
Abstract
Methylmalonic acidemia (MMA) is a severe, and sometimes lethal, monogenic metabolic disorder in need of improved treatments. A number of new genomic therapies, which include canonical adeno-associated virus gene addition, genome editing, and systemic mRNA therapy, have shown great promise in murine models of MMA. Each approach has unique advantages and disadvantages for treating genetic disorders like MMA. This article reviews traditional viral gene therapy experiments that have provided enabling proof of concept studies in animal models, and newer approaches that may emerge as effective treatments for MMA and related disorders of organic acid metabolism.
Introduction
Isolated methylmalonic acidemia (MMA) is an autosomal recessive inborn error of metabolism caused by defects in the metabolism of methylmalonate and/or 5′-deoxyadenosylcobalamin that result in elevation of methylmalonic acid in all the body fluids. 1 Deficiency of the mitochondrial localized enzyme methylmalonyl-CoA mutase (MMUT, formerly designated MUT) causes isolated MMA, with mutations in the MMUT most commonly identified as the cause of the disorder. The MMUT enzyme has an obligate requirement for 5′-deoxyadenosylcobalamin, a form of vitamin B12, and catalyzes a critical step in the metabolism of branched chain amino acids, odd chain fatty acid, gut-derived propionate, and cholesterol. Because the intracellular transport and metabolism of cobalamin requires many enzymes, MMA exhibits genetic heterogeneity. An overview of methylmalonyl-CoA metabolism is given in Fig. 1.
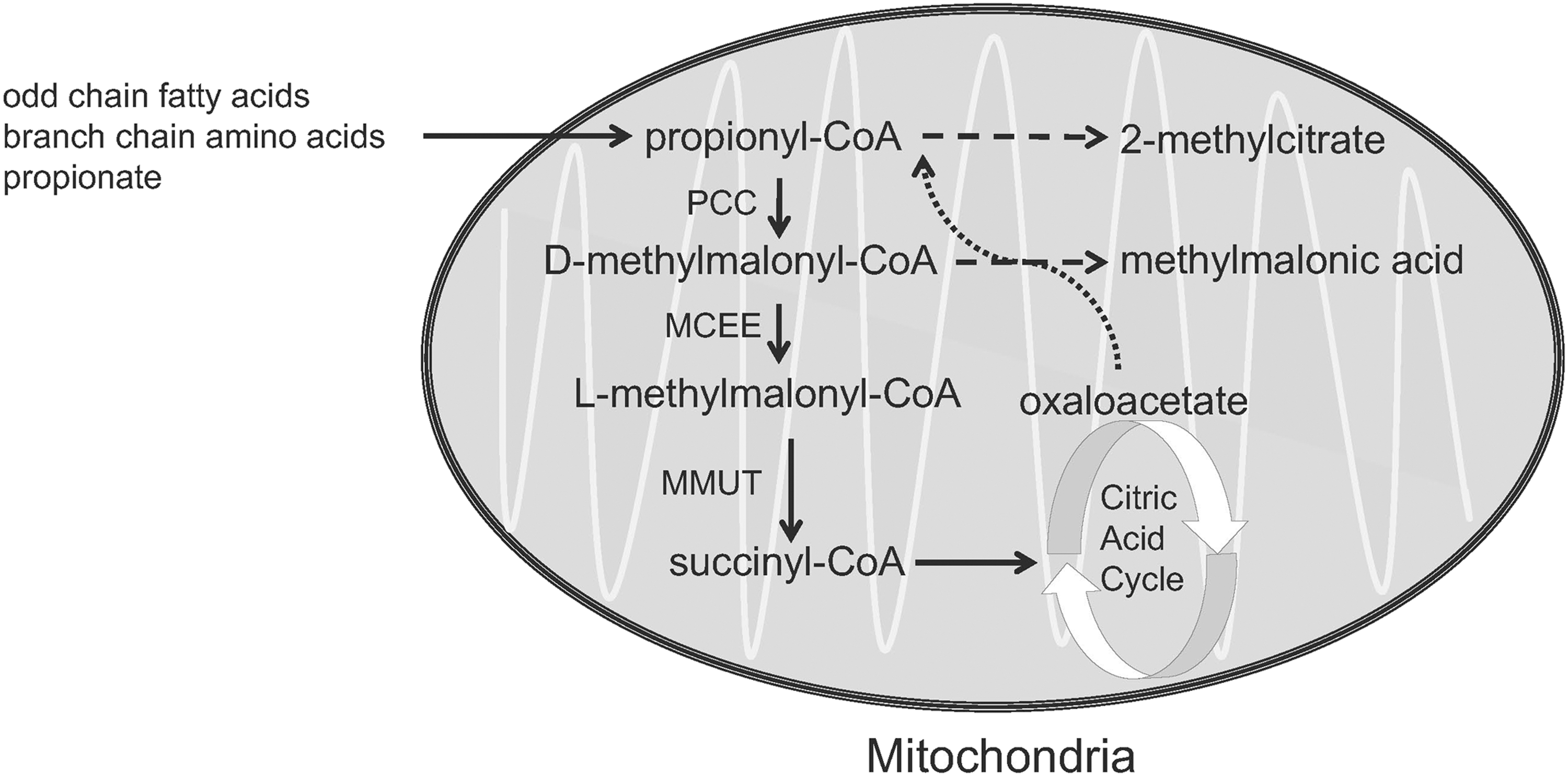
Major pathway of the conversion of propionyl-CoA into succinyl-CoA, which occurs in the mitochondria. The biotin-dependent enzyme PCC converts propionyl-CoA into
Patients with the most severe form of MMUT deficiency, sometimes described by the complementation class as mut 0, typically experience lethargy, vomiting, hypothermia, respiratory distress, severe ketoacidosis, and hyperammonemia in the newborn period. If successfully rescued from such an initial metabolic decompensation, the patients are typically treated with a protein-restricted diet, assessed for responsiveness to cobalamin, and treated symptomatically. However, despite meticulous medical and dietary management, patients with all forms of isolated MMA, especially MMUT deficiency, can experience severe and potentially lethal metabolic instability, and require frequent hospitalizations for emergency support. The multisystemic complications of MMA add additional challenges to medical management, and increase the risk for acute mortality and long-term morbidity.
The recalcitrant nature of mut MMA to conventional management with dietary restriction of protein and cofactor support has led to the use of elective liver transplantation as a surgical treatment. Although the MMUT enzyme is expressed and present in many tissues, the hepatic activity of MMUT is the most important source of enzyme in the body, as demonstrated by the observations that metabolic stability is restored in patients after successful liver transplantation. Numerous animal models and proof-of-concept gene therapy experiments provide addition support for the critical role of the liver in cobalamin and methylmalonyl-CoA metabolism. 2,3 Despite the persistence of elevated levels of metabolites after successful organ transplantation, several studies have reported clinical improvement of MMA patients after liver replacement. 4 –6 Because liver transplantation is an invasive procedure, can be associated with significant procedural morbidity, and requires life-long immunosuppression for the recipient, the community has pursued alternative approaches to the treatment of MMA. In this article, we review the various preclinical gene-based therapies that have been tested in cellular systems and murine models, and highlight future approaches that will further expand the scope of new treatments for MMA.
Proof of Principal in Vitro Gene Transfer
Gene therapy has been postulated as a potential treatment for monogenic disorders, including MMA, for decades. 7,8 Early proof of principal experiments delivered murine Mmut or human MMUT by chemical transfection or viral infection to human and murine cell lines deficient in methylmalonyl-CoA mutase activity. 7,9,10 In fact, two decades ago, Chang et al. transduced MMUT-deficient T cells with a recombinant retrovirus encoding the human MMUT cDNA, and noted correction of propionate metabolism and proposed haematopoietic cell-based metabolic sink therapy for mut MMA by T lymphocyte/haematopoietic stem cell-directed gene transfer. 10 Although this approach has not been more formally tested, recent in vitro studies have demonstrated that antisense morpholino oligonucleotides can correct splicing defects in MMUT-deficient patient fibroblasts, expanding the scope of gene modulation approaches as a treatment for MMA. 11 In aggregate, the cellular experiments clearly show that in cellular systems restoring methylmalonyl-CoA mutase expression, by a number of approaches, can restore enzymatic activity in vitro. However, the question of whether in vivo gene therapy could be beneficial in MMA, a multisystem disease caused by the loss of a ubiquitously expressed mitochondrial enzyme, remained unexplored until the first murine models of MMA were developed.
Murine Models of MMA
The first knockout MMA mouse models had similar phenotypes, and replicate the most severe end of the human spectrum of mut 0 type MMA. Mmut −/− mice are born normally, in Mendelian proportions, and indistinguishable from healthy littermates at birth, but become lethargic with gasping breath after the first day of life, with uniform lethality by the second day of life. 12,13 Mmut −/− mice uniformly perished by 48 h of age, and lack methylmalonyl-CoA mutase mRNA, enzyme and activity. Of great significance, the animals had massive increases of methylmalonic acid and methylcitric acid in the blood and urine. At the end of life, the measured methylmalonic acid content in organs of Mmut −/− mice was highest in the liver, followed by skeletal muscles, kidney, and brain. 12 These measurements highlight the potential additional role of skeletal muscle in production of metabolites, and provided motivation for the further creation of tissue-specific transgenic Mmut animal models.
To improve the survival of the mut 0 model, a background-modified Mmut −/− knockout mouse model was developed. 14 The generation of triply mixed [(C57BL/6x129Sv/Ev) × FVB/N] Mmut −/− mice with increased survival compared with the parental strain made it possible to generate rare Mmut −/− mice that could survive until weaning. Ultrastructural studies of hepatocytes in surviving Mmut −/−mice revealed megamitochondria in the hepatocytes, proximal tubules, but not in the heart or skeletal muscle. Mitochondrial abnormalities similar to those seen in the mouse livers and kidneys were also noted in the livers and kidneys of patients with mut 0 MMA. 2,14 –18 Megamitochondria formation is therefore a characteristic feature of the ultrastructural pathology seen in MMA, and exhibits cell specificity.
To critically examine the benefits of liver-specific expression of methylmalonyl-CoA mutase in MMA, a liver-directed transgenic MMA model where mice express hepatic Mmut from the mouse albumin promoter was generated. 2 Mmut −/−;TgINS-Alb-Mut mice were protected from the neonatal lethality that characterized the Mmut −/− mice; low-level Mmut expression in liver restored growth to levels comparable with that seen in healthy heterozygous littermates. After challenge with a high-protein diet, Mmut −/−;TgINS-Alb-Mmut mice manifested decreased kidney function and formed megamitochondria in the proximal tubules but transgenesis corrected the mitochondrial abnormalities seen in the face of massively elevated plasma methylmalonic acid concentrations, suggesting that the hepatic mitochondriopathy of MMA can be rescued by low-level expression of Mmut.
To study the hepatorenal pathology of MMA and create a model for the testing of liver-targeted therapies, a mouse model expressing methylmalonyl-CoA mutase in the skeletal muscle was next generated. 15 Mmut −/−;TgINS-MCK-Mmut are protected from the neonatal lethality associated with Mmut deficiency because of transgenic overexpression of Mmut in the skeletal and cardiac muscles, but are fragile and remain significantly smaller than their littermates throughout their life span. These MMA mice display normal activity, but are sensitive to stress and can exhibit substantial postweaning mortality. 19 In addition to serving as a tool for liver-directed gene therapy, Mmut −/−;TgINS-MCK-Mmut mice have been instrumental in the discovery and validation of a number of hepatic biomarkers of MMA, including the key metabolic regulator, fibroblast growth factor 21 (Fgf21). 20 –22
The murine mouse models described previously, as well as several others, 23 have provided a platform to test the efficacy of systemic- and hepatic-targeted therapies for MMA that are given in Table 1.
Gene therapy experiments in murine models of methylmalonic acidemia
AAV, adeno-associated virus; CBA, cytomegalovirus enhancer chicken beta-actin promoter; CMV, cytomegalovirus immediate-early enhancer and promoter; hAAT, APOE enhancer human alpha-1 antitrypsin; HIV-1, human immunodeficiency virus type 1; LNP, liponanoparticle; MCK, muscle creatine kinase; Mmut, murine methylmalonyl-CoA mutase; MMUT, human murine methylmalonyl-CoA mutase; pMMA, plasma methylmalonic acid; PFUs, plaque-forming units; rAAV, recombinant adeno-associated virus; rAAV8, rAAV serotype 8; vg, vector genomes; TBG, thyroid hormone-binding globulin.
Adenoviral Gene Therapy
The first successful in vivo gene therapy experiments were performed using an early generation (E1, E3 deleted) adenovirus type 5 (Ad5). 24 These studies showed that an adenovirus, configured to express the murine Mmut cDNA (2.3 kb) under the control of the cytomegalovirus (CMV) promoter, when delivered as an intrahepatic dose of 3.3 × 108 plaque-forming units of Ad5 at birth, could rescue the neonatal lethal phenotype of the MMA knockout mice. More than 50% of the Ad5-treated Mmut −/− mice survived for 15 days, with one mouse surviving for 8 months. Of interest, very little Mmut expression was needed to stabilize the treated Mmut −/− mice; hepatic Mmut RNA expression in Mmut −/− mice was ∼5% of wild-type expression in mice 19–25 days post-treatment. The treated MMA mice exhibited a temporal loss of transgene expression and eventual died, possibly because of dilution of episomal Ad transgenes during hepatic growth after neonatal delivery. The Ad gene delivery experiments provided a convincing demonstration of the in vivo potential for viral gene delivery as a treatment for MMA and were accompanied by a separate in vitro gene therapy study using hepatocytes from a patient with mut 0 MMA. However, the recognized toxicity associated with Ad-based gene therapy in humans, and transient expression noted in the MMA mice, stopped further development of Ad vectors to treat MMA. 25
Canonical Adeno-Associated Virus Gene Therapy
The promising but transient success observed with adenoviral gene delivery for MMA led to exploration of recombinant adeno-associated virus (rAAV) as a vector for MMA gene therapy. rAAVs were selected because they were successfully used as gene delivery vehicles in numerous animal models of human disease, and have yielded long-term transgene expression without vector-related toxicity. 26 Because rAAV vector genomes (vg) remain largely episomal and have a very low genomic integration frequency, the risk of insertional mutagenesis by rAAVs is theoretically low compared with integrating vectors, such as retroviral vectors. In addition, numerous natural and engineered AAV serotypes, some with distinct tissue tropism, further expand this vector as a platform for preclinical studies.
The positive outcomes experienced by MMA patients after liver transplantation, and the earlier success of treating MMA mice with intrahepatic injections of Ad5, led to the selection of an rAAV serotype 8 (rAAV8), which has a strong liver tropism, for the first rAAV gene therapy experiments (Table 1). 27,28 In addition, rAAV8 is known to efficiently transduce skeletal muscle, which generates substantial amounts of methylmalonic acid in MMA. 12,15 The first therapeutic transgene packaged into the rAAV8 used a combination CMV enhancer and a ubiquitous chicken-beta actin promoter to express the murine Mmut cDNA. These experiments, performed exactly as the Ad5 studies, showed that an rAAV8 vector could rescue the neonatal mouse model of MMA for up to 2 years, lowering methylmalonic acid levels and improving the growth of treated versus untreated Mmut −/− mice. Although methylmalonic acid levels were lower than those of untreated Mmut −/− mice, the serum metabolites in the treated MMA mice were still markedly elevated compared with wild-type littermates. However, the older treated mice had robust whole body 1- 13 C propionate oxidation, and Mmut RNA expression in a broad spectrum of tissue types that included the liver, skeletal muscle, cardiac muscle and brain. Of importance, AAV8 proved superior to Ad5 at treating the neonatal lethal model of MMA.
A later report of rAAV gene therapy in the Mmut −/− mice tested an rAAV9 gene delivery to treat MMA. 29 Like rAAV8, rAAV9 has a strong liver tropism but is more efficient than rAAV8 at transducing extrahepatic tissues such as the brain and kidney. 28 The rAAV9 vector utilized the identical cassette as in the initial rAAV8 gene therapy proof of principal study to treat MMA. The results of this study indicated that rAAV9 was as effective as rAAV8 at providing a sustained therapeutic benefit. Of note, this study showed that hepatic Mmut protein expression reached wild-type levels of expression levels at 24 h after rAAV9 delivery and levels >50-fold that of wild-type expression at 72 h post-rAAV9 treatment. Hepatic Mmut diminished to wild-type levels by 2 months post-rAAV9 injection, most likely caused by the dilution of episomal AAV transgene during liver growth after neonatal gene delivery. 30 Of interest, rAAV9 was capable of transducing the kidney and produced low levels of Mmut expression in the kidney. Achieving renal Mmut expression may be beneficial for the treatment of MMA as patients can develop kidney failure. This study also showed that redosing of rAAV9 1 year after neonatal rAAV9 treatment increased Mmut expression and activity, suggesting that the neonatal rAAV9 treatment of Mmut −/− mice tolerized them to rAAV9.
The second rAAV8 gene delivery study in Mmut −/− mice tested a liver-specific expression approach to treat the MMA mice. 3 The rAAV8 was packaged with a transgene that used a microglobin/bikunin enhancer with a liver-specific thyroid hormone-binding globulin to express the murine Mmut cDNA and was effective at rescuing the neonatal model of MMA, providing long-term survival and reduction of metabolites. Although not directly compared, the efficacy of rAAV8 using a liver-specific promoter was similar to the results observed after rAAV8 treatment of Mmut −/− mice with a ubiquitous promoter. 27
The third reported rAAV8 gene delivery experiments examined dosing in newborn Mmut −/− pups. 31 This study was conducted using rAAV8 to deliver the human MMUT coding sequence expressed under the control of a ubiquitous chicken beta-actin promoter. Mmut −/− mice that received a dose of 1 × 109 vg/pup (∼7 × 1011 vg/kg) at birth escaped neonatal lethality but died between 1 and 2 months of age, whereas Mmut −/− mice that received a dose of 1 × 1010 vg/pup (∼7 × 1012 vg/kg) at birth survived for over a year. These findings show that a dose of ∼7 × 1011 vg/kg of rAAV8 approaches the lower limit of AAV required for the rescue of Mmut −/− mice from neonatal lethality, and suggest that a dose closer to 7 × 1012 vg/kg will be required for a sustained therapeutic effect. This study also reported the first rAAV gene therapy experiments that used the human MMUT coding sequence to treat Mmut −/− mice, and established that neither a species barrier to mitochondrial processing nor an apparent immune response to MMUT impedes the use of MMA mouse models to test the efficacy of human gene therapy vectors for MMA.
The most recent report of liver-specific rAAV8 gene delivery tested gene delivery in juvenile transgenic mouse model of MMA, Mmut −/−;TgINS-MCK-Mmut. 15 The rAAV8 was configured to express a codon-optimized human MMUT cDNA under the control of the ApoE-enhanced, human α 1 antitrypsin promoter/enhancer combination. rAAV8 was systemically delivered through a retro-orbital injection at a dose of 5 × 1012 vg/kg to Mmut −/−;TgINS-MCK-Mmut mice at weaning. The treated Mmut −/−;TgINS-MCK-Mmut mice showed substantial weight gain, an increase in whole body 1- 13 C propionate oxidation, and a prolonged metabolic stability after gene therapy, relative to untreated mutant controls. A novel MMA-associated serum biomarker, Fgf21, was found to be significantly decreased in the Mmut −/−;TgINS-MCK-Mmut mice after rAAV8 treatment. Fgf21 is a peptide hormone that is synthesized mainly by the liver, and is involved in the regulation energy homeostasis and oxidative stress. The results support the use of serum Fgf21 along with whole body 1- 13 C propionate oxidation as additional biomarkers to measure the efficacy of successful hepatic correction of MMUT deficiency.
Limitations and Caveats of Canonical r AAV Gene Therapy
Although a number rAAV gene delivery studies for MMA in murine models have shown impressive efficacy, rAAV gene delivery for MMA has not been without complications or limitations. Genotoxicity in the form of hepatocellular carcinoma (HCC) formation was observed after rAAV neonatal treatment of MMA mice and wild-type controls, similar to that observed in other murine neonatal gene delivery studies. 32 –35 A genome-wide survey of integrations in the rAAV HCC compared with control tissue confirmed a previously identified association between insertional mutagenesis of the Rian locus by the rAAV vector and upregulation of genes proximal to the rAAV insertion site by the enhancer–promoter of the rAAV. Of interest, we observed that not all enhancer–promoter combinations produced the same level of genotoxicity, suggesting that vector design could reduce the genotoxicity associated with rAAV integration. Furthermore, the rAAV HCC integrations tended to cluster in a specific microRNA, MiR341, which is present only in mice and rats. A genomic analysis of this sequence showed a simple quadranucleotide repeat, and had a very high CpG content. Whether this element can act as a nidus or substrate for AAV recombination remains to be further studied, but it seems possible given the uniqueness of the small genomic area.
Another consideration in the generalization of post-treatment sequalae after neonatal murine gene therapy surrounds how the physiology of the neonatal mouse liver may influence the translation of the results. For example, the AAV HCC sites frequently targeted fall into a locus (Rian) that is highly expressed in the neonatal mouse liver, but not at later points. 36 Whether a similar expression pattern will be seen in patients seems unlikely. Studies of rAAV integrations in humanized mice might help resolve this issue.
Furthermore, rAAV gene delivery in the neonatal period for MMA is followed by a rapid temporal decease in the vector copy number and transgene expression, which can result in a complete loss of efficacy as we documented in the dosing studies with the rAAV8 MMUT vector. 30,31 As early treatment of MMA will afford patients the best possible outcome, overcoming the difficulties associated with rAAV gene delivery early in life to provide life-long transgene expression are paramount for the treatment of MMA, and many other inborn errors of metabolism.
A final limitation is that rAAV gene therapy experiments for MMA have been performed with capsids that are known to display preexisting immunity. Although studies have shown that the MMA patient population has a low incidence of seropositivity to AAV2, -8 and -9 capsids, some have preexisting immunity, and therefore may not be candidates for systemic AAV gene therapy with these serotypes. 37 The use of immunomodulation and novel capsids may circumvent immune responses to AAV, and expand the eligible patient population.
Lentiviral Gene Therapy
Unlike AAVs, lentiviruses are integrating vectors and enable persistent transgene expression at stable levels in growing tissues, which could be advantageous for gene delivery in the neonatal period. In the setting of MMA, Wong et al. constructed a human immunodeficiency virus type 1 (HIV-1) lentivirus to deliver a human codon-optimized MMUT sequence under the transcriptional control of the EF1-α gene promoter. 38 A mouse model of methylmalonic aciduria (Mut −/− MUT h2) 23 was injected intravenously at 8 weeks of age with a dose of 130 μg p24 equivalent of HIV-1. HIV-1-treated Mut −/− MUT h2 mice showed improved growth, increased MMUT protein expression, and reduced plasma methylmalonic acid levels supporting the potential use of integrating vectors to treat MMA.
mRNA-Based Treatments for MMA
Unlike, rAAV gene delivery, lipid nanoparticle (LNP)-mRNA gene therapy is not limited by preexisting immunity and has a very low risk of genotoxicity because the cargo does not reach the nucleus. In addition, LNP-mRNA can be redosed, unlike rAAV. An et al. used LNP to deliver pseudoU-modified codon-optimized mRNA-encoding human methylmalonyl-CoA mutase (hMMUT) to two murine models of MMA. Mmut −/−;TgINS-MCK-Mmut mice were treated with a systemic dose of 0.2 or 0.5 mg/kg LNP-MMUT mRNA. 19 The LNP-mRNA–treated Mmut −/−;TgINS-MCK-Mmut mice had decreased plasma methylmalonic acid plasma levels compared with untreated mice for up to 10 days, but by 14 days postinjection the methylmalonic acid plasma levels rebounded. Repeat dosing of LPN-MMUT mRNA in the Mmut −/−;TgINS-MCK-Mmut was able to increase growth, reduce plasma methylmalonic acid levels, and increase survival in comparison with untreated controls. Of importance, repeated intravenous dosing did not increase markers of liver toxicity or inflammation in heterozygote MMA mice. Although redosing makes it possible to maintain methylmalonyl-CoA mutase activity, the half-life of methylmalonyl-CoA mutase activity after LNP-mRNA delivery will need to be at an interval of 10–14 days based on the animal studies, and delivered by an intravenous route.
Future Directions
In this section, we discuss the on-going research that has been presented at scientific meetings and published in abstract form. Given hereunder is a summary of some of the different gene delivery approaches being tested to treat MMA and a brief summary of some of the preliminary results that have been disclosed.
There are currently studies underway examining new approaches using systemic mRNA therapy, AAV gene addition, and genome editing to improve upon, and refine, currently available treatment approaches. 39 –43 A proprietary LNP formulation and mRNA expression cassette have been used with success in Mmut −/−;TgINS-MCK-Mmut mice. 19 Treatment with a novel LNP mRNA nanoparticle was demonstrated to restore hepatic MMUT, drop circulating metabolites by ∼90% within 3 days of treatment, and enable significant MMUT expression as long as 2 weeks after a single treatment.
Several promising novel capsids have also been used with success. A newly isolated natural rAAV serotype, rAAV44.9, was shown to be efficacious at treating murine models of MMA. 42 Adult Mmut −/−;TgINS-MCK-Mmut mice and neonatal Mmut −/− mice treated with rAAV44.9-CBA-MMUT displayed increased weights and decreased plasma methylmalonic acid levels relative to untreated controls. In addition, rAAV44.9-CBA-MMUT–treated Mmut −/−mice had increased the survival.
Anc80, a capsid designed using in silco modeling to create an ancestral AAV capsid, has been tested as a gene delivery vector in MMA. 44,45 These studies compared rAnc80 and rAAV8 delivery for MMA. Both rAAVs used the liver-specific APOE-hAAT enhancer–promoter combination to express a codon-optimized MMUT. Adult Mmut −/−;TgINS-MCK-Mmut were treated by a systemic retro-orbital injection of 5 × 1012 vg/kg of rAAV8 or rAnc80. Treatment of with rAAV8 or rAnc80 resulted in similar improvements in growth 1- 13 C propionate oxidation and plasma methylmalonic acid levels in comparison with untreated Mmut −/−;TgINS-MCK-Mmut.
An immunological barrier to systemic rAAV delivery in pediatric patients stems from the inability to redose patients because of de novo formation of vector-induced neutralizing antibodies against the rAAV capsid. Selecta Biosciences has developed tolerogenic nanoparticles encapsulating rapamycin (ImmTOR™ or SVP-Rapamycin), which if coadministered with AAV-based vectors provide a dose-dependent suppression of humoral and T cell responses against the rAAV capsid enabling rAAV redosing. 46,47 Preliminary testing of Anc80 in combination with SVP-Rapamycin in Mmut −/−;TgINS-MCK-Mmut has recently been reported. 40,41 These studies showed that Mmut −/−;TgINS-MCK-Mmut can be redosed with Anc80 and SVP-Rapamycin with therapeutic benefit and complete inhibition of IgG antibodies to Anc80.
A new AAV-based technology termed GeneRide™, which has the potential to minimize the potential of vector-related insertional mutagenesis and to preserve MMUT expression after gene therapy even after neonatal delivery, has also been tested in murine models of MMA. 48 –51 The GeneRide vector is promoterless and utilizes homologous recombination to achieve site-specific gene addition of a human codon-optimized MMUT into the mouse albumin (Alb) locus. This new vector, AAV-Alb-2A-MMUT, contains arms of homology flanking a 2A-peptide coding sequence proximal to the MMUT gene, and generates MMUT expression from the endogenous Alb promoter after integration. 52 Treatment of Mmut −/−and Mmut −/−;TgINS-MCK-Mmut pups with AAV-Alb-2A-MMUT, delivered at a dose of 8.6 × 1011–2.5 × 1012 vg/pup at birth, reduced disease-related metabolites, and increased growth and survival in murine models of MMA. RNAscope of older AAV-Alb-2A-MMUT-treated MMA mice revealed robust MMUT expression, and MMUT-positive hepatocytes appeared as distinct and widely dispersed clusters, consistent with a pattern of clonal expansion. In addition, the RNAscope studies revealed that the MMUT expression was present in ∼5–40% of the hepatocytes in treated MMA mice versus 1% as noted in wild-type controls. Finally, hepatic MMUT protein expression and the number of Alb-integrations were also observed to increase overtime in treated MMA mice. The aggregate findings suggest that a selective advantage for corrected hepatocytes can be achieved in the murine models of MMA after treatment using MMUT GeneRide. The selection of the corrected hepatocytes in MMA should improve the efficacy of this platform that relies on in vivo homologous recombination that occurs rarely in hepatocytes after rAAV delivery.
Conclusions and future Perspectives
A constellation of findings from mouse studies and supporting patient observations clearly demonstrate that hepatic MMUT expression, even at very low levels and provided in small numbers of cells, can drastically influence the phenotype of MMA in mice and human patients. It should also be emphasized that expression of the Mmut enzyme in extrahepatic tissues can also mitigate the metabolic phenotype of MMA—this is proven most elegantly by the Mmut −/−;TgINS-MCK-Mmut mice, where expression of the Mmut enzyme in only the skeletal and cardiac muscle can fully rescue the lethal effects that accompany whole body Mmut deficiency.
The preclinical studies have also identified metabolic, peptide, and isotopic biomarkers that accompany the hepatic response to increased methylmalonyl-CoA mutase activity. These include small molecules and hormones that reliably decrease, such as methylmalonic acid and Fgf21, and whole body 1- 13 C propionate oxidation, which increases when hepatic mMUT activity is restored. Thus, it appears that a suite of new gene-based therapies and accompanying biomarkers are emerging for the treatment of MMA that will provide, for the first time, definitive therapies for this grave inborn error of metabolism.
Footnotes
Acknowledgments
R.J.C. and C.P.V. were supported by the Intramural Research Program of the National Human Genome Research Institute.
Author Disclosure
The NIH has filed patents on behalf R.J.C. and C.P.V.