruvoyltetraphydrobiopterin synthase.
RSV-LTR, Rous sarcoma virus long-terminal repeat; CBA, CMV (cytomegalovirus) early enhancer/chicken β actin promoter; EF, human elongation factor 1-α promoter; CMV, cytomegalovirus promoter; LPS; liver-specific promoter is a combination of two copies of a human a1-microglobulin/bikunin enhancer and the promoter from the human thyroid hormone-binding globulin gene 48 ; LP1 promoter consists of the human apolipoprotein E/C-I hepatic control region and the human α1-antitrypsin promoter 50 ; P3, synthetic liver-specific promoter 61 ; U6, RNA polymerase III promoter.
i,p., intraportal vein infusion; i.v., intravenous tail vein infusion; i.m., intramuscular (M. gastrognemius); i.p, intraperitoneal injection; HTV, hydrodynamic tail vein injection.
Physiologic Requirements of Successful Liver-Directed Gene Therapy in PKU
As already mentioned, the liver is not affected by high
Liver Gene Therapy with Viral Vectors
An early approach of using a recombinant retrovirus vector was able to induce PAH activity in PAH-deficient mouse hepatocytes in vitro, but no in vivo studies have been reported thereafter.
41
Two independent studies showed that liver-directed gene therapy using an adenovirus vector corrected serum
Recombinant adeno-associated virus vector for PKU
rAAV is currently the favored vector system for safe and effective liver-directed gene addition, with active clinical trials ongoing for adults with Hemophilia A or B, ornithine transcarbamylase deficiency, glycogen storage disease type 1A, acute intermittent porphyria, Crigler–Najjar syndrome, and others.* rAAV-mediated liver-directed gene therapy for PKU has been explored preclinically by several investigators using the Pahenu2/2 mouse model for ∼20 years. A complete review of the history of rAAV liver gene therapy is beyond the scope of this review, so we will necessarily focus upon the development of this treatment approach specifically for PKU.
Attempts at liver-directed gene therapy using early rAAV vectors were likely impaired by the problem of poor transduction frequency and consequently incomplete correction of HPA. Administration of a PAH-expressing rAAV2 serotype 244 or serotype 545 yielded correction of blood
Pseudotyping of rAAV2 genomes with capsid proteins from alternative rAAV serotypes alters tissue tropism and enhances therapeutic gene expression.
47
The discovery and implementation of rAAV2 serotype 8 vectors were a major breakthrough for AAV-mediated liver-directed gene therapy as the AAV8 capsid shows the greatest affinity for and yields the best transduction frequency in rodent liver of any available AAV serotype. rAAV2/8 vectors expressing either murine or human PAH-cDNAs have been administered by either tail vein, portal vein, or intraperitoneal injection to successfully treat murine PKU by 3 different laboratories.
48
–50
Administration of as few as 5 × 1011 vg rAAV2/8 expressing the murine (m) Pah-cDNA under the transcriptional control of a strong liver-specific promoter through portal vein injection yielded complete correction of blood
In all reported successful trials, the hypopigmentation associated with chronic HPA in the mice has been fully reversed after rAAV2/8-mediated liver-directed gene therapy (see Fig. 1). 51 Later, direct comparison of serotypes 1, 2, and 8 vectors demonstrated the unequivocal superiority of serotype 8 vector for the treatment of Pahenu2/2 model through liver-directed gene therapy and, furthermore, it was demonstrated that even simple intramuscular injection of rAAV2/8 vector can lead to trafficking of vector to and transduction of liver with robust PAH expression and correction of HPA. 52
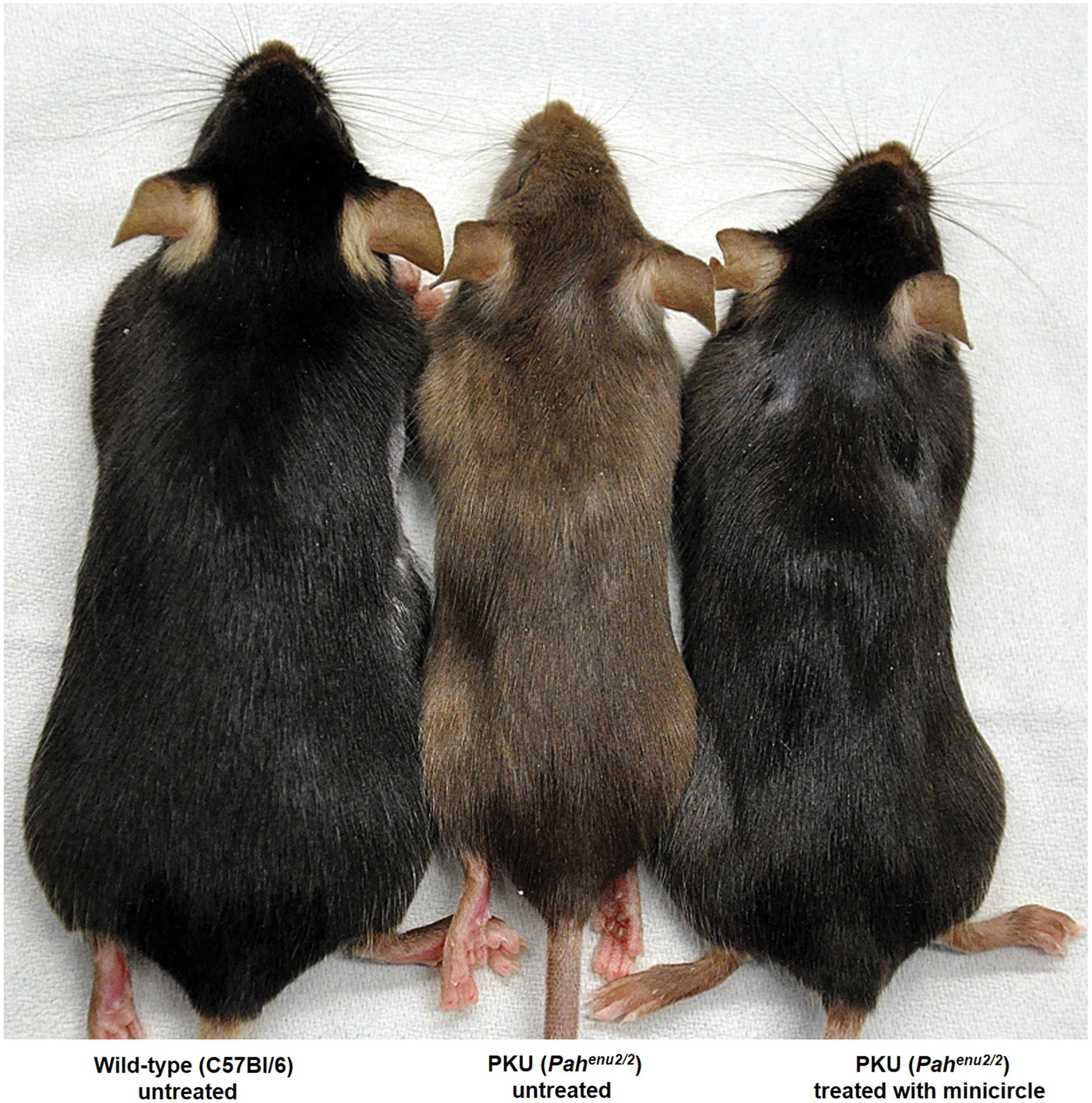
Phenotype reversion of fair hair upon gene therapy of the PKU mice. Reversion from brown to black coat of treated (C57Bl/6) PKU mice as depicted in the figure is a phenomenon that can be seen from gene therapy or any other treatment, including dietary or pegvaliase treatment. Not only mice but also patients with untreated PKU exhibit blond hair and fair skin. Tyrosine that is needed to make melanin, the pigment that gives skin and hair its coloring, is limited under untreated PKU conditions and the corresponding enzyme, tyrosinase, is competitively inhibited by elevated phenylalanine levels. The figure is reproduced with permission from Viecelli et al. 57 wherein mice were treated with nonviral naked DNA vector expressing phenylalanine hydroxylase on the right, wild type mice untreated on the left, and PKU mice untreated in the middle. PKU, phenylketonuria. Color images are available online.
The Kume laboratory constructed a self-complementary (sc) AAV2/8 vector that expressed the mPah-cDNA using a compact enhancer/promoter cassette (a human apolipoprotein E/C-I hepatic control region fused to the human alpha-1-antitrypsin promoter) and demonstrated improved efficacy at lower doses than a comparable single-stranded AAV2/8 vector that included the cytomegalovirus (CMV) immediate–early enhancer/chicken β-actin promoter.
50
Blood
A novel set of proprietary AAV vectors derived from human CD34+ hematopoietic stem cells (AAVHSCs) from Homology Medicines, Inc. (Bedford, MA) containing a promoter-less human (h) PAH-cDNA has recently been reported to correct HPA in Pahenu2/2
mice after intravenous tail vein injection.
54
The hPAH-cDNA was integrated into the mPah locus through homologous recombination. As a result, blood
An entirely different approach is the targeting of skeletal muscle for PKU gene therapy. The systemically elevated
Liver Gene Therapy with Nonviral Vectors
The development of nonviral gene delivery systems as an alternative to viral vectors has gradually gained attention due to the potentially better safety profile and low production costs. The recent FDA approval of patisiran (Onpattro™; Alnylam Pharmaceuticals, Cambridge, MA), the first siRNA drug to use RNA interference to downregulate protein as a treatment for polyneuropathy in adults with hereditary transthyretin-mediated amyloidosis, is a breakthrough in the drug discover field. This drug is encapsulated in lipid nanoparticles and N-acetylgalactosamine for efficient and specific delivery to the liver. 56 DNA-mediated gene transfer into tissues or organs is more preferable for long-term treatment for monogenic metabolic diseases but generally suffers from lower rates of cell transduction and transient therapeutic responses.
The Thöny laboratory has reported on persistent and efficacious treatment of PKU mice through hydrodynamic delivery of nonviral naked minicircle (MC)-DNA vectors without adverse effects.
57
MC-DNA vectors are a form of supercoiled DNA for use in nonviral gene transfer that contain a minimal expression cassette and from which all bacterial DNA is removed by intravector recombination.
58
Studies have shown that bacterial DNA contributes to biological safety problems and transgene silencing.
59,60
MC-DNA vectors (3 × 1013 vg/mouse) expressing murine PAH from a synthetic liver-specific promoter (designated as P361) yielded therapeutic PAH activity accompanied by complete revision of hypopigmentation in the PKU mouse model (Fig. 1) for 1 year after liver gene transfer by a single hydrodynamic tail vein injection.
57
At the same time, there was no reduction of
Since there is no limitation of package capacity of the DNA insert, the expression cassette consisting of a 3.6-kb native endogenous Pah promoter/enhancer sequence, codon-optimized mPah–cDNA, and a truncated intron was included in the MC-DNA vector. 62 The efficacy of this improved expression cassette of MC-DNA vectors resulted in a significant reduction in vector dosage (6 × 1012 vg/mouse) required for successful treatment. Analysis of treated mice upon sacrifice revealed that statistically <1 copy of MC vector was present per diploid hepatocyte genome in a whole liver. The loss of MC-DNA vectors and PAH activity after partial hepatectomy and liver regeneration confirmed that MC-DNA vectors do not integrate into the genome and remain episomal. However, a limiting factor for human usage and treatment of PKU remains delivery of naked DNA vectors to liver (and other tissues).
In summary, naked DNA-based therapeutic tools are still in its infancy compared with the much more advanced AAV vector field, but nonviral alternatives for gene delivery are receiving growing attention and it is anticipated that this trend will continue in the future.
Liver Gene Therapy by Gene Editing
In recent years, a number of studies have implemented genome editing for the treatment of animal models of genetic liver diseases. 63 It has the advantage compared with gene addition approaches that endogenous loci can be corrected. Thus, temporary intervention enables permanent repair and expression of the functional gene under its endogenous promoter.
The most widespread genome editing tool is the CRISPR/Cas9 system, where a programmable chimeric guide RNA targets the Cas9 endonuclease to the locus of interest.
64,65
If generated DNA double-stranded breaks (DSBs) are restored by homology-directed repair (HDR) from an exogenous template DNA, mutations are repaired with single base precision. However, although precise repair is essential for correcting autosomal recessive disorders such as PKU, hepatocytes in the adult liver predominantly repair DSBs through nonhomologous end-joining pathways, resulting in deletion or insertion (indel) mutations.
66,67
Thus, when CRISPR/Cas9 was applied to adult mouse models for ornithine transcarbamylase deficiency, hereditary tyrosinemia, and hemophilia B, repair rates were extremely low, with 2%, 0.4%, and 1%, respectively.
68
–70
Unfortunately, such low repair rates would likely not be sufficient to reduce blood
Recently a novel CRISPR-based genome editing tool that enables direct conversion of single bases has been developed. These so-called base editors are chimeric proteins that consist of nuclease-impaired Cas9 fused to catalytic domains that enable DNA deamination. Cytidine base editors enable single C·G to T·A base pair conversions through uracil intermediates, and adenine base editors enable single A·T to G·C base pair conversions through inosine intermediates. 75,76 Since base editors enable precise exchange of single base pairs independently of DSBs formation and HDR, they also enable efficient base editing in postmitotic cells such as adult hepatocytes. 77 –79
In a recent study, cytidine base editors have been employed to correct the G to A missense mutation in the Pahenu2/2
mouse model.
80
rAAV-mediated delivery of base editors into the liver resulted in repair rates of the target base >20%, and a reduction of blood
Trends in the Pharmaceutical Industry for Clinical Gene Therapy Trials in PKU Patients
As of this writing (May, 2019), no gene transfer into humans for the purpose of treating PKU has yet been attempted, but several pharmaceutical companies have publicly announced their intentions to do so in the very near future employing either rAAV- or lentivirus-mediated gene addition strategies.
Homology Medicines, Inc. has presented preclinical data in abstract form demonstrating sustained correction of blood
BioMarin Pharmaceutical, Inc. has publicly announced its plans to investigate gene addition in adults with PKU using an rAAV2/5 vector expressing human PAH. BioMarin scientists are completing the preclinical validation of their PAH-expressing rAAV2/5 vector and will be applying for an IND soon.
In any rAAV-mediated gene addition trial, the majority of therapeutic gene expression will be from episomal vector genomes residing within hepatocytes. Any stimulus to hepatocyte regeneration would threaten the long-term stability of expression, and truthfully the ultimate duration of therapeutic gene expression after a single IV infusion of a liver-directed rAAV2 vector to an adult human is unknown, although robust Factor IX expression has been measured in scAAV2/8-treated individuals >10 years from their initial injection. In addition, the efficacy of this gene addition approach is only temporary in juvenile animals during rapid hepatocyte proliferation due to episomal dilution. 85 –87
As detailed elsewhere, direct gene editing or the use of integrating vectors avoids this issue, but these approaches all struggle to achieve editing or integration frequencies (in the absence of a selective growth advantage for PAH expressing hepatocytes) sufficient to yield a physiologically relevant PAH positive cell population. However, American Gene Technologies, Inc. (Rockville, MD) has publicly announced that it is developing a liver-directed gene therapy strategy based upon a proprietary integrating lentivirus-based vector system. Its vector has been granted Orphan Drug status by the FDA (no. DRU-2018-6572), but the company has not released any preclinical data or other information regarding its progress toward a clinical trial.
Outlook
Gene addition is for some rare monogenic disorders where no good alternative treatment is available, becoming a reality for patients with severe genetic defects. Furthermore, for in vivo approaches to target, for example, the liver, AAV vectors are currently the most effective choice. Owing to the long history of successful dietary treatment, if started early in life, PKU was not considered to be a primary target for a genetic treatment. On top of that, newborn screening programs implemented today in most countries for PKU guarantee early diagnosis and intervention. It is acknowledged today that treatment from early in life with dietary therapy has protected adults with PKU from permanent neuropathologic damage associated with chronic HPA. Adherence to diet to keep blood
Since most of these symptoms should be reversible upon successful correction of blood
Whether gene therapy for PKU with the current technology and experience available is also an option in other countries such as in Europe or in Asia remains to be seen. Intravenous administration of AAV vectors has proven safe in several clinical trials and evidence of efficacy has also been seen for many. A critical question to be answered is which serotype will be most efficacious at which minimally effective dose, and whether the treatment effect will be permanent. The goal of course is sustained disease correction over many years without the need for redosing. However, in children with PKU, this approach will likely not be successful, as episomal AAV genomes are known to not be stably maintained in growing liver of juvenile mammals. 85,86
Solutions for permanent cure in, for example, pediatric patients with a growing liver are either integration or addition of replication-competent gene copy, or correction of the disease-causing mutation. Such future approaches may include integration of a PAH-cDNA through AAV-directed homologous recombination, 54 random integration of PAH-cDNA through liver-directed lentiviral vectors, 89 –91 hepatocyte transfection with replicating nonviral vectors, 92 –94 or genome editing with base editors through liver-directed AAV vectors or lipid-nanoparticles for mRNA delivery. 80,94 Although some of these technologies have proven therapeutic in the PKU mouse model, they are still at its developing level and need to be improved for efficacy and/or assessed for safety profiles. We look forward to a series of robust clinical trials, which hopefully will address pitfalls of current PKU therapies.
Footnotes
Author Disclosure
C.O.H. has received consulting fees or funds in support of clinical or preclinical research from BioMarin Pharmaceutical, Inc., Ultragenyx, Inc., Horizon Pharmaceutical, Synlogic, Inc., Rubius Therapeutics, Cydan, Inc., StrideBio, Inc., Voyager Therapeutics, and Pfizer, Inc. All other authors have no conflict of interest.
Funding Information
Our study was funded by Forschungszentrum für das Kind (to H.M.G-C.), the Swiss National Science Foundation (3100A0-105250 and CRSII5_180257/1 to B.T., 31003A_160230 to G.S.), Wolfermann Nägeli Stiftung (to B.T.), and NIH (R01NS080866 to C.O.H.).