
Abstract
Clinical translation of DNA-based administration of monoclonal antibodies (mAbs) is uncertain due to lack of large animal data. To bridge the clinical gap, we evaluated a panel of novel plasmid DNA (pDNA)-encoded mAbs in 40–70 kg sheep with a clinical intramuscular electroporation protocol. Injection of 4.8 mg of pDNA, encoding ovine anti-human CEA mAb (OVAC), led to peak plasma mAb titers of 300 ng/mL. OVAC remained detectable for 3 months and was boosted by a second pOVAC administration. Hyaluronidase muscle pretreatment increased OVAC concentrations up to 10-fold. These higher plasma titers, however, led to anti-drug antibodies (ADAs) toward the OVAC variable regions, resulting in loss of mAb detection and of adequate redosing. Transient immune suppression avoided ADA formation, with OVAC peaking at 3.5 μg/mL and remaining detectable for 11 months after pOVAC injection. DNA-based delivery of ovine anti-human EGFR mAb (OVAE), identical to OVAC except for the variable regions, preceded by hyaluronidase, allowed for at least three consecutive administrations in an immune-competent sheep, without ADA response. When tripling the pOVAE dose to 15 mg, transient ADAs of limited impact were observed; plasma OVAE peaked at 2.6 μg/mL and was detected up to 7 months. DNA-based anti-HER2 trastuzumab in sheep gave no detectable mAb concentrations despite previous validation in mice, highlighting the limitations of relying on small-rodent data only. In conclusion, our results highlight the potential and caveats of clinical DNA-based antibody therapy, can expedite preclinical and clinical development, and benefit the field of gene transfer as a whole.
Introduction
Recombinant monoclonal antibodies (mAbs) are part of the mainstay of treatments in several indications. A wider accessibility and implementation, however, is hampered by the high production cost and prolonged need for frequent administration. The surge in more effective but costly mAb combination therapies further adds to the financial burden. These issues highlight the need for innovations in conventional mAb production and delivery.
In vivo antibody gene transfer seeks to administer the mAb-encoding nucleotides, rather than the mAb protein. This allows the site of administration, for example, the muscle, to produce the therapeutic in a cost- and labor-effective manner for a prolonged period of time. 1 Applied expression platforms include viral vectors, 2 messenger RNA (mRNA), 3 and plasmid DNA (pDNA). 4,5 For the latter, electroporation is the standard clinical delivery approach, allowing efficient and safe uptake into the target tissue. Our laboratory previously demonstrated proof of concept for intramuscular DNA-based trastuzumab electrotransfer in mice, 6 complementing a mounting number of preclinical studies in small rodents that demonstrate promise in oncology, infectious diseases, and inflammatory disorders. 4 The advances in the past 15 years, however, have yet to translate to the clinic. No products are on the market, and clinical evaluation of DNA-based mAbs is currently limited to a completed Phase I–II for treatment of melanoma (NCT01138410) and a recently initiated first-in-human (FIH) Phase I for prevention of Zika virus (NCT03831503).
This slow clinical entry is in no small part related to the lack of pharmacokinetic (PK) and pharmacodynamic (PD) data in animal models that resemble humans in terms of body weight, musculature, and blood volume. Indeed, one of the main challenges for clinical DNA-based antibody therapy is “scalability,” that is, the ability to produce sufficient mAb in vivo to allow for therapeutic concentrations in circulation. It is currently uncertain whether such mAb exposure can be attained through intramuscular pDNA electrotransfer in human subjects, and what factors play a defining role. To bridge the clinical gap for DNA-based antibody therapy and compile a robust preclinical data set, we used 40–70 kg sheep as animal model, generated a panel of DNA-based ovine mAbs to match the host species, and implemented a clinical intramuscular electroporation protocol. Experiments were performed in both immune-competent and immune-suppressed sheep, to take into account the host immune system. mAb PK and formation of anti-drug antibodies (ADAs) were assessed using dedicated immunoassays.
Materials and Methods
Cell culture
FreeStyle 293-F suspension cells (purchased from Thermo Fisher Scientific in 2015) were maintained in FreeStyle 293 Expression Medium on a CO2-resistant orbital shaker (Thermo Fisher Scientific) in a 37°C humidified incubator at 8% CO2. Cell line identity was confirmed using short tandem repeat analysis at the Laboratory of Forensic Biomedical Sciences, KU Leuven, most recently in June 2018.
Animals
Female Swifter sheep of ∼1 year old and 40–45 kg were purchased from the KU Leuven Zootechnical Center (Lovenjoel, Belgium). Animals were housed on wood shavings and received hay and water ad libitum, and pellets twice daily. Six- to 8-week-old immune-deficient C57BL/6J RAG1 dko mice were purchased from the division Translational Research in Gastrointestinal Disorders (Department of Chronic Diseases, Metabolism and Ageing) at the KU Leuven. All animal experiments were approved by the KU Leuven Animal Ethics Committee (projects P163/2013, P211/2015, and P157/2017).
Antibody-encoding plasmids
Four plasmid constructs, each encoding a different mAb, were generated and evaluated. pOVAC encodes a fully ovine anti-human carcinoembryonic antigen (CEA) immunoglobulin G (IgG)1. pOVAE expresses a fully ovine anti-human epidermal growth factor receptor (EGFR) IgG1 and is identical to pOVAC except for the variable regions. The IgG1 constant heavy chain (HC) and constant lambda light chain (LC) complementary DNA (cDNA) sequences were derived from IMGT (X69797) and GenBank (AY734681.1), respectively. The variable HC and LC cDNA sequences of OVAC and OVAE were obtained from Bioventix under material transfer agreement. pTras encodes the humanized trastuzumab and was previously validated in vitro and in mice. 6 pOCT encodes an ovine chimeric version of trastuzumab, with the trastuzumab cDNA variable regions 7 grafted onto an ovine IgG1 framework. The constant kappa LC was derived from GenBank (CAA38046.1). The Kozak sequence preceded the start codon of each transgene followed by an appropriate signal peptide. OVAC, OVAE, and OCT cDNA sequences were codon-optimized for sheep and synthesized by Genewiz. Icosagen cloned the cDNAs into a single plasmid in which each mAb chain is driven by a ubiquitous CAG promoter. 6 The plasmid backbone includes an ampicillin-resistance gene and pUC origin of replication. Synthesis and cloning were verified via restriction analyses and sequencing (LGC). pDNA was produced in Escherichia coli, purified using the NucleoBond Xtra Maxi EF Kit (Macherey-Nagel) according to the manufacturer's instructions, and eluted in sterile D-PBS (no magnesium, no calcium, 14190144; Thermo Fisher Scientific). Plasmid purity and integrity were assessed via spectrophotometry and agarose gel electrophoresis. In vitro mAb expression was verified by enzyme-linked immunosorbent assay (ELISA) and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Intramuscular pDNA electrotransfer in mice
In mice, DNA electrotransfer was performed as described previously. 6 In brief, intramuscular injection of 30 μL of 2 μg/μL pDNA, formulated in sterile D-PBS, was followed by in situ electroporation using an NEPA21 Electroporator (Sonidel) with CUY650P5 tweezer electrodes at a fixed width of 5 mm. The applied electroporation protocol consisted of three series of four 20 ms square-wave pulses of 120 V/cm with a 50 ms interval between the pulses and polarity switching after two of the four pulses. The targeted tibialis anterior muscle was injected with 40 μL of 0.4 U/μL hyaluronidase from bovine testes (H4272; Sigma; reconstituted in sterile saline), ∼1 h before pDNA electrotransfer. Blood was collected via retro-orbital bleeding with capillary tubes coated with trisodium citrate dihydrate, processed to plasma via differential centrifugation, and stored at −20°C until analysis.
Intramuscular antibody gene electrotransfer in sheep
Intramuscular electrotransfer in sheep was based on a protocol that has been validated in swine and human subjects. 8,9 Sheep were sedated with 0.4–0.6 mg/kg xylazine intramuscularly (Xyl-M 2%; VMD). Induction was performed by 3.5–4 mg/kg ketamine intravenously (Nimatek 100 mg/mL; Dechra) or 3.5–5 mg/kg propofol (Diprivan; Aspen). Animals were intubated and ventilated with a tidal volume of 8–10 mL/kg at a frequency of 12 to keep normocapnia, and anesthesia was maintained with isoflurane (Iso-Vet Piramal Healthcare) or 16–24 mg/(kg·h) propofol. A gastric tube was placed and 0.9% NaCl was given continuously through an intravenous line in the saphenous vein. The left hind limb was shaven, disinfected and draped, and an incision of ∼20 cm was made 2–3 cm cranial of the virtual line connecting hip to knee. Unless stated otherwise, sheep received 12 injections of pDNA in a solution of sterile D-PBS, 200 μL each at a concentration of 2 μg/μL, totaling 4.8 mg of pDNA. The injections were done either in the musculus biceps femoris or musculus quadriceps. These muscles were selected for their accessibility, size, and thickness, all relevant parameters to accommodate the procedure. Sites of injection were marked with nonresorbable polypropylene sutures (Prolene; Covidien). pDNA injection speed was <2 s. Injections were typically divided across two parallel rows across the muscle, with at least 2–3 cm between the targeted sites to allow sufficient space and avoid overlap in electrical field. Any pDNA redosing was performed on the opposite flank or neighboring muscle, never on the sites of the past injections. In most animals (Table 1), electrotransfer of a muscle site was preceded by an injection of 400 μL of 0.4 U/mL hyaluronidase (H4272; Sigma), 1 h before pDNA injection. Approximately 10–15 s following pDNA injection, electrical pulses were applied using a Cliniporator generator (EPS01 FULL, serial number 00530107) and N-20-4B or A-15-4B needle arrays, both kindly provided by IGEA Medical. The needle array was placed perpendicular to the muscle fiber direction. The pulse protocol consisted of one high voltage pulse of 700 V/cm for 100 μs and one low voltage pulse of 80 V/cm for 400 ms with a 1 s interval. When the procedure was concluded, the incision was closed with resorbable sutures (Vicryl 3-0) and the animal was allowed to recover from anesthesia.
Overview of the treated sheep and regimen
CP, cyclophosphamide; i.m., intramuscular; i.v., intravenous.
Transient suppression of the sheep immune system
We implemented a previously reported sheep immune-suppression protocol. 10 Animals received an intravenous infusion of 37 mg/kg cyclophosphamide (CP) (Endoxan; Baxter), resolved in sterile water as per the manufacturer's instruction, on the day of intramuscular pDNA electrotransfer. To limit infection risk, the animal was housed solitary in a separated room of the animal facility. A Tyvek suit, shoe covers, gloves, a hairnet, and a mouth mask were worn when entering the room. Body temperature, appetite, activity, and skin turgor were routinely assessed. Prophylactic 5 mg/kg lincomycin–spectinomycin (VMD) were given intravenously at the time of the CP infusion and continued via intramuscular injection for another 2 days. To avoid the risk of sterile hemorrhagic cystitis, a known side effect of CP therapy in humans and dogs, sheep received 10 mg/kg furosemide (Dimazon; Intervet) daily for 2 weeks after CP injection: intravenously on the day of CP administration and intramuscularly afterward. Blood was collected intravenously and transferred to ethylenediaminetetraacetic acid (EDTA)-coated tubes (Greiner Bio-One). One tube was processed to plasma via centrifugation for plasma, and another tube was maintained as whole blood to monitor white blood cells (WBC) on an ADVIA 120 Hematology System (Siemens) (MedVet). WBC count included neutrophils, monocytes, basophils, eosinophils, and lymphocytes. The latter carried the highest relevance and are therefore included in the article.
In vitro mAb production and purification
mAbs were produced in vitro in FreeStyle 293-F cells and purified from the supernatant. Transfection of the encoding pDNA was carried out with X-tremeGENE HP DNA Transfection Reagent (Roche) in Freestyle media (Thermo Fisher Scientific) following the manufacturer's protocol. Five days after transfection, cells and media were collected. Supernatant was obtained via centrifugation and 0.2 μm filtration and stored at −20°C. Subsequent purification of the expressed mAbs from the supernatant was carried out on ÄKTAprime plus (GE Healthcare Life Sciences), using a 1 mL prepacked column with the Protein A affinity resin Amsphere A3 (JSR Life Sciences), according to the manufacturer's protocol. Following elution with 100 mM sodium acetate pH 3.5 and neutralization with 1M Tris pH 9, fractions were pooled and dialyzed to 20 mM sodium phosphate, 150 mM NaCl pH 7.5, aliquoted, and stored at −80°C. Glassware was rinsed with H2O2 30% to minimize endotoxin contamination. Batches of purified mAb were evaluated for consistency via an antigen-specific ELISA, SDS-PAGE, and ultraviolet spectrophotometry, as previously reported. 6
Monoclonal antibody ELISA
Ninety-six-well plates were coated overnight at 4°C with 4 μg/mL CEA in PBS (#11077-H08H; Sino Biological) for OVAC quantification, 1 μg/mL EGFR in PBS (10001-H08H; Sino Biological) for OVAE quantification, and 500 ng/mL human epidermal growth factor receptor 2 (HER2) in PBS (10004-H08H; Sino Biological) for trastuzumab or OCT quantification. Plates were blocked with 1% bovine serum albumin (BSA) in PBS for 2 h at room temperature (RT). Samples were diluted in PTAE (PBS 0.1% BSA, 0.002% Tween 80, 5 mM EDTA) and incubated on the blocked antigen-coated plates for 1 h at RT. Serial twofold dilutions of the purified mAb were used as calibration curve. Detection of the ovine mAbs and trastuzumab was performed with the rabbit anti-sheep IgG (Fc-specific), conjugated with horseradish peroxidase (HRP) (1:10,000 dilution in PTA, RASh/IgG(Fc)/PO; Nordic-Mubio), and the goat anti-human IgG (Fc-specific), conjugated with HRP (1:10,000 dilution in PTA, GAHu/IgG(Fc)/PO; Nordic-Mubio), respectively, and incubated for 1 h at RT. Each incubation step was preceded by a washing step with PBS 0.05% Tween 20. Plates were developed for 30–45 min using o-phenylenediamine and H2O2 in citrate buffer. The reaction was stopped with 4 M H2SO4. Absorption was measured at 490 nm using an ELx808 Absorbance Microplate Reader (BioTek Instruments). Plasma trastuzumab in sheep #4409 and #0999 was also quantified with a commercial HER2-coated SHIKARI Q-TRAS ELISA (Matriks Biotek) following the manufacturer's instructions. Sample concentrations were calculated based on the calibration curve using a nonlinear regression fit (GraphPad Prism 7.0).
Anti-drug antibody ELISA
The presence of antibodies against the in vivo expressed mAb was assessed via a drug-sensitive bridging or a drug-tolerant affinity capture elution (ACE) ELISA, setup as previously described. 11 For trastuzumab and OCT, the drug-sensitive bridging ELISA sufficed to pick up ADAs. In brief, 96-well plates were coated overnight with either trastuzumab (1 μg/mL) or OCT (0.5 μg/mL) in PBS and blocked with 1% BSA in PBS for 2 h. Diluted plasma samples were added to the plate (1 h of incubation), followed by the addition of the biotin-labeled mAb of interest as primary conjugate (1 h of incubation) and HRP-labeled streptavidin as secondary conjugate (30 min of incubation). For OVAC and OVAE, drug-tolerant ACE ELISAs were used, as these were more sensitive in detecting the ADAs. In brief, plates were coated overnight with 5 μg/mL OVAC or OVAE in bicarbonate buffer and blocked with 1% BSA in PBS for 1 h. Plasma samples were first diluted 1:2 in PTAE (PBS 0.1% BSA, 0.002% Tween 80, 5 mM EDTA), then further diluted 1:10 in 300 mM acetic acid (pH 3), and incubated 5–15 min to allow dissociation of complexes of ADA with the mAb of interest. Then, samples were transferred to the mAb-coated plate, prefilled with 1 M Tris buffer, and incubated for 1 h. Subsequently, the plate was washed and mAb-bound ADA was eluted using 300 mM acetic acid followed by transfer of the samples to noncoated plates containing 1 M Tris buffer and incubated 1 h allowing to coat the plate with the eluted ADA. Plates were blocked with 1% BSA in PBS for 1 h, and plate-bound ADAs was detected using the biotin-labeled mAb of interest (1 h of incubation) and HRP-conjugated streptavidin (30 min of incubation). All steps were performed at RT, except for the overnight coating, which was performed at 4°C. For both ADA assays, plates were developed using o-phenylenediamine and H2O2 and the reaction stopped with H2SO4. Absorption at 490 nm was measured using an ELx808 Absorbance Microplate Reader (BioTek Instruments).
Statistics
Sheep mAb PK and ADA data are reported in a descriptive manner. All data were based on a minimum of two independent assays, in which samples where ran in duplicate at different dilutions. When relevant, data were presented as mean + standard error of the mean and analyzed using Student's t-test, analysis of variance, and Tukey's multiple comparison test. Two-sided p values <0.05 were considered significant. Statistical analyses and figure drawing were carried out using GraphPad Prism 7.0 (GraphPad Software).
Results
Ovine anti-CEA mAb gene transfer in sheep
Data of a total of 12 sheep are reported in this study (Table 1). For the first experiments, we used the fully ovine anti-human CEA mAb (OVAC) as model. Sheep #0969 received an intramuscular electrotransfer of 4.8 mg pOVAC, divided across 12 injections of 200 μL at 2 μg/μL each. The resulting plasma OVAC concentrations peaked after 4 weeks at ∼300 ng/mL and remained above the ELISA limit of detection (50 ng/mL) for ∼3 months (Fig. 1A). A second 4.8 mg pOVAC dose, delivered 4 months after the first under identical conditions, led to a similar PK profile. Throughout follow-up, no anti-OVAC antibodies were detected, using both drug-tolerant or drug-sensitive assays. To increase the plasma OVAC concentrations, we pretreated the targeted muscle sites with hyaluronidase, an enzyme that transiently degrades hyaluronic acid and increases tissue permeability. In sheep #4520, this led to 6- to 10-fold higher plasma OVAC concentrations in the first 2 weeks, peaking at ∼1.7 μg/mL. In contrast to the hyaluronidase-naive sheep #0969, plasma OVAC decreased over the course of the next 3 weeks and was no longer detected 6 weeks after gene transfer (Fig. 1B). A second 4.8 mg pOVAC dose, 6.5 weeks after the first dose, failed to raise a similar mAb PK profile. The observed decline in plasma OVAC corresponded with the emergence of ADAs against OVAC, which was aggravated by the second pOVAC dose (Fig. 1C). Immune response reactivity was directed against the OVAC variable regions, as the ADAs did not bind OVAE, an ovine IgG1 identical to OVAC except for the variable regions (Fig. 1C). The impact of circulating ADAs was further illustrated by an intravenous injection of 3 mg of purified OVAC protein (Fig. 1D). In sheep #4520, the intravenous injection amplified the ADA response (Supplementary Fig. S1) and OVAC detection was lost after a week, likely due to ADA-associated binding and clearance. In contrast, in nontransfected control sheep #7262, OVAC remained detectable in circulation for up to 3 weeks after intravenous injection and no ADAs were detected.
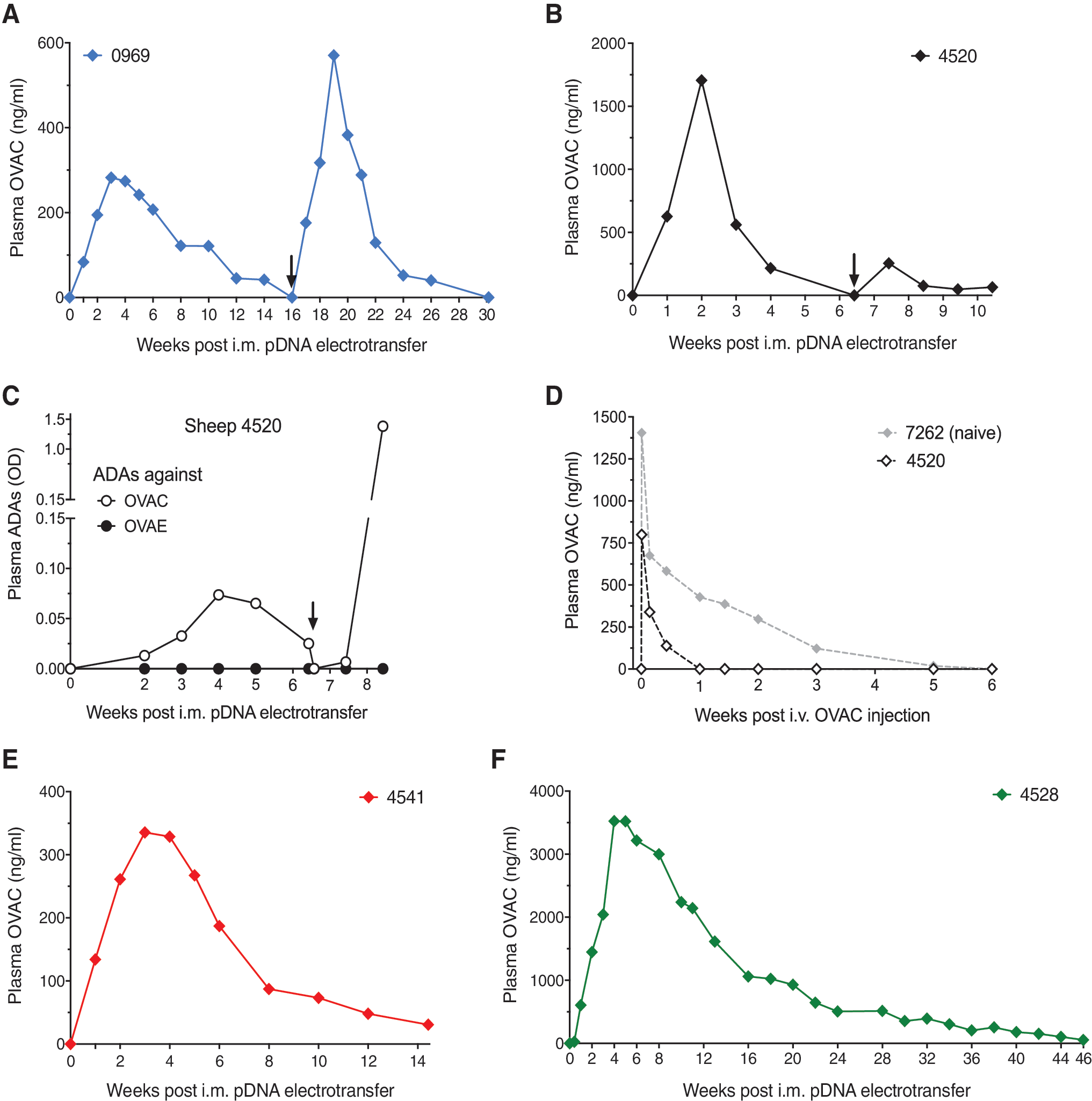
OVAC pharmacokinetics and ADAs in immune-competent and immune-suppressed sheep.
We subsequently assessed whether the ADA response in sheep #4520 was linked to the hyaluronidase pretreatment or to the magnitude of the OVAC titers. In sheep #4541, we lowered the pOVAC dose sixfold (two injections instead of the original 12) while hyaluronidase use was maintained. Plasma OVAC concentrations were similar to those in sheep #0969 (4.8 mg pOVAC and no hyaluronidase) (Fig. 1E vs. Fig. 1A), and similarly did not trigger an ADA response. These results suggest that the magnitude of plasma OVAC concentrations triggered the immune response, and not the use of hyaluronidase. Two strategies were subsequently pursued to overcome the ADA response: (1) immune suppression of the host and (2) modification of the delivered DNA-based mAb.
Impact of sheep immunosuppression on OVAC gene transfer
We applied a previously reported immune suppression protocol for sheep, in which animals receive a single intravenous dose of 37 mg/kg CP. 10 CP was administered at the time of the intramuscular electrotransfer of 4.8 mg pOVAC, which was preceded by hyaluronidase treatment. In sheep #4528, lymphocytes decreased 44% in the first week and restored to baseline levels within 2 weeks after CP injection (Supplementary Fig. S2A). CP side effects included a few days of appetite loss and transient alopecia, in line with clinical observations. No anti-OVAC antibodies were detected throughout the entire follow-up, even after lymphocyte levels returned to baseline. Plasma OVAC continued to increase for several weeks, peaking after 4 to 5 weeks at ∼3.5 μg/mL and remaining detectable for 11 months (Fig. 1F). These data demonstrate that a transient immune suppression avoided an ADA response against OVAC, enabling plasma mAb concentrations that were higher and detected for a longer period of time than in the immune-competent sheep.
Impact of mAb modifications via OVAE gene transfer
A second strategy to address the observed ADA response focused on mAb design. Because the ADA response was directed against the OVAC variable region, we swapped the latter for that of an ovine anti-human EGFR mAb (OVAE). The resulting pOVAE is thus identical to pOVAC except for the variable region. Intramuscular electrotransfer of 4.8 mg of pOVAE in sheep #0406, preceded by hyaluronidase, led to peak titers of ∼600 ng/mL 2 weeks after pDNA administration. OVAE remained detectable for close to 3 months (i.e., >50 ng/mL) (Fig. 2A). A second and third pOVAE dose, administrated under identical conditions with a lag period of ∼16 weeks, each time led to a plasma PK profile similar to the first dose. The weight of sheep #0406 increased during the course of the study, from 43 kg (first dose) to 60 kg (second dose) and 70 kg (third dose). For the last administration, the pDNA dose was adapted for the increased weight and 7.6 mg pOVAE was administered instead of 4.8 mg (Fig. 2A). Overall, peak plasma OVAE concentrations were approximately threefold lower than those obtained with pOVAC (Fig. 2A vs. Fig. 1B). Throughout follow-up, no anti-OVAE antibodies were detected using either a drug-sensitive or a drug-tolerant assay.
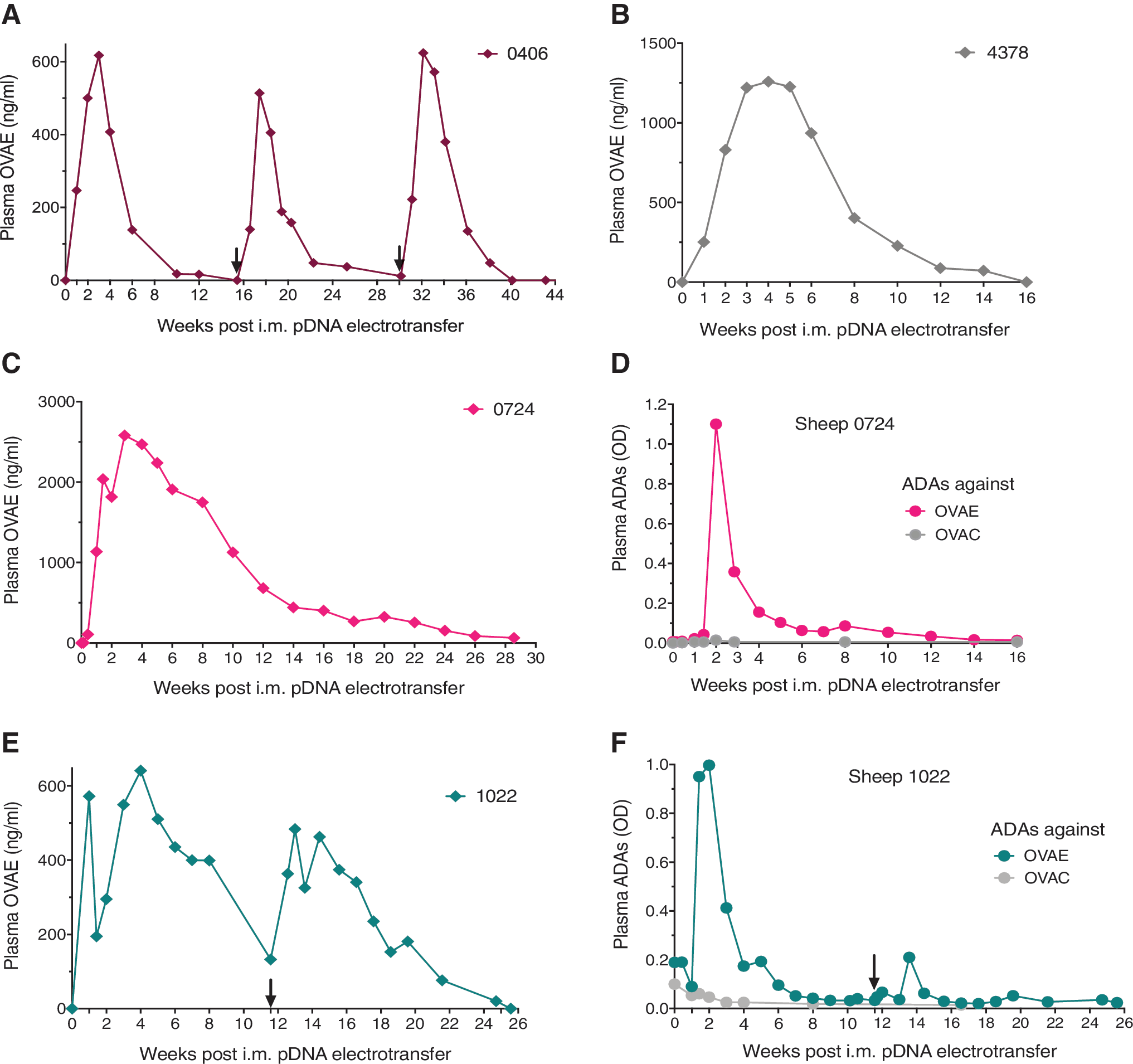
OVAE pharmacokinetics and ADAs in immune-competent and immune-suppressed sheep.
To evaluate the impact of the host immune system on OVAE expression, sheep #4378 received 4.8 mg pOVAE via intramuscular electroporation, preceded by hyaluronidase, with a concomitant intravenous injection of 37 mg/kg CP. Lymphocytes demonstrated a maximal drop of 34% and returned to baseline within a week after CP administration (Supplementary Fig. S2B). Compared with the immune-competent and CP-naive sheep #0406, OVAE titers showed a more moderate decrease over time, but the overall duration of detection was comparable (Fig. 2B). The limited difference in OVAE PK between the immune-competent and immune-suppressed sheep further supports the absence of ADAs.
To assess whether the lack of ADAs with pOVAE was linked to the lower plasma OVAE concentrations, we increased the pOVAE dose from 4.8 to 15 mg in an immune-competent sheep, #0724, maintaining hyaluronidase pretreatment and pDNA concentration. This increased plasma OVAE titers up to fourfold, with peak plasma concentrations of ∼2.6 μg/mL at week 3 (Fig. 2C). The minor and brief drop in OVAE (10%) at week 2 corresponded to a transient spike in anti-OVAE antibodies (Fig. 2D). Immune response reactivity was directed against the OVAE variable regions, as the ADAs did not bind OVAC. The ADA emergence had no further obvious impact, and OVAE remained detectable for ∼7 months. This is likely due to the transient nature and low OVAE ADA response, which nevertheless was detected by our sensitive drug-tolerant assays. Similar observations have been reported with conventional mAb protein therapy, where transient ADA responses show little to no impact on mAb PK. 12 This is different from what was previously observed with pOVAC (Fig. 2C vs. Fig. 1B), where OVAC ADAs led to loss of detection of the ability to adequately redose. To further explore this observation, another sheep, #1022, received 15 mg pOVAE. To reduce the number of injections (i.e., from 38 to 25), dose concentration was increased from 2 to 3 μg/μL. This, however, had undesired consequences. Compared with sheep #0724, plasma OVAE concentrations were about threefold lower, the drop in plasma OVAE (65%) around week 2 was more pronounced with a rebound that took a few weeks longer (Fig. 2E). Since the timing and magnitude of the ADA response was similar in sheep #1022 and #0724 (Fig. 2F vs. Fig. 2D), the more obvious impact of ADAs in the former could be linked to the lower plasma mAb titers; indeed, for the same amount of ADAs, less free OVAE would remain in circulation. High pDNA concentrations are subject to a higher viscosity and are known to impact in vitro cell viability and immune responses, which could all play a role in the lower titers that were observed at 3 μg/μL. About 12 weeks after the initial 15 mg pOVAE, the animal was redosed under identical conditions. The PK profile was similar as observed with the first dose, but the ADA response and, possibly as a consequence thereof, drop in OVAE titers were less pronounced. pOVAE redosing did not amplify the ADA response, as opposed to pOVAC (Fig. 2F vs. Fig. 1C).
Overall, pOVAE appeared somewhat less immunogenic than pOVAC but was not exempt from triggering ADAs. These data reveal that several parameters, including mAb sequence, plasma mAb titers, and pDNA concentrations can impact the host immune response.
Trastuzumab gene transfer in sheep
To broaden the application of our sheep model, we evaluated the intramuscular electrotransfer of pTras, a plasmid that encodes the clinically approved humanized anti-HER2 trastuzumab. This construct was previously generated and validated in mice, demonstrating prolonged mAb expression and significant antitumor responses. 6 To address the anticipated immune response against expressed trastuzumab, we immune-suppressed the sheep using the CP protocol. In sheep #4409, intramuscular electrotransfer of 4.8 mg pTras, preceded by hyaluronidase, did not lead to detectable plasma trastuzumab. Despite the immune suppression, a marked increase in anti-trastuzumab antibodies was detected as early as 11 days after pDNA electrotransfer (Fig. 3A). A second pTras administration 10 weeks later, at double the amount of pDNA and without CP administration, again failed to result in trastuzumab detection. It did induce a surge in ADAs, similar to the earlier observations for pOVAC (Fig. 1C). These data indicate that a single CP dose was unable to avoid an ADA response against trastuzumab. The applied pTras dose was also insufficient to reach detectable mAb concentrations in the first week following electrotransfer, before ADAs emerged. In sheep #0999, we attempted to address both issues by implementing the following modifications. The applied pTras dose was increased to 15 mg. To maintain the immune-suppressed state for a more prolonged period of time, a second CP dose was administered 8 days after the first dose (Supplementary Fig. S2C). These modifications failed to overcome either issue; no plasma trastuzumab was detected during the first week and an ADA response was again obvious 10 days after intramuscular electrotransfer (Fig. 3B). Follow-up of the animal was therefore discontinued. These data demonstrate that the increased pDNA dose only led to a more robust ADA response and that modifications to the immune suppressive protocol are required to overcome the immunogenicity of humanized mAbs. A third or fourth CP dose was previously found to be too toxic for sheep and was therefore not applied. The absence of mAb detection but the presence of ADAs indicates that the mAb is likely expressed at concentrations below the detection range of the commercial trastuzumab ELISA (reportedly 2 ng/mL).

Plasma trastuzumab, OCT, and ADAs in sheep.
Ovine chimeric trastuzumab gene transfer in sheep
We evaluated whether the ADA response against trastuzumab could be avoided by engineering an ovine chimeric trastuzumab construct (pOCT), which implied replacing the human IgG1 constant region by an ovine IgG1 constant region. Intramuscular electrotransfer of 4.8 mg pOCT, preceded by hyaluronidase, in sheep #0492 led to detectable but low peak plasma concentrations of ∼110 ng/mL, 10 days after electroporation (Fig. 3C). Plasma OCT concentrations dropped to nondetectable levels shortly after, which correlated with the emergence of anti-OCT antibodies (Fig. 3D). The ADA signal was detected against OCT and trastuzumab, but not against OVAE. This indicated that the ADA response was directed towards the variable trastuzumab regions of the chimeric OCT. Overall, chimeric ovine engineering of trastuzumab increased the mAb expression somewhat, but the humanized variable region remained highly immunogenic in sheep.
DNA-based mAb expression in vitro and in mice
Among the different DNA-based mAbs, the resulting PK profile in sheep varied considerably. Trastuzumab was not detected, even before an ADA response, and OCT had a fairly limited expression. In contrast, at the same pDNA dose, OVAC and OVAE plasma concentrations attained already several hundred nanograms per milliliter in the first week, with OVAC having about twofold higher peak concentrations than OVAE in the weeks after. We explored whether these differences were also obvious in vitro and in mice. In vitro expression was evaluated by transfecting FreeStyle 293-F cells with the different plasmids. The highest mAb concentrations were detected in the supernatant of the pOVAC transfected cells, respectively 2.8-, 13-, and 27-fold higher than OVAE, trastuzumab, and OCT (Fig. 4A). pOVAC, pOVAE, and pTras were further evaluated in immune-deficient RAG1 dko mice, via a single intramuscular electrotransfer of 60 μg pDNA. This resulted in peak plasma concentrations of 24 μg/mL OVAC, 13 μg/mL OVAE, and 3.5 μg/mL trastuzumab (Fig. 4B). In a separate experiment in RAG1 dko mice, plasma OVAC concentrations up to 5 μg/mL were maintained for over 1 year after intramuscular electrotransfer (Supplementary Fig. S3). Overall, the differences among the DNA-based mAbs at sheep level were reflected in vitro and in mice. The less efficient expression of trastuzumab in vitro and in mice may consequently explain the lack of detectable levels in sheep. Other variables might also contribute, including mAb half-life, target cross-reactivity, and expression efficiency of fully or chimeric ovine mAbs versus foreign mAbs in sheep.

Differential mAb expression in vitro and in mice.
Discussion
To support the clinical translation of intramuscular DNA-based antibody gene transfer, we generated an extensive data package in sheep. The translational nature of our study is supported by the following elements.
First, sheep resemble humans in terms of body weight, blood volume, and musculature, providing a relevant model to assess clinical feasibility. The 40–70 kg sheep in this study present the largest animal model in which this approach has been reported to date. In addition to mice, DNA-based antibody gene transfer has previously been evaluated in 15–17 kg lambs 5 and 5–6 kg nonhuman primates (NHPs), 13 but the size of these animals still limited clinical relevance. Second, we generated a panel of DNA-based ovine mAbs to match the host species, as would typically be the case in the clinic. Third, the mAb-encoding DNA was administered via a clinical intramuscular electroporation protocol, 9 providing a direct path forward to use in human subjects. Fourth, both plasma mAb concentrations and ADAs were assessed and quantified using dedicated immunoassays.
This allowed us to gain insight in the magnitude and duration of in vivo mAb expression, two key translation factors for DNA-based mAb therapy, and in the impact of parameters such as mAb construct, dosing, administration, and ADAs.
In a first set of experiments, the expressed OVAC was found in circulation for close to 3 months after pOVAC electrotransfer, significantly longer than when administered intravenously as a protein. A second pOVAC dose resulted in a similar mAb PK profile, illustrating our ability to successfully retreat. Hyaluronidase pretreatment increased plasma OVAC concentrations up to a 10-fold, reaching therapeutically relevant concentrations in the single-microgram per milliliter range. Hyaluronidase has previously shown to improve intramuscular transgene electrotransfer in mice. 6,14 This enzyme is also routinely used in various applications, 15,16 including mAb formulation for subcutaneous administration, and could support clinical translation of antibody gene electrotransfer. The higher OVAC titers did trigger an ADA response against the variable fragment of OVAC, which led to the loss of mAb detection and of ability to successfully retreat.
To address this pitfall, two strategies were pursued. First, we suppressed the sheep immune system with CP, a clinically approved immune suppressant that indiscriminately kills proliferating cells, including T and B cells, by interfering with DNA replication. 10,17 A single CP dose given concomitantly with pOVAC avoided the formation of ADAs, even after the lymphocyte cell count returned to normal 1 to 2 weeks later. This enabled OVAC to reach peak plasma concentrations of up to 3.5 μg/μL and remain detectable in circulation for close to 1 year. Compared with immune-competent sheep, immune suppression with CP led to a longer mAb accumulation in the first weeks following pDNA administration. One possible explanation is that CP avoids or delays the innate and cell-based immune responses directed against the mAb-expressing muscle cells. This is subject to further research. The resulting higher OVAC concentrations in the immune-suppressed sheep also take more time to be cleared, adding to the longer detection in circulation. Although CP was highly effective in avoiding an ADA response against the ovine mAbs, the trastuzumab studies showed that further modifications to the immune suppressive protocol are warranted to broaden its use. One antibody gene transfer study, using viral vectors, previously reported the immune suppression of NHPs. 18 Monkeys received cyclosporine A every other day to avoid ADAs against an rAAV-expressed simianized mAb. Although effective while on treatment, cessation of cyclosporine A led to a rapid ADA emergence and decrease in mAb titers. 18,19 The difference with our observations is likely linked to the use of different immune suppressants and/or expression platforms.
Despite the prolonged and robust mAb expression, DNA-based mAb delivery in immune-suppressed subjects does not represent the preferred clinical scenario. A second strategy to address the ADAs focused on modifying the mAb variable regions, which elicited the response, via OVAE—identical to OVAC except for the variable region. The initial evaluation of pOVAE did not lead to ADA responses, although the resulting plasma OVAE concentrations where consistently lower than with pOVAC. Only when increasing the pOVAE dose to 15 mg, a transient ADA response was observed, although the impact was less pronounced than with pOVAC.
Overall, a complex picture emerged from these data, one in which several parameters, including mAb sequence, plasma mAb titers, and pDNA concentrations, appear to play a role in humoral antibody immune response. These observations require further scrutiny in a drug development context. Indeed, ADAs are frequently observed with conventional clinically approved mAbs 20 and are equally, if not more, critical for in vivo expressed mAbs. A better understanding of the underlying mechanisms is thus warranted. Possible routes forward include the use of plasmids less likely to trigger an immune response, for example, by eliminating the bacterial backbone or reducing CpG content 21,22 or by using muscle-specific rather than ubiquitous promoters. 23
The data in sheep also exposed the limitations of small-rodent studies, carrying relevance toward preclinical development of DNA-based mAbs. The added value of sheep was obvious at different fronts. First, the expressed mAb PK profile differed between mice and sheep. In immune-competent sheep, expressed OVAC and OVAE were detected for a prolonged but also defined period of time, typically 3 to 4 months. In immune-competent mice, we previously found that the expression of a murine mAb lasts at least 9 months. 6 The underlying reason for this difference is currently unclear but carries translational relevance. If replicated in human subjects, a more defined mAb expression could allow for a better control and have a higher biosafety profile, broadening the application scope. Second, promising PK/PD data in mice are no guarantee for clinical translation. We previously demonstrated that intramuscular electrotransfer of DNA-based trastuzumab (pTras) gave high mAb titers and was highly effective in a breast tumor mouse model. 6 pTras in sheep, however, gave no detectable plasma mAb concentrations, even before an ADA response emerged. These findings are a critical reminder of the limited translational value of small-animal models without a proper reference window.
In the current study, data from 12 sheep that received a total of 17 intramuscular electrotransfer procedures were included. Sheep received between 2 and 38 pDNA doses per treatment cycle, with even the smallest number of injections leading to detectable mAb expression. Due to the ethical considerations with the use of these large animals, not all experimental setups were performed in multiple sheep. The observed reproducibility and consistency between different experiments, however, support the robustness of our data set. pDNA administrations typically gave similar PK profiles in different as well as in individual animals, in which the redosed animals acted as their own control and allowed to confirm observations. We also observed a clear dose–response for both pOVAC and pOVAE, further supporting the consistency of our procedures.
Pending further validation, our setup in sheep can play different roles to advance DNA-based mAb development. It can help to further elucidate the mechanisms that drive PK/PD of in vivo expressed mAbs. It can also be used to assess and anticipate the clinical impact of modifications to the electroporation procedure, pDNA dosing, and construct design. In addition to the myriad of criteria mAb proteins are submitted to, the development of DNA-based mAbs could be complemented with a sequence of expression screens, in which the best in vitro expressing candidates are advanced for PK/PD evaluation in both small and large animal models for lead selection. This approach could allow entering FIH trials with a more robust data set and de-risked DNA-based lead. The application of the sheep model also goes beyond DNA-mediated antibody gene transfer. Other expression platforms, for example, viral vectors or mRNA, and transgenes (antibody fragments, cytokines, etc.), could be evaluated for translatability.
In conclusion, our translational sheep model highlights both the potential and caveats of clinical DNA-based mAb therapy, serves to steer and expedite preclinical and clinical development, and can benefit the field of gene transfer as a whole.
Footnotes
Acknowledgments
The authors wish to thank Dr. Mattia Ronchetti (IGEA Medical) for providing a Cliniporator generator and needle electrodes, Inez Feyton and Hans Dierckx (KU Leuven) for taking care of the sheep, and Gerald Platteau (JSR Life Sciences) for his support regarding protein A-based antibody purification.
Authors' Contributions
K.H., N.G., and P.D. contributed to study design. K.H., S.D.V., E.D.S., and G.V. collected material or performed experiments. K.H., S.D.V., E.D.S., N.G., and P.D. interpreted results. K.H. wrote the article. All authors reviewed and approved the article for publication.
Author Disclosure
K.H. received consulting fees from OncoSec Medical. All other authors declare no conflict of interest.
Funding Information
This research is funded by Research Foundation—Flanders (FWO: postdoctoral mandate 1278114N and research project G0E2117N), KU Leuven (C2 grant: C22/15/024), and Flanders Innovation and Entrepreneurship (VLAIO: IWT.150743).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.