
Abstract
The pro-renin receptor (PRR) is an important novel component of the renin–angiotensin (Ang) system that has multiple functions, which are not yet completely understood. In this study, we aimed to explore the effect of PRR on the formation of Ang II–induced abdominal aortic aneurysm (AAA) in apolipoprotein E-knockout mice. We used Ang II (1.44 mg/kg/day) infusion to induce AAA followed by a treatment of saline, telmisartan, no treatment, Ad-EGFP, Ad-PRR, or Ad-PRR plus telmisartan. The incidence of AAA was 35%, 60%, 65%, 90%, and 55% in the Telmisartan, Vehicle, Ad-EGFP, Ad-PRR, and Ad-PRR+Telmisartan groups, respectively. Compared with the Vehicle and Ad-EGFP groups, PRR overexpression markedly increased macrophage infiltration; levels of proinflammatory cytokines, including monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α); the expression and activity of MMP2 and MMP9; NOX2 and NOX4 protein and mRNA expression; nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity; extracellular-signal-regulated kinase (ERK) and P38MAPK expression; but decreased smooth muscle cells content in AAA. However, telmisartan reversed the adverse effects of PRR. In addition, ERK inhibitor PD98059 eliminated the acceleration of Ang II–induced AAA formation by PRR, and coadministration of telmisartan and PD98059 further abolished the adverse effects of PRR on Ang II–induced AAA formation. Thus, PRR plays an important role in the pathological development of AAA via both Ang II–dependent and Ang II–independent activation of ERK pathways. These results suggest that inhibition of PRR activation may be a promising approach to the treatment of AAA.
Introduction
Abdominal aortic aneurysm (AAA) is a common, often asymptomatic, potentially lethal vascular degenerative disease, which is characterized by expansion of all layers of the arterial wall as a result of vascular smooth muscle cell (VSMC) apoptosis, 1 loss of elastin, and oxidative stress. 2 Inflammation and matrix degradation by matrix metalloproteinases (MMPs) in the vasculature is crucial for AAA formation. 3,4
Numerous studies have suggested that the renin–angiotensin system (RAS) plays a crucial role in the pathogenesis of AAA. In experimental studies, a widely accepted animal model of AAA is subcutaneous infusion of angiotensin II (Ang II) for 28 days in apolipoprotein E-knockout (ApoE−/− ) mice. Ang II is an important family member of RAS that induces matrix degradation, inflammation, and aortic remodeling. Furthermore, administration of AT1 receptor antagonist, losartan, prevented Ang II–induced AAA formation. 5 In addition, recent research revealed that Ang (3–8), also known as Ang IV, has a bidirectional effect on Ang II–induced AAA in ApoE−/− mice. 6
The pro-renin receptor (PRR), an important novel component of RAS, is a specific receptor for renin and prorenin that on ligand binding not only increases local generation of Ang I from angiotensinogen but also induces Ang II–independent intracellular signaling. 7 Previous studies demonstrated that PRR overexpression induced oxidative stress through both Ang II–dependent and Ang II–independent mechanisms. 8 Moreover, PRR triggered extracellular matrix remodeling and deterioration of cardiac function via Ang II–independent activation of extracellular-signal-regulated kinase1/2 (ERK1/2) phosphorylation and the Ang II–dependent pathway. 7 Aliskiren, a direct renin inhibitor, limits the progression of AAA in an Ang II–infused mouse model via downregulation of PRR overexpression. 9 Based on these observations, we hypothesized that PRR overexpression may promote AAA formation via Ang II–dependent and Ang II–independent pathways. In this study, we sought to explore the effects of adenovirus vector–mediated overexpression of PRR on AAA formation and to elucidate the underlying mechanisms mediating these effects in an Ang II–infused mouse model.
METHODS
Detailed Materials and Methods section are available in the Supplementary Data.
Adenoviral vector construction
A full-length coding region of murine (P)RR cDNA was subcloned into SalI and HindIII sites of the pShuttle-CMV vector (Qbiogene, Inc., Illkirch, Cedex-France). The pShuttle-CMV contains CMV promoter and the SV40 polyadenylation signal. The PRR cDNA sequence in the pShuttle-CMV-PRR plasmid was confirmed by sequencing. Recombinant adenoviruses (Ads) carrying the murine PRR (Ad-PRR) or a control transgene (Ad-EGFP) were prepared as previously described with the AdMax system (Microbix Biosystems, Toronto, ON, Canada). 7,10
Generation of shRNA construct and adenovirus production
Based on the protocol from Signaling Gateway, the shRNA cassette containing target sequence of PRR (GCTCCGTAATCGCCTGTTTCA) was designed. The cassette was subcloned into pEN-hH1c vector as described previously. 11
Animal protocol
Male ApoE−/− mice were injected with Ad-SC-shRNA, Ad-PRR-shRNA, Ad-EGFP, and Ad-PRR via tail vein injection and repeated 2 weeks later. Mice received telmisartan administration (10 mg/kg/day) by intragastric intubation or PD98059 administration (20 mg/kg/day) by intraperitoneal injection. The model of AAA was established by implanting an Alzet osmotic minipump, which delivered Ang II (1.44 mg/kg/day, for 28 days) as described previously. 12 At the end of experiment, all mice were sacrificed under anesthesia. The whole aortas including thoracic and abdominal aortas were collected for morphological and histological analysis of AAA.
Blood pressure measurements
Blood pressures and heart rates were measured in conscious mice using a noninvasive tail-cuff system specifically designed for mice (Softron BP-98A, Tokyo, Japan).
Serum lipid assay
Serum levels of total cholesterol, triglycerides, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and glucose were determined by a commercially available enzymatic assay using a biochemistry automatic analyzer (HITACHI 7170A; Hitachi, Tokyo, Japan).
Analysis and quantification of AAA
To quantify AAA incidence and size, the adventitia was removed postmortem and the maximum width of the abdominal aorta was measured with Image-Pro Plus software (Media Cybernetics, Inc.).
Histological and morphological analysis
Aortic sections were obtained from animals in each experimental group and were stained by hematoxylin and eosin for morphological assessment. Elastic fiber integrity of the abdominal aorta was visualized by use of a Verhoeff–Van Gieson staining kit (Gemed Scientific, Inc.) according to the manufacturer's instruction.
Immunohistochemistry
Formaldehyde-fixed paraffin sections were incubated with primary antibody overnight at 4°C. The primary antibodies used were α-smooth muscle actin, monocyte+macrophage antibody (MOMA-2), MMP2, MMP9, monocyte chemoattractant protein-1 (MCP-1), and tumor necrosis factor-α (TNF-α). As a negative control, species- and isotype-matched IgG were used in place of the primary antibody.
Immunofluorescence
Following antigen retrieval, corresponding sections from each experimental group were placed on separate slides and then washed three times (5 min per wash) with phosphate-buffered saline (PBS). The sections were then blocked with 10% normal donkey serum in PBS and coincubated with the antibodies against GFP and PRR at 4°C overnight. After washing, sections were coincubated with green-conjugated secondary antibody and red-conjugated secondary antibody for 45 min. Sections were then imaged using a fluorescence microscope.
Nicotinamide adenine dinucleotide phosphate oxidase activity
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity was measured in aortas of ApoE−/− mice or cultured mouse vascular smooth muscle cells (mVSMCs) as previously described. 13
Cell cultures
mVSMCs were from ATCC. mVSMCs were divided into six groups: Control, Telmisartan, Ang II, Ad-EGFP+Ang II, Ad-PRR+Ang II, and Ad-PRR+Ang II+Telmisartan groups. To further study the effect of PRR on mVSMCs, cells were divided into three groups: Control, Ang II, and Ang II+PRO20 groups. The levels of proteins in the media were determined by Western blot.
Adenoviral infection
mVSMCs were infected with adenovirus expressing a PRR as described previously. 14 A replication-defective adenoviral vector expressing GFP was used as control.
Detection of reactive oxygen species
Reactive oxygen species (ROS) production in cultured cells was detected using the fluorescent probe dihydroethidium (DHE) as described previously. 15
Real-time polymerase chain reaction
The messenger ribonucleic acid expression of NOX2 and NOX4 was quantitated using real-time polymerase chain reaction (RT-PCR).
Western blot analysis
The protein expression of MMP2, MMP9, TNF-α, MCP-1, PRR, NOX2, NOX4, phosphorylated P38MAPK (pP38), P38, phosphorylated ERK (pERK), and ERK was assayed by Western blot analysis.
Gelatin zymography
The activity of MMP2 and MMP9 was evaluated by gelatin zymography.
Statistical analysis
Data are reported as mean ± SEM. Comparisons of parameters among more than two groups were made by one-way analysis of variance with least significant difference post hoc analysis. Chi-square test was applied to comparisons of AAA incidence and mortality. A p-value of <0.05 was considered statistically significant.
RESULTS
Serum lipids and blood pressure
Serum lipid levels did not differ significantly across experimental animal groups (Supplementary Tables S1–S4). Ang II infusion significantly increased systolic blood pressure (sBP) in ApoE−/− mice compared with sBP measured in the Vehicle group. However, PRR, telmisartan and PD98059 had no effect on sBP.
PRR overexpression increased the formation of Ang II–induced AAA, and Telmisartan partially abolished its effect
The control mice did not exhibit AAA when infused with saline (Fig. 1A). However, Ang II infusion for 4 weeks significantly increased the incidence of AAA as evidenced by enlarged abdominal aortas morphologically (Fig. 1A, B) as previously reported. 16,17 To determine the effects of PRR on AAA formation, we generated PRR overexpression mice by infecting adenovirus containing PRR to upregulate PRR protein expression in ApoE−/− mice (Supplementary Fig. S1B, C). We found that the incidence of AAA was 35%, 60%, 65%, 90%, and 55% and the mortality was 10%, 25%, 20%, 45%, and 20% in Telmisartan, Vehicle, Ad-EGFP, Ad-PRR, and Ad-PRR+Telmisartan groups, respectively (Fig. 1B). The incidence of AAA and the mortality of mice were substantially higher in the Ad-PRR group than in the Vehicle and Ad-EGFP groups but did not differ significantly between the Vehicle group and the Ad-EGFP group (Fig. 1A, B). Compared with the Ad-PRR+Telmisartan group, AAA incidence and mortality were significantly increased in the Ad-PRR group but were markedly decreased in the Telmisartan group. In addition, the maximal diameter of the abdominal aorta was significantly larger in the Ad-PRR group than in the Vehicle, Ad-EGFP, and Ad-PRR+Telmisartan groups but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group (Fig. 1D). Moreover, the maximal diameter of the abdominal aorta was significantly decreased in the Telmisartan group compared with the Ad-PRR+Telmisartan group. Therefore, PRR overexpression increased the formation of Ang II–induced AAA, and Telmisartan partly abolished its effect.
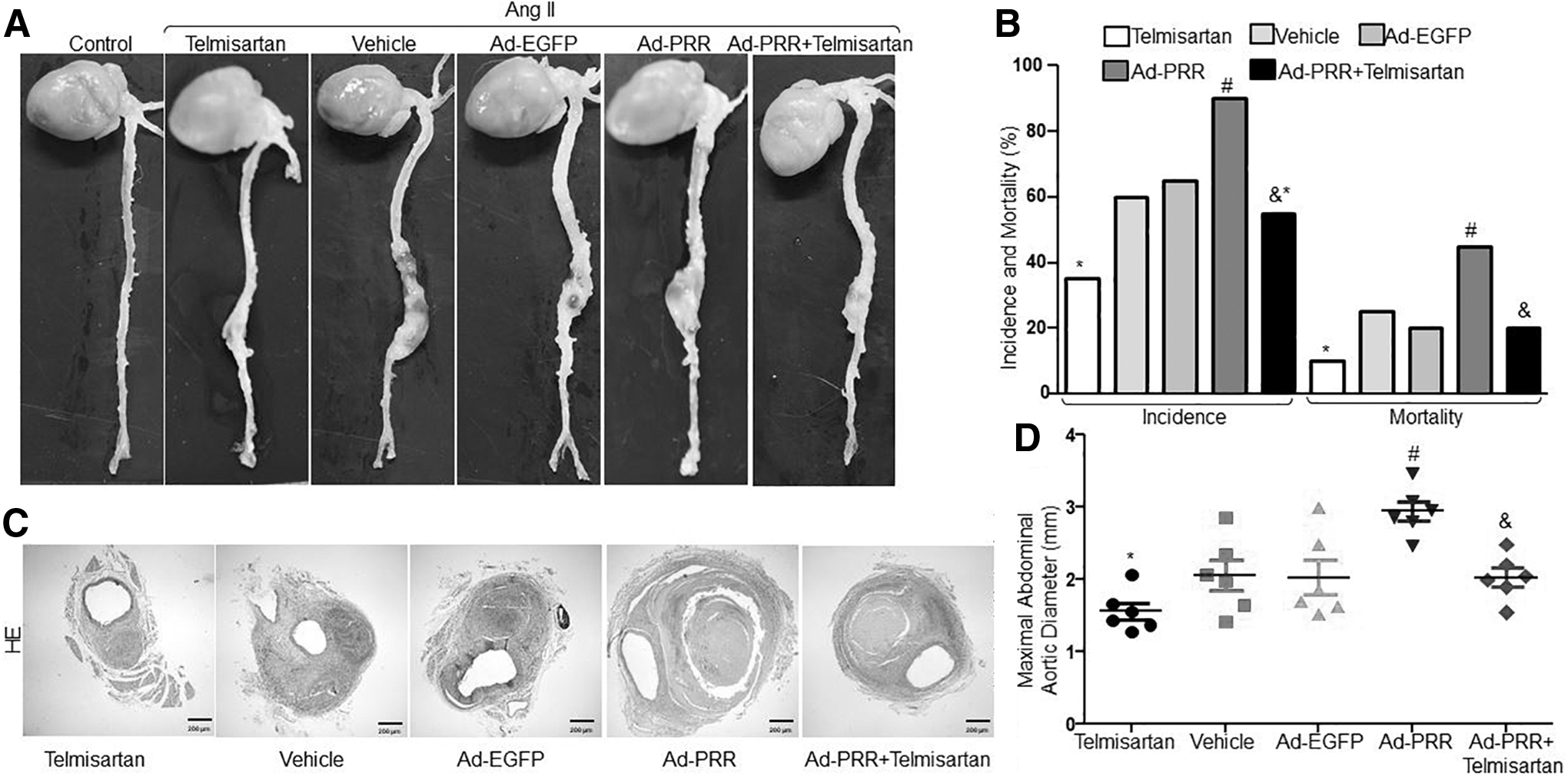
PRR promoted the formation of Ang II–induced AAA in ApoE−/−
mice.
Effect of PRR on Ang II–induced histological and morphological changes in ApoE−/− mouse aortas
Hematoxylin and eosin and Verhoeff–Van Gieson staining revealed that Ang II infusion gave rise to breakdown of the aortic media and adventitia, intraluminal thrombus formation, hypertrophy of the adventitia, and destruction and discontinuity of elastin fibers in the aortic wall. Compared with the Vehicle, Ad-EGFP, and Ad-PRR+Telmisartan groups, these pathological changes were largely increased in the Ad-PRR group but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group (Figs. 1C and 2A). In addition, these pathological changes were significantly decreased in the Telmisartan group compared with the Ad-PRR+Telmisartan group.
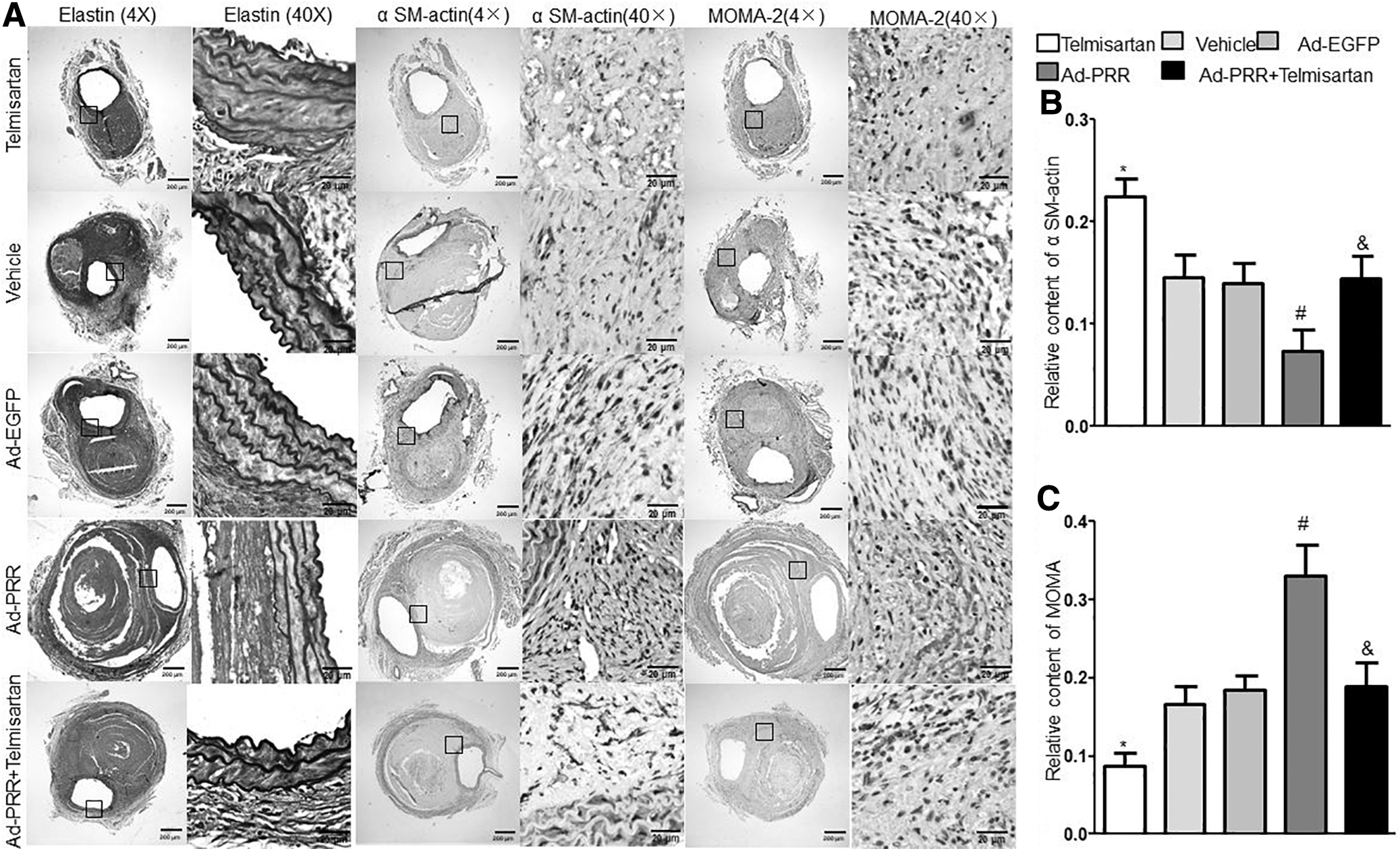
Effect of PRR on aortic wall compositions in Ang II–infused ApoE−/−
mice.
Specifically, the content of smooth muscle cells (SMCs) in the aortic wall as revealed by immunohistochemical staining was substantially decreased, whereas the relative content of macrophages was increased in the Ad-PRR group relative to the Vehicle, Ad-EGFP, and Ad-PRR+Telmisartan groups but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group (Fig. 2A–C). Compared with the Ad-PRR+Telmisartan group, SMCs content was markedly increased, whereas macrophage content was significantly decreased in the Telmisartan group.
Effect of PRR on the expression of MMP2 and MMP9 in vivo and in vitro
MMPs are crucial for the extracellular matrix degradation in the initiation and progression of AAA. 14 In particular, VSMC-derived MMP2 and macrophage-derived MMP9 are key enzymes in AAA formation. 18 Thus, we next determined the relative content of MMP2 and MMP9 in Ang II–infused ApoE−/− mice. Compared with the Vehicle and Ad-EGFP groups, PRR overexpression significantly increased both the protein expression level and activity of MMP2 and MMP9 in aorta, whereas telmisartan blunted such effects of PRR on MMP2 and MMP9 but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group (Fig. 3A–C, F–H and Supplementary Fig. S2A–C). What is more, these measurements were significantly lower in the Telmisartan group than in the Ad-PRR+Telmisartan group.
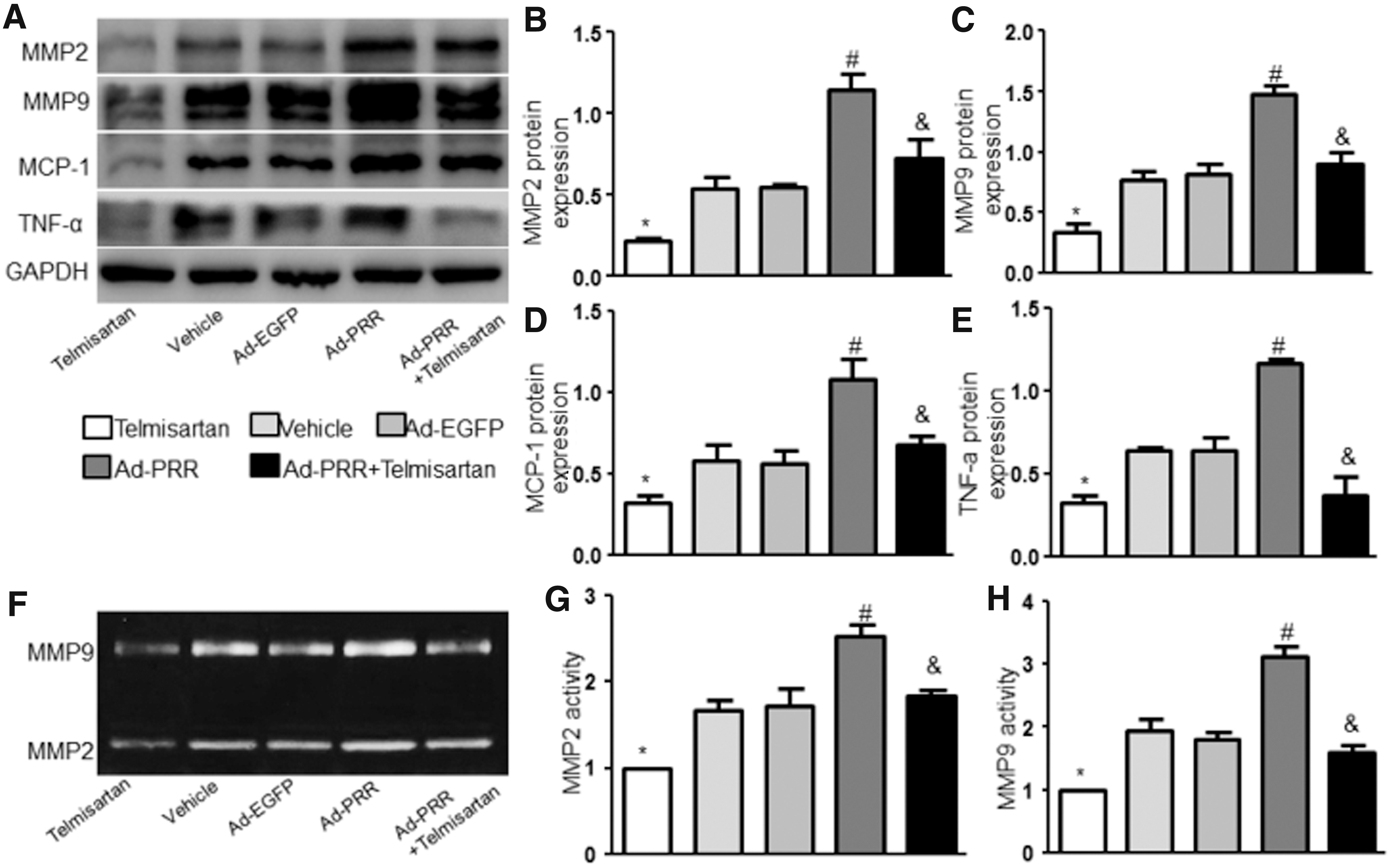
Effect of PRR on MMPs and proinflammatory cytokines in Ang II–infused ApoE−/−
mice.
To further determine the effect of PRR on Ang II–induced MMPs, we transfected mVSMCs with Ad-PRR to upregulate PRR protein expression (Supplementary Figs. S3D and S3F), and the result also showed that Ang II significantly increased the level of PRR protein expression. In vitro study showed that Ang II significantly increased the levels of MMP2 and MMP9 protein expression, and PRR overexpression aggravated Ang II–induced upregulation of MMP2 and MMP9 expression in mVSMCs as previously observed in vivo (Supplementary Figs. S3A and S3B), whereas telmisartan reversed the effect of the Ad-PRR on MMP2 and MMP9 expression but did not differ between the Ang II group and the Ad-EGFP+Ang II group or Ad-PRR+Ang II+Telmisartan group. Compared with Ad-PRR+Ang II+Telmisartan, the levels of MMP2 and MMP9 protein expression were markedly lower in the Ang II+Telmisartan group.
PRR increased proinflammatory cytokines expression in vivo and in vitro
Previous studies demonstrated that proinflammatory cytokines, such as MCP-1 and TNF-α were upregulated in Ang II–infused ApoE−/− mice. 19,20 Both Western blot and immunostaining analysis showed that the levels of MCP-1 and TNF-α protein expression in aortic tissues were significantly increased by Ad-PRR treatment relative to the Vehicle and Ad-EGFP groups but did not differ between the Vehicle group and the Ad-EGFP group (Fig. 3A, D, E and Supplementary Fig. S2A, D, E). Compared with the Ad-PRR+Telmisartan group, the levels of MCP-1 and TNF-α protein expression were significantly increased in the Ad-PRR group but were markedly decreased in the Telmisartan group. Results obtained in vitro in mVSMCs closely aligned with results obtained in vivo (Supplementary Fig. S3A, C).
PRR affected oxidative stress in vivo and in vitro
Oxidative stress is involved in Ang II–induced AAA development. 21 We measured the levels of NOX2 and NOX4, two markers of oxidative stress, in the aortic tissues of ApoE−/− mice by Western blot and RT-PCR and found that the levels of NOX2 and NOX4 protein and mRNA expression were dramatically increased in the Ad-PRR group compared with the Vehicle and Ad-EGFP groups but did not differ between the Vehicle group and the Ad-EGFP group (Fig. 4A–E). Furthermore, the levels of NOX2 and NOX4 were significantly lower in the Ad-PRR+Telmisartan group than in the Ad-PRR group and were markedly decreased in the Telmisartan group compared with the Ad-PRR+Telmisartan group. Compared with the Vehicle, Ad-EGFP, and Ad-PRR+Telmisartan groups, NADPH oxidase activity were largely increased in the Ad-PRR group but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group (Fig. 4F). In addition, NADPH oxidase activity was significantly decreased in the Telmisartan compared with the Ad-PRR+Telmisartan group.
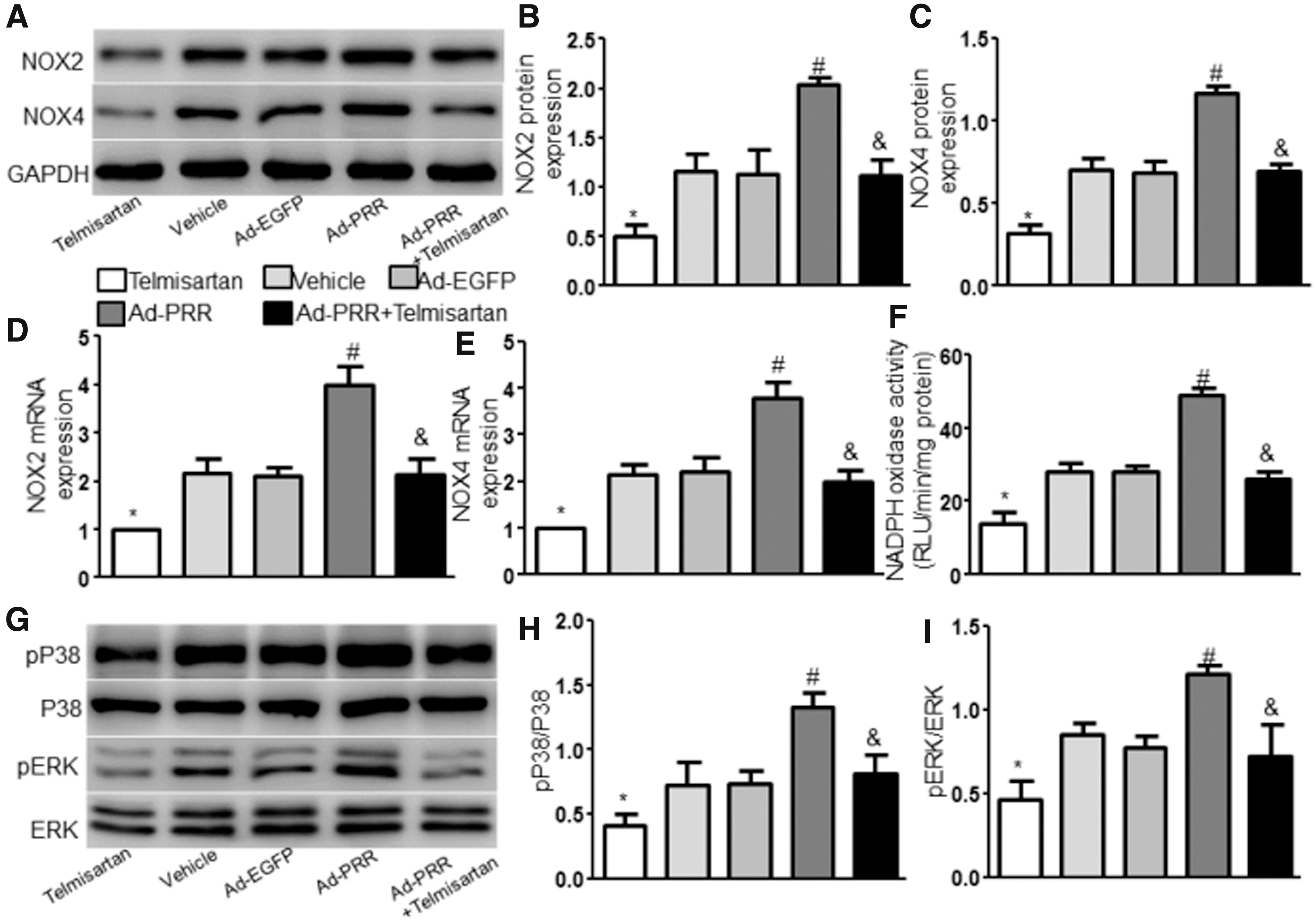
Effect of PRR on oxidative stress and signaling proteins in Ang II–infused mice.
In mVSMCs, Ad-PRR treatment dramatically increased the levels of Ang II-induced NOX2 and NOX4 protein and mRNA expression, NADPH oxidase activity, and ROS productions measured by DHE, whereas telmisartan eliminated the effect of PRR on Ang II–induced oxidative stress but did not differ between the Ang II group and the Ad-EGFP+Ang II group or Ad-PRR+Ang II+Telmisartan group (Supplementary Fig. S3D, E, I–L). What is more, oxidative stress was significantly inhibited in the Telmisartan group compared with the Ad-PRR+Telmisartan group.
P38 and ERK signaling was involved in the effect of PRR on Ang II–induced AAA
To test the signaling proteins involved in the effect of PRR on Ang II–induced AAA, we examined the levels of P38 and ERK protein expression in vivo and in vitro. In Ad-PRR–treated mice and mVSMCs, phosphorylated P38 and pERK levels but not total P38 and ERK were significantly increased compared with the Vehicle, Ad-EGFP, and Ad-PRR+Telmisartan groups of mice and the Ang II, Ad-EGFP+Ang II, and Ad-PRR+Ang II+Telmisartan groups of cells but did not differ between the Vehicle group and the Ad-EGFP group or Ad-PRR+Telmisartan group of mice and between the Ang II group and the Ad-EGFP+Ang II group or Ad-PRR+Ang II+Telmisartan group of cells (Fig. 4G–I and Supplementary Fig. S3D, G, H). In addition, pP38 and pERK levels were significantly decreased in the Telmisartan or Telmisartan+Ang II group compared with the Ad-PRR+Telmisartan or Ad-PRR+Ang II+Telmisartan group of both mice and cells.
Deficiency of PRR affects MMPs, proinflammatory cytokines, oxidative stress, and signaling proteins in vitro and Ang II–induced AAA formation in ApoE −/− mice
To further study the effect of PRR on Ang II–induced MMPs, proinflammatory cytokines, oxidative stress, and signaling proteins, we used the PRR inhibitor PRO20 to cotreat mVSMCs. We found that blunted PRR signaling eliminated the upregulation of MMP2 and MMP9 protein expression and activity, NOX2 and NOX4 protein and mRNA expression, MCP-1, TNF-α, pP38, and pERK protein expression, NADPH oxidase activity, and ROS productions by Ang II (Fig. 5). In addition, the effects of PRR knockdown on AAA formation were also evaluated. We generated PRR-knockdown mice by infecting adenovirus containing PRR-shRNA to downregulate PRR protein expression in ApoE−/− mice. As shown in Supplementary Fig. S4D, the expression of PRR was significantly increased in the Vehicle and Ad-SC-shRNA groups than in the Control group and decreased in the Ad-PRR-shRNA group than in the Vehicle and Ad-SC-shRNA groups. As depicted in Supplementary Fig. S4A, Ad-PRR-shRNA treatment significantly inhibited the Ang II–induced AAA formation in ApoE−/− mice. The incidence and mortality of AAA (Supplementary Fig. S4B) and maximal diameter (Supplementary Fig. S4C) were decreased compared with vehicle and Ad-SC-shRNA–treated mice. Thus, deficiency of PRR inhibited the formation of Ang II–induced AAA in ApoE−/− mice and downregulated MMPs expression, proinflammatory cytokines, oxidative stress, and signaling proteins in mVSMCs.
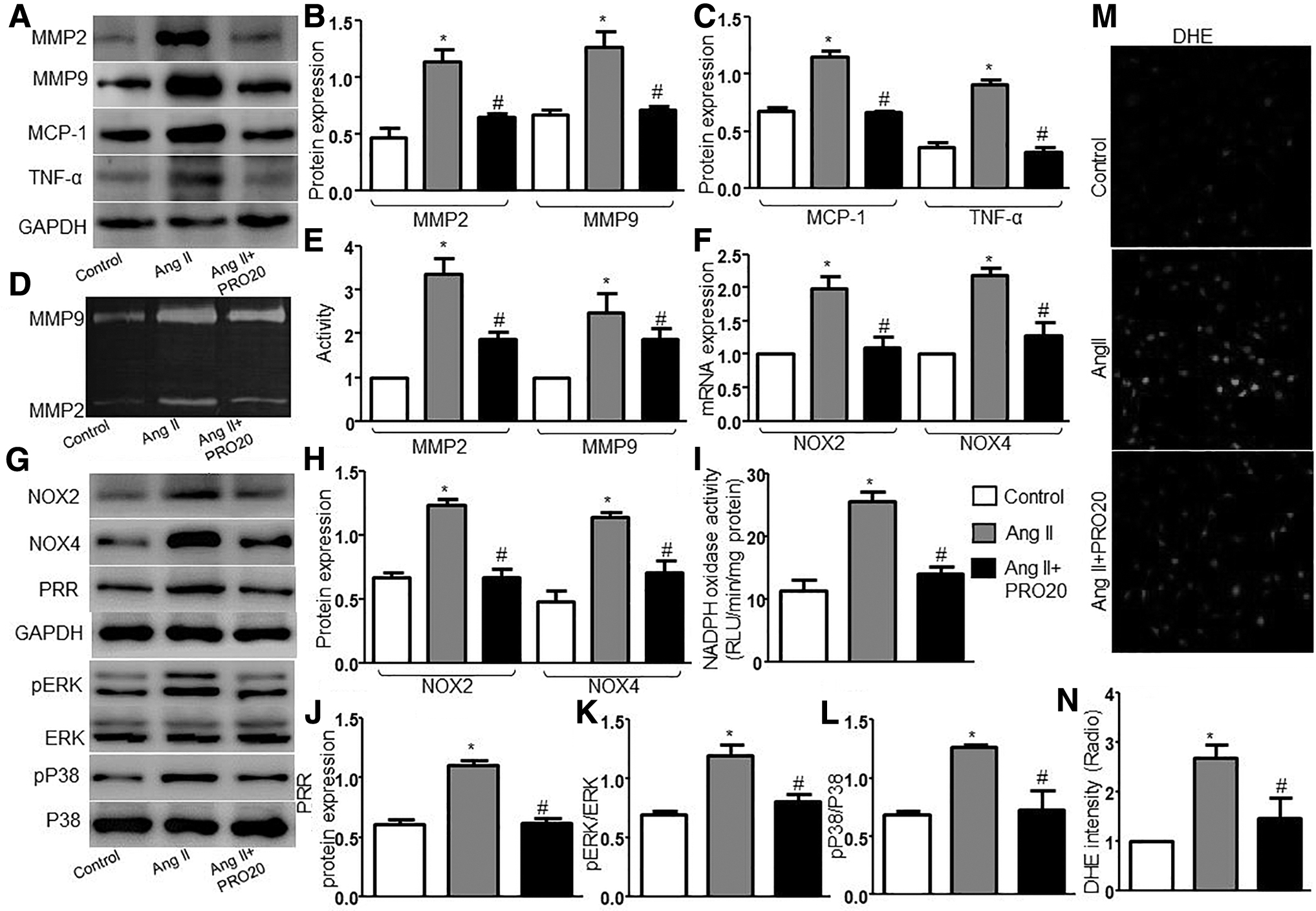
Effects of PRR inhibitor PRO20 on MMPs, proinflammatory cytokines, oxidative stress, and signaling proteins in cultured mVSMCs.
ERK inhibitor PD98059 blunted the effect of PRR on Ang II–induced AAA formation
To further understand whether PRR exerted its effect via ERK, we cotreated mice with ERK inhibitor PD98059 and Ad-PRR. As shown in Supplementary Fig. S5A–C, the expression of PRR was significantly increased in the Ad-PRR and Ad-PRR+PD98059 groups than in the PD98059 and Ad-EGPP groups, and pERK protein expression was inhibited by PD98059 (Supplementary Fig. S5B, D). As depicted in Fig. 6A, Ad-PRR treatment significantly accelerated the Ang II–induced AAA formation in ApoE−/− mice. The incidence and mortality of AAA (Fig. 6B), maximal diameter (Fig. 6C), MOMA-2 content (Fig. 6D, F), MMP2 and MMP9 protein expression and activity (Fig. 7A–C, F–H and Supplementary Fig. S6A–C), inflammation (Fig. 7A, D, E and Supplementary Fig. S6A, D, E), NOX2 and NOX4 protein and mRNA expression (Supplementary Fig. S5B, E–H), and NADPH oxidase activity (Supplementary Fig. S5I) were increased, whereas SMC content was decreased (Fig. 6D, E), compared with vehicle-treated mice. However, all these effects induced by PRR were reversed in ApoE−/− mice if they were cotreated with PD98058 but did not differ between the Vehicle group and the Ad-PRR+PD98059 group. In addition, compared with the Ad-PRR+PD98059 group, PD98059 treatment only significantly decreased MMPs, inflammation, and oxidative stress. Together, ERK may play an important role in the promoting effect of PRR on Ang II–induced AAA.
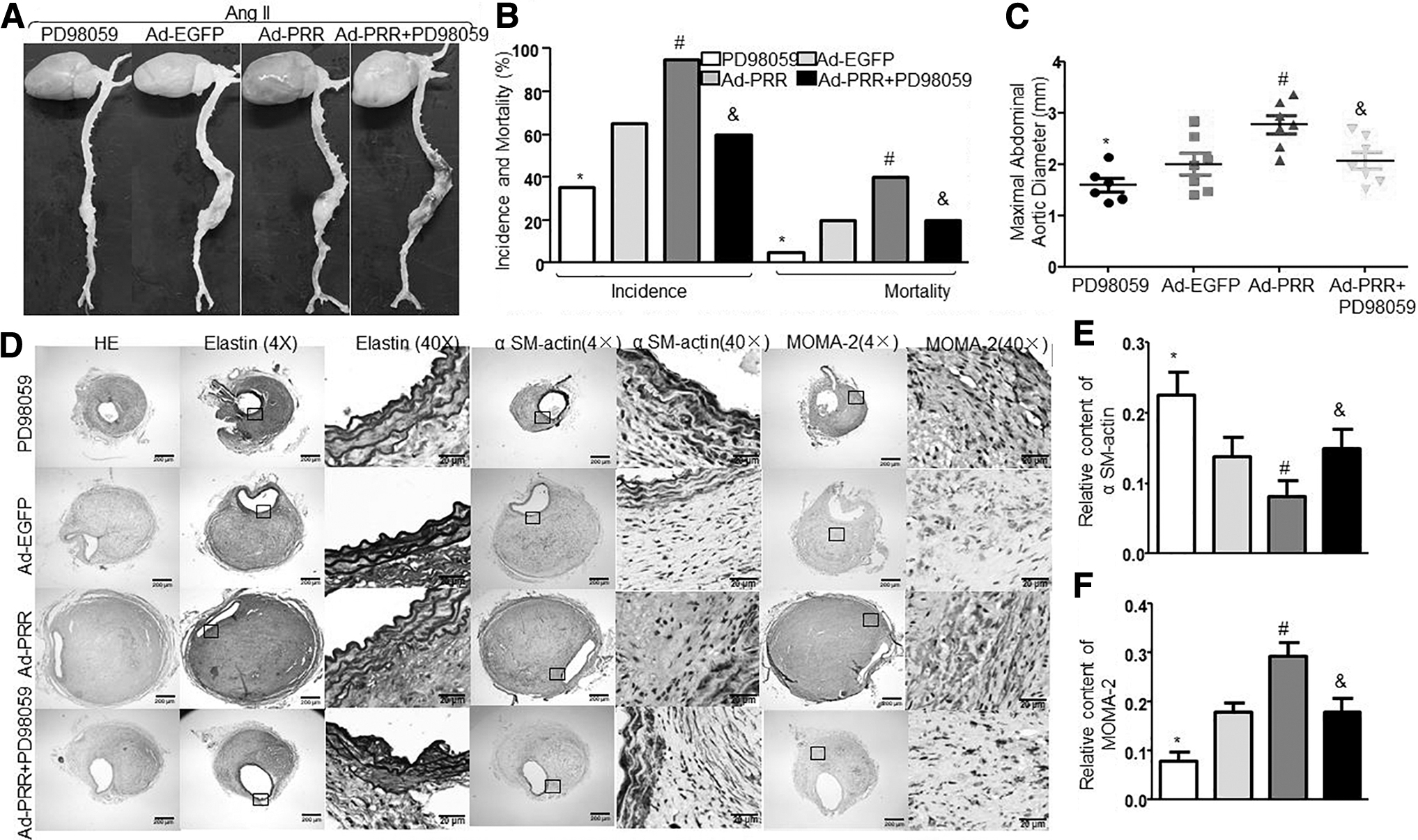
Effects of ERK inhibitor PD98059 on Ang II–induced AAA formation and aortic wall compositions in Ang II–infused ApoE−/−
mice.
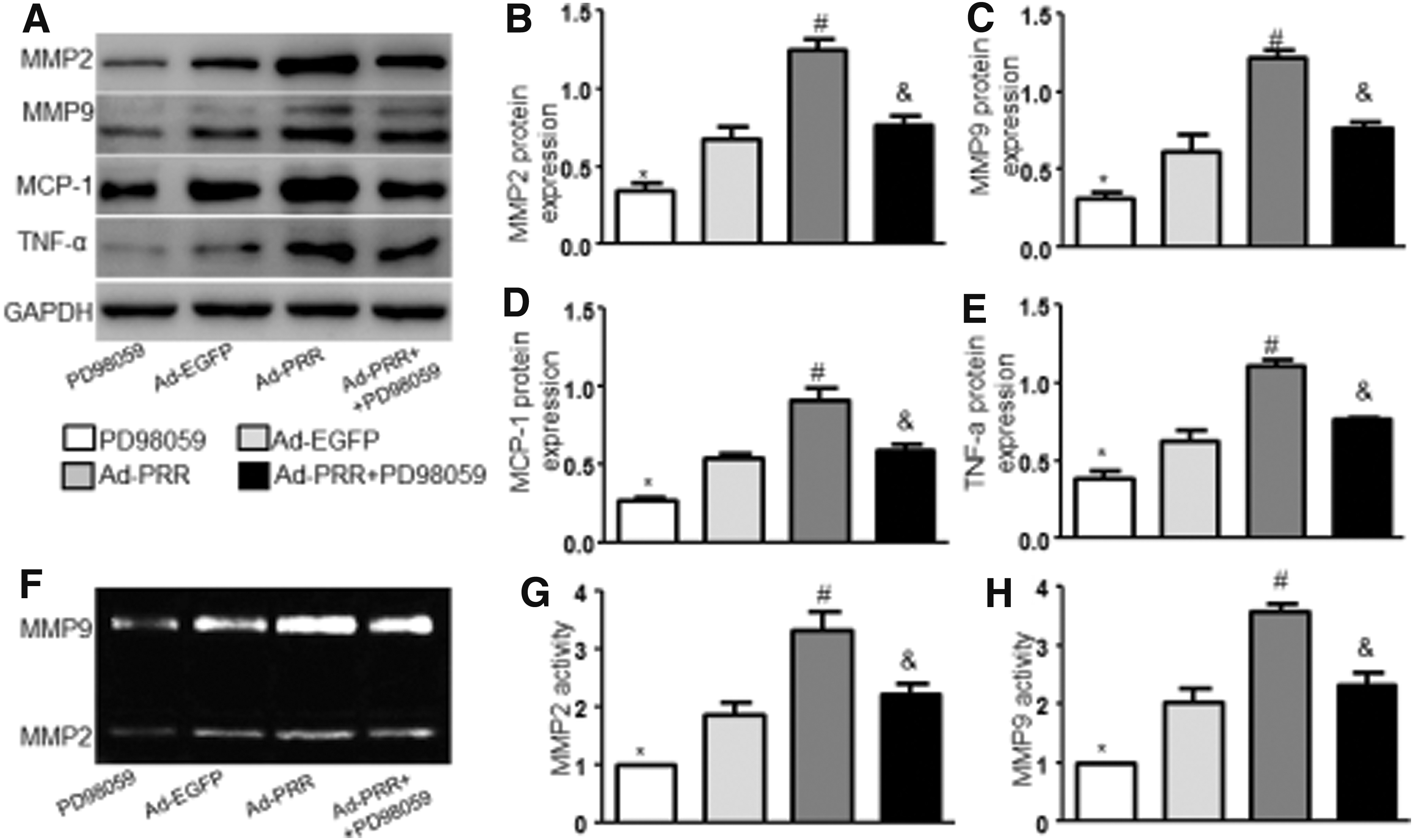
Effects of ERK inhibitor PD98059 on MMPs and proinflammatory cytokines in Ang II–infused ApoE−/−
mice.
ERK inhibitor PD98059 abolished an effect of PRR on Ang II–induced AAA formation independent of Ang II pathway
To further verify whether ERK exerted an effect PRR on Ang II–induced AAA formation independent of Ang II pathway, we cotreated mice with ERK inhibitor PD98059 and Ad-PRR and telmisartan. The expression of PRR had no significant difference among the three groups, whereas pERK protein expression was inhibited by PD98059 compared with the Ad-PRR+Telmisartan group (Supplementary Fig. S7G, J, K). As shown in Fig. 8A–C, compared with the Ad-PRR+Telmisartan group, PD98059 treatment significantly reduced the incidence and mortality of AAA in Ad-PRR+Telmisartan+PD98059 group. As well, it markedly abolished the suppression of SMC content and the upregulation of inflammation, MMP2 and MMP9 protein expression and activity, NOX2 and NOX4 protein and mRNA expression, macrophage infiltration, and NADPH oxidase activity by Ad-PRR plus telmisartan (Fig. 8D, E and Supplementary Figs. S7A–I, S8). Thus, ERK may be responsible for the promoting effect of PRR on Ang II–induced AAA independent of Ang II pathway.
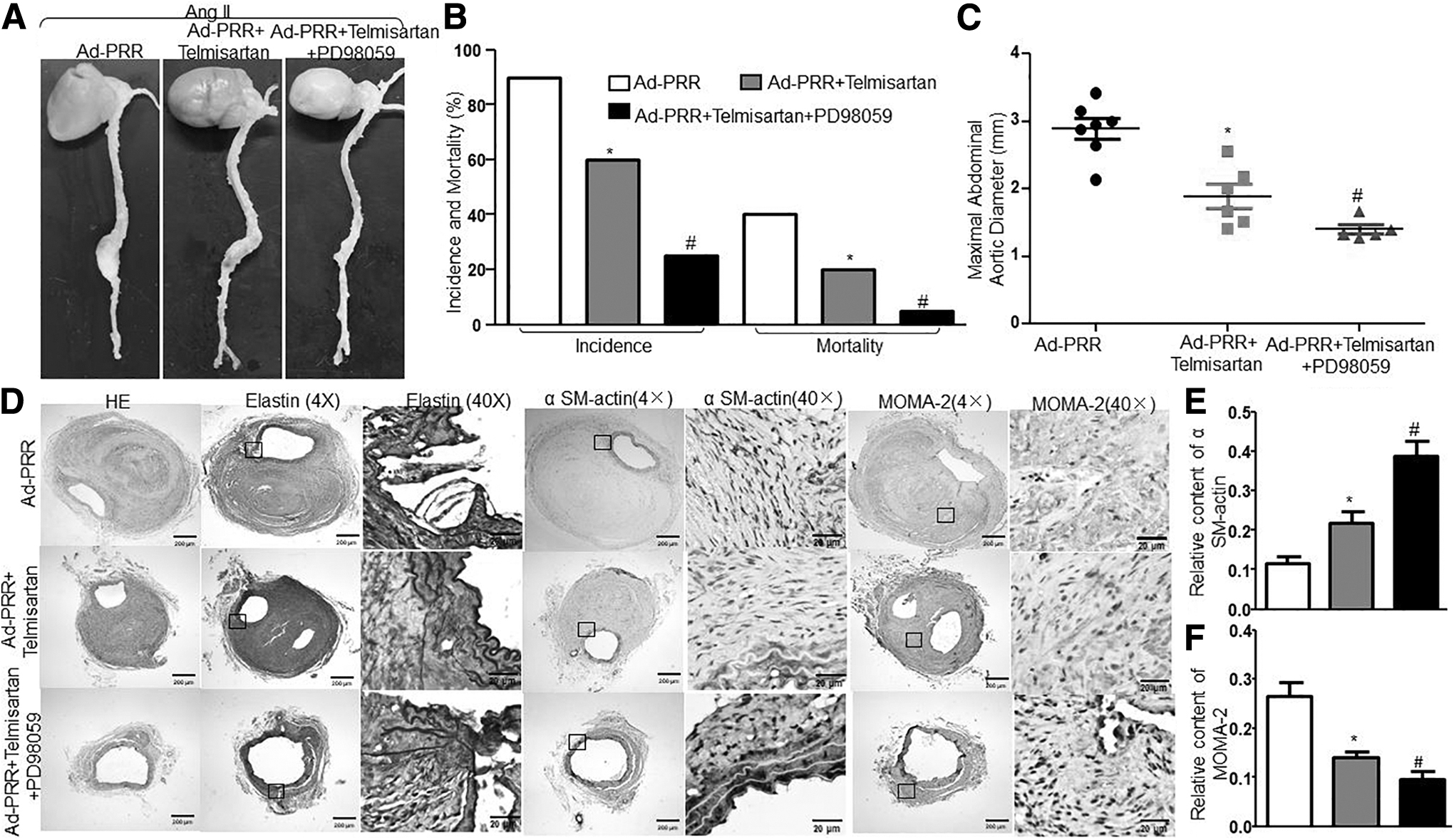
Effects of ERK inhibitor PD98059 on Ang II–induced AAA formation and aortic wall compositions independent of Ang II pathway in Ang II–infused ApoE−/−
mice.
DISCUSSION
In the present study, we provided the first evidence that PRR overexpression promoted Ang II–induced AAA formation in ApoE−/− mice. The mechanisms underlying these deteriorative effects included increased MMPs expression and activity, decreased SMC content, enhanced oxidative stress, increased inflammation, and activation of ERK and P38MAPK signaling. First, we found that both AT1 receptor antagonist, telmisartan, and ERK inhibitor PD98059 eliminated the acceleration of Ang II–induced AAA formation by PRR. Then, the PRR-induced AAA incidence was further reduced by co-treatment of telmisartan and PD98059. Therefore, PRR promoted Ang II–induced AAA formation via both Ang II–dependent and Ang II–independent activation of ERK pathways.
An important finding of this study was that the acceleration effects of PRR on AAA were independent of serum lipid and blood pressure. First, the serum lipid levels were not markedly different among all animal groups. Second, although Ang II infusion induced a marked elevation of blood pressure in ApoE−/− mice, PRR treatment exerted no noticeable effects on the elevated blood pressure in our mouse model. These findings are in line with previous studies that demonstrated blood pressure did not play an important role in Ang II–induced AAA formation in ApoE−/− mice. 22 Thus, the effect of PRR on AAA was blood pressure-independent.
MMPs play a crucial role in the extracellular matrix degradation and vascular remodeling that underlie the development of AAA. 14 In particular, the expression and activity of VSMC-derived MMP2 and macrophage-derived MMP9 were significantly increased in human aneurysmal tissues. 18 Experimental studies demonstrated that MMP2 and MMP9 also play a key role in AAA progression, whereas MMP2 and MMP9 deficiency could prevent AAA development. 18 In the present study, our results show that PRR overexpression markedly increased the expression and activity of MMP2 and MMP9 induced by Ang II in mouse aortas and in mVSMCs, which may be responsible for extracellular matrix degradation and vascular rupture in our animal model.
Chronic inflammation of the aortic wall due to the infiltration of macrophages, lymphocytes, and mast cells results in the production of inflammatory cytokines, loss of medial VSMC, and destruction of elastin, which has been considered an instigating mechanism underlying Ang II–induced AAA. 23 In our study, we found that PRR overexpression significantly increased macrophage infiltration and the expression of proinflammatory cytokines, such as MCP-1 and TNF-a in mouse aortas. Similar results were obtained in mVSMCs. These data indicate that the deleterious effects of PRR on AAA were associated with the extent of inflammation in the aortic wall.
Oxidative stress plays an important role in the pathogenesis of Ang II–induced AAA. 21,24 Previous studies reported that ROS generation is markedly upregulated in AAA tissues, whereas genetic and pharmacological inhibition of ROS production suppressed AAA formation. 25 –27 A major source of ROS in vascular tissue is the NADPH oxidase, which includes transmembrane subunits such as Nox1–5 and p22phox as well as cytosolic subunits such as p47phox, p67phox, and small GTPase Rac1 (16, 28, 29). NADPH oxidase activity was significantly increased in human aneurysmal aortas. 28 Oxidative stress was associated with proinflammatory cytokines and MMPs expression in the development of AAA. 2 In the present study, PRR overexpression substantially increased NOX2 and NOX4 protein and mRNA expression and NADPH oxidase activity in mouse aortas and in vitro, and upregulated ROS production in mVSMCs, suggesting that oxidative stress may be an important mechanism in promoting AAA formation by PRR overexpression in our animal model.
ERK and P38MAPK, main family members of mitogen-activated protein kinase, are markedly increased in human and mouse AAA tissues and were associated with activated inflammation and MMP activity in the pathogenesis of aneurysms. 29,30 Previous study demonstrated that not only simvastatin prevented Ang II–induced AAA formation via the inhibition of ERK activation but also treatment of mice with Ang II plus CI1040, an ERK inhibitor, inhibited AAA formation. 31 In our study, the activation of ERK and P38MAPK was markedly upregulated by PRR overexpression. In addition, our in vivo study also demonstrated that blockade of ERK signaling by PD98059 abolished the adverse effects of PRR, and the deleterious effects of co-administration of Ad-PRR and PD98059 were more serious than only PD98059 treatment, indicating that the deleterious effects of PRR on AAA were associated with the activation of ERK and P38MAPK.
Previous studies demonstrated that PRR could elicit a series of adverse effects via Ang II–dependent and Ang II–independent pathways. 7,8 Telmisartan, a AT1 receptor antagonist, prevents experimental aneurysms formation. 32 In the present study, we found that telmisartan reversed the adverse effects of PRR on Ang II–induced pathological changes in vivo and in vitro, whereas the adverse effects of co-administration of Ad-PRR and telmisartan were more serious than only telmisartan treatment, suggesting that PRR promoted Ang II–induced AAA formation via both Ang II–dependent and Ang II–independent pathways.
PRR triggered the worsening of cardiac function via Ang II–independent activation of ERK1/2 phosphorylation. 7 To determine the effect of PRR via Ang II–independent activation of ERK, we made the further research. We found that telmisartan decreased the incidence and mortality of Ang II–induced AAA. More importantly, the incidence and mortality of AAA were further decreased by co-treatment with telmisartan and PD98059. In addition, in vitro study, blockade of PRR expression with PRO20, a newly developed, highly specific PRR decoy inhibitor, abolished the adverse effects of Ang II on MMP expression, inflammation, and oxidative stress. In view of PRR as the upstream mediator of Ang II, the result of PRO20 treatment seemed controversial. Previous studies demonstrated that the inhibition of ERK significantly attenuated Ang II pathological effects in AAA. 31 Our study confirmed the PRO20 suppression of ERK phosphorylation, which might contribute to PRO20 elimination of the Ang II effect. We obtained similar results from PRR-knockdown mice. These data indicate that PRR triggers Ang II–induced AAA formation via Ang II–independent activation of ERK.
Conclusions
PRR overexpression promoted Ang II–induced AAA formation via both Ang II–dependent and Ang II–independent pathways in ApoE−/− mice. The mechanisms underlying these deteriorative effects included increased MMPs expression and activity, decreased SMC content, enhanced oxidative stress, increased inflammation, and activation of ERK and P38MAPK signaling. ERK inhibitor PD98059 eliminated the acceleration of Ang II–induced AAA formation by PRR, and co-administration of telmisartan and PD98059 further abolished the adverse effects of PRR on Ang II–induced AAA formation. Thus, PRR plays an important role in the pathological development of AAA via both Ang II–dependent and Ang II–independent activation of ERK pathways, and inhibition of PRR activation may be a promising approach to the treatment of AAA.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (Nos. 81870283, 81570729, 81170207), and Program of Taishan Scholars (No. ts 20190979).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.