
Abstract
Liver fibrosis is a chronic liver disease that could further develop to cirrhosis and liver carcinoma. Hepatic stellate cells (HSCs) are primary effector cells to initiate liver fibrosis. We aimed to explore the function and underlying mechanisms of mitochondrial fusion protein Mitofusin-2 (MFN2) in liver fibrosis. First, we utilized an alpha-smooth muscle actin promoter to overexpress MFN2 specifically in HSCs using adeno-associated virus (AAV) vector (AAV-MFN2). Overexpression of MFN2 was specifically achieved in HSC-T6 cells, but not in murine bone marrow-derived macrophages or hepatocyte AML-12 cells. We found that high expression of MFN2 induced apoptosis of HSC-T6 cells. Mechanistically, we demonstrated that high level of MFN2 inhibited TGF-β1/Smad signaling pathway, triggered downregulation of type I, type III, and type IV collagen, and antagonized the formation of factors associated with liver fibrosis. Furthermore, we found that overexpression of MFN2 using AAV-MFN2 ameliorated CCl4-induced liver fibrosis in vivo with significantly decreased immune cell infiltration. Taken together, our findings indicate that MFN2 is critical in regulating apoptosis and liver fibrosis in HSCs, which might be a useful therapeutic target to treat liver fibrosis.
Introduction
Liver fibrosis is a chronic liver disease with excessive extracellular matrix protein accumulation such as collagens. 1 It can further develop and process into severe diseases such as cirrhosis and hepatocellular carcinoma and finally death. 2,3 The causes for liver fibrosis include hepatitis C virus (HCV) infection, alcohol abuse, and nonalcoholic steatohepatitis. 4 The understanding of pathogenesis and the underlying mechanisms of liver fibrosis are greatly improved during the past decades. Emerging clinical and experimental evidence suggests that liver fibrosis can be reversed by suppressing the accumulation of extracellular matrix proteins. 5,6 Therefore, it is crucial to develop efficient therapeutic approaches to treat liver fibrosis.
Multiple types of cells including hepatocytes, activated hepatic stellate cells (HSCs), and immune cells cooperate to initiate liver fibrosis. 7 HSCs are primary effector cells of liver fibrosis and activated HSCs differentiate into fibroblasts, producing a large number of extracellular matrices. 7 –10 Liver leukocytes (NK cells, Kupffer cells, T and B lymphocytes, and so on) have been identified as critical players in liver fibrosis by inducing the transition of activated HSCs to myofibroblast. 11 A variety of treatments have been utilized to cure liver fibrosis including anti-inflammatory treatment and removal of HCV infection. 10 Although targets to prevent liver fibrosis have been identified in mouse studies, the efficacy of treatments has been proven in humans and there is no standard liver fibrosis treatment in clinical settings. 1 Inhibition of HSC activation has been demonstrated to be a promising strategy to treat liver fibrosis. 12,13 Extensive studies have been performed to understand the mechanisms underlying regulation of HSC activation.
Mitochondrion is a multifunctional organelle that is highly correlated with the state of cellular function and plays an essential function in cell cycle, metabolism, proliferation, and apoptosis. 14 The mitochondrial fusion proteins (Mitofusin, MFN), including MFN1 and MFN2, are required for mitochondrial outer membrane fusion. 15 MFN2 has multiple functions in regulating various cellular processes, including cell proliferation, apoptosis, ER stress, and autophagy. 16,17 Yongbiao Chen et al. reported that MFN2-protected hepatocyte mitochondrial function against the liver damage induced by glycochenodeoxycholic acid. 18 MFN2 has positive effect on insulin sensitivity and overexpression of MFN2 improves high-fat diet-induced insulin resistance. 19 However, whether MFN2 plays essential functions in liver fibrosis is not fully elucidated.
In this study, we constructed an adeno-associated virus (AAV) vector that could overexpress MFN2 (AAV-MFN2) in HSCs specifically using alpha-smooth muscle actin (α-SMA) promoter to examine the biological functions of MFN2 in the regulation of liver fibrosis.
Materials and Methods
Animals and CCl4-induced liver fibrosis model
C57BL/6 mice (male) were obtained from Vital River Laboratory (Beijing, China). The animal experiments were approved by The Institutional Animal Care and Use Committee of Hangzhou First People's Hospital. To establish CCl4-induced liver fibrosis model, mice were administrated with 10% CCl4 in olive oil twice a week for 4 weeks. In total, 2.5 × 1011 vg/mouse AAV-Ctrl or AAV-MFN2 was injected intravenously twice a week after the first CCl4 administration. Animals were euthanized by CO2 inhalation at the end of experiments.
Cell culture
Bone marrow-derived macrophage (BMDM) was prepared from C57BL/6 as previously described. 20 Hepatocytes AML12 and HSCs HSC-T16 were from Cell Bank, Institute of Biochemistry and Cell Biology (Shanghai, China). Cells were cultured in DMEM with 10% fetal bovine serum (Gibco, Grand Island, NY) and 1% penicillin/streptomycin (Life Technologies, Pleasanton, CA) in a humid incubator with 5% CO2 at 37°C. In total, 2 × 105 cells were seeded into a 12-well plate and transduced with 1 × 105 vg/cell AAV-Ctrl or AAV-MFN2 12 h later. The green fluorescent protein (GFP) reporter was examined 48 h later using a confocal microscope.
Vector construction and AAV package
MFN2 or null control was cloned into the backbone of pAAV-CAG-GFP vector and the CAG promoter was replaced with an α-SMA promoter. For AAV package, HEK293T cells were transfected with the backbone vector and packaging vector at a ratio of 2:1 using Lipofectamine 3,000 (Life Technologies). AAVs were concentrated and titrated with a commercial kit (ThermoFisher, Waltham, MA). The primer sequence of MFN2 for real-time PCR was forward sequence: CATTGCTGACAGGATGCAGAAGG, reverse sequence: TGCTGGAAGGTGGACAGTGAGG.
Reverse transcription-polymerase chain reaction
Total RNA was prepared from liver tissues or cultured cells using Trizol (ThermoFisher) and reverse transcribed to cDNA. Real-time qPCR was conducted using SYBR Green Master mix (Takara, Dalian, China). All primers were obtained from Origene. The relative mRNA expression levels of target genes were normalized to GAPDH control using the 2−ΔΔCT method.
Western blot
Western blotting was conducted following standard protocols and the primary antibodies for Western blot were anti-MFN2, antitransforming growth factor (TGF)-β1, anti-Smad2/p-Smad2, anti-Smad3/p-Smad3, anti-Bax, anti-Bcl-2, and anticleaved-caspase-3 (all from Cell Signaling Technology, Danvers, MA).
Cell apoptosis assay
HSC-T6 cells were transduced with AAV-Ctrl or AAV-MFN2. Seventy-two hours later, cells were stained with Annexin V/PI Kit (BD Science, San Diego, CA) and analyzed by flow cytometry to determine the apoptosis rate.
Enzyme-linked immunosorbent assay
Cell culture supernatants were collected 48 h after transduction. Commercial enzyme-linked immunosorbent assay (ELISA) kits for type I, type III, and type IV collagen were purchased from LianKe Biotech (Hangzhou, China) and ELISA was performed following the manufacturer's manual.
Serum alanine aminotransferase
Serum samples were collected and serum alanine aminotransferase (ALT) was assessed using a commercial kit (Rongsheng, Shanghai, China).
Hematoxylin and eosin and sirius red staining
Hematoxylin and eosin (H&E) staining was conducted as previously described. 21 In brief, liver tissues were fixed and embedded in paraffin. Tissue sections (6 μm) were deparaffinized and stained with H&E to analyze the morphological changes and liver damage, or stained with sirius red to evaluate liver fibrosis.
Flow cytometry analysis
Liver mononuclear cells were prepared as described previously. 21 The antibodies used for staining were all from BioLegend, San Diego, CA: APC-CD45, FITC-CD11b, PE-Cy7-CD19, Percp-Cy5.5-CD3, PE-Cy5-F4/80, and BV510-Gr-1. The stained cells were analyzed using BD LSR II and the data were analyzed with Flowjo (BD, San Jose, CA).
Statistics
Results were statistically analyzed using Graphpad Prism 7 (Prism, San Diego, CA). Data were analyzed using one-way ANOVA with a Bonferroni correction and presented as mean ± standard deviation. A p < 0.05 was considered statistically significant.
Results
Construction and validation of AAV-MFN2 for HSC-specific MFN2 overexpression
To study the function of MFN2 in HSCs, we first constructed an AAV vector overexpressing MFN2 (Fig. 1a). α-SMA was reported to be specifically expressed in activated HSCs. 22 So, we incorporated the α-SMA promoter in the AAV vector to achieve the HSC-specific MFN2 overexpression and a GFP reporter to visualize the transduced cells (AAV-MFN2) (Fig. 1a). AAV-MFN2 was transduced into murine BMDMs, hepatocyte AML-12, and HSC line HSC-T6. The GFP reporter was specifically expressed only in HSC-T6 cells (Fig. 1b). We also confirmed the HSC-specific MFN2 overexpression in HSC-T6 cells, not in BMDMs and AML-12 cells, by real-time PCR and Western blotting (Fig. 1c–e). Thus, we successfully constructed and validated the AAV-MFN2 for HSC-specific MFN2 overexpression.

Construction and validation of AAV-MFN2 for HSC-specific MFN2 overexpression.
MFN2 overexpression promotes HSC apoptosis
To explore the MFN2 function on HSCs, we transduced HSC-T6 cells with AAV-MFN2 or AAV-Ctrl and analyzed apoptosis-related targets. As shown in Fig. 2a and b, MFN2 overexpression significantly enhanced the expression levels of proapoptosis molecules such as Bax and cleaved-caspase 2, while dramatically inhibited the expression of antiapoptosis Bcl-2. Annexin-V/PI staining also revealed that overexpression of MFN2 remarkably increased the cell apoptosis (Fig. 2c, d). Thus, MFN2 overexpression greatly promoted HSC apoptosis.
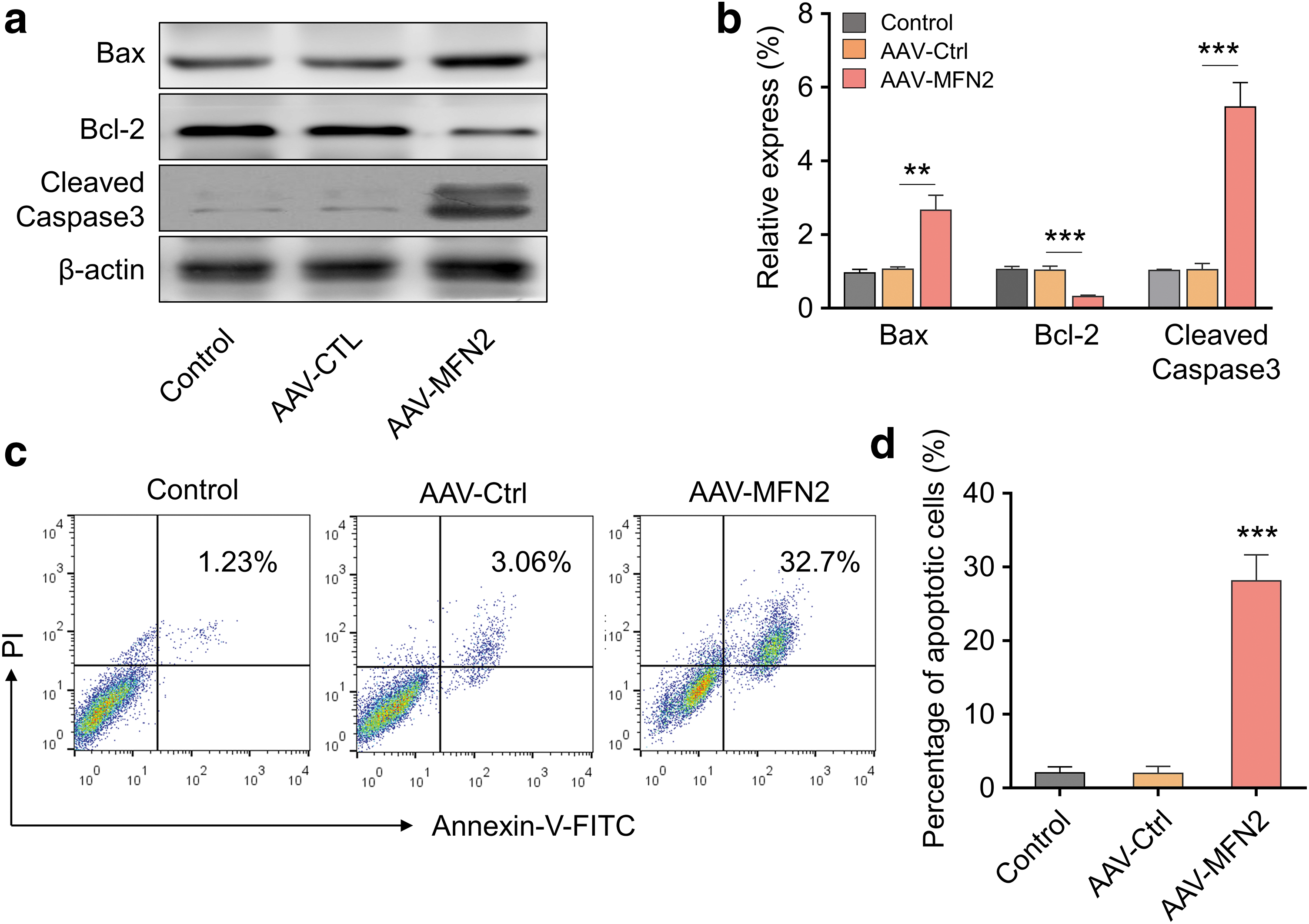
MFN2 overexpression promotes HSC apoptosis.
MFN2 overexpression suppresses TGF-β1/Smad signaling and inhibits collagen production in HSCs
Emerging evidence documented that TGF-β1/Smad signaling is critical for liver fibrosis progression. 23,24 We then explored the function of MFN2 overexpression on TGF-β1/Smad signaling in HSC-T6 cells. The results suggested that MFN2 overexpression decreased the protein levels of TGF-β1, and the phosphorylated Smad2/Smad3 (Fig. 3a). Furthermore, we found that type I, type III, and Type IV collagen in the culture supernatant of HSC-T6 cells transduced with AAV-MFN2 were drastically decreased in comparison with those in AAV-Ctrl groups (Fig. 3b). These findings indicated that MFN2 overexpression might ameliorate liver fibrosis.
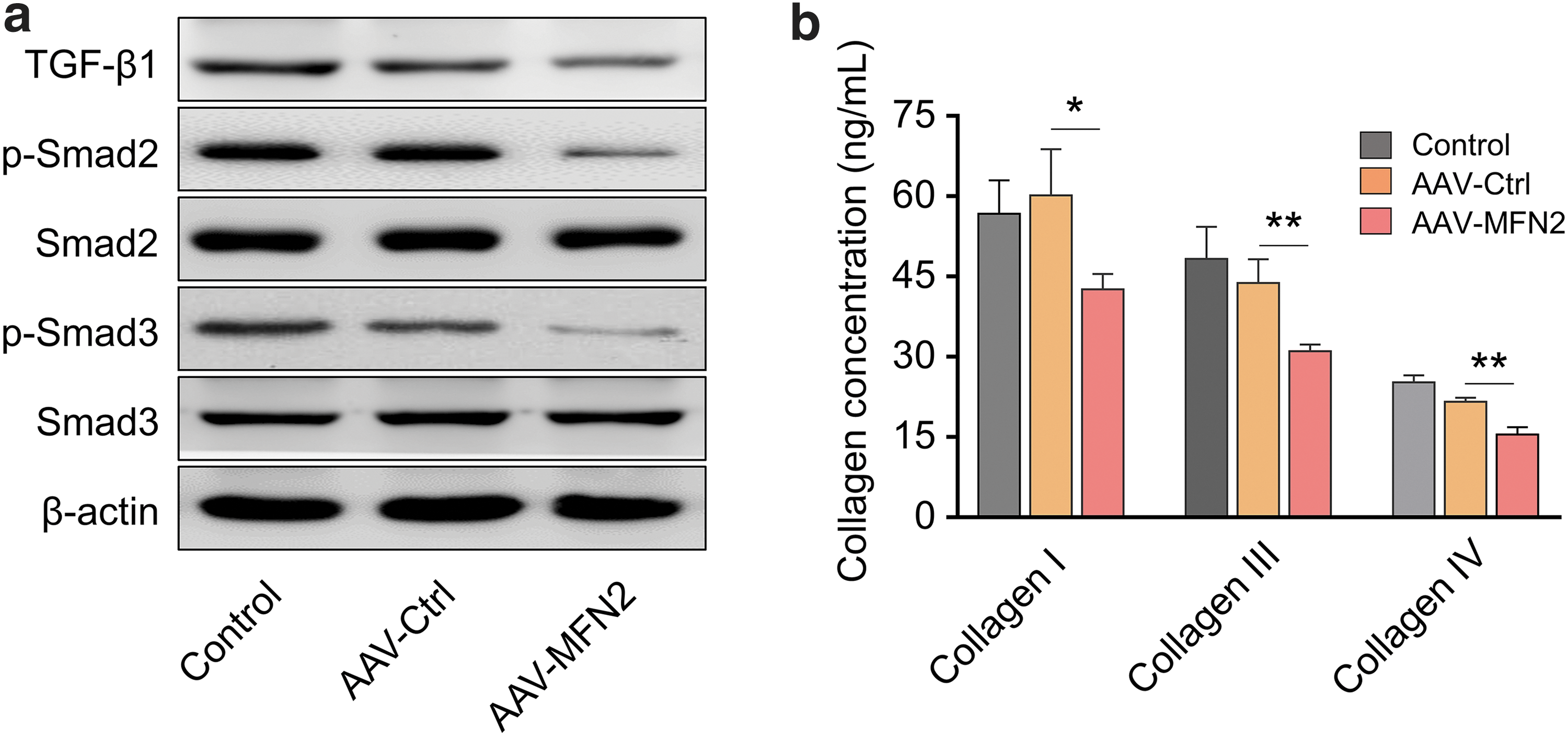
MFN2 overexpression suppresses TGF-β1/Smad signaling and inhibits collagen production in HSCs.
MFN2 overexpression ameliorates liver fibrosis in CCl4-treated mice
To further evaluate the function of MFN2 on liver fibrosis in vivo, we treated the mice with CCl4 to induce liver fibrosis, together with AAV-Ctrl or AAV-MFN2 (Fig. 4a). MFN2 overexpression significantly protected liver from liver injury induced by CCl4 treatment, with a much lower ALT level in AAV-MFN2 group (Fig. 4b). Key genes involved in liver fibrosis such as Mmp2, Colla1, Acta2, and Timp1 were analyzed in liver tissues from different groups and the results demonstrated that MFN2 overexpression markedly decreased the expression of Mmp2, Colla1, Acta2, and Timp1 (Fig. 4c). In addition, sirius red staining also showed that MFN2 overexpression ameliorated liver fibrosis with significantly less sirius red positive area in live tissue slice (Fig. 4d, e).

MFN2 overexpression ameliorates liver fibrosis in CCl4-treated mice.
MFN2 overexpression inhibits immune cell infiltration in the liver of CCl4-treated mice
We further investigate the function of MFN2 overexpression on immune cell infiltration in the liver of CCl4-treated mice. As shown in Fig. 5a, MFN2 overexpression greatly ameliorated the liver damage and immune cell infiltration as demonstrated by H&E staining. Moreover, we found that the numbers of total leukocytes, macrophages, T cells and B cells were all significantly decreased in the livers of CCl4-treated mice with AAV-MFN2 treatment in comparison with those in control groups (Fig. 5b–f). In summary, MFN2 overexpression inhibited immune cell infiltration and ameliorated liver damage in mice treated with CCl4.

MFN2 overexpression inhibits immune cell infiltration in the liver of CCl4-treated mice.
Discussion
Liver fibrosis is a chronic liver disease involving various cells including immune cells, hepatocytes, and HSCs. 25 HSCs are liver-specific mesenchymal cells that have critical functions in liver fibrogenesis. 26 Thus, HSCs are key targets to treat the liver fibrosis. In this study, we developed an AAV vector that could specifically express MFN2 in HSCs using α-SMA promoter. Overexpression of MFN2 promoted HSC apoptosis, suppressed TGF-β1/Smad signaling, and inhibited collagen production in HSCs. Moreover, we revealed that MFN2 overexpression could decrease the immune cell infiltration in liver and ameliorate liver fibrosis in mice treated with CCl4.
α-SMA promoter was explored as a useful tool to achieve fibroblast-specific expression in multiple studies. 27,28 α-SMA is upregulated in activated HSCs and marks the early progression of liver fibrosis. 29 –31 Thus, we evaluated the HSC-specific expression of MFN2 by using α-SMA promoter. The results showed that AAV-MFN2 significantly increased MFN2 expression in HSC line HSC-T6, but not in BMDMs (mimic liver kupffer cells) or AML-12 (liver hepatocyte cell line) (Fig. 1). Thus, AAV-MFN2 with α-SMA promoter provides us an ideal tool to specifically study the function of MFN2 in HSCs. MFN2 was reported to regulate cell proliferation, apoptosis, ER stress, and autophagy. 16,17 Although the HSC apoptosis is important for the reversal of liver fibrosis, we demonstrated that overexpression of MFN2 promoted HSC apoptosis (Fig. 2). Ma et al. reported that MFN2 promotes breast cancer cells MCF-7 apoptosis and inhibits cell proliferation through PI3K/Akt signaling pathway. 32 MFN2 can also suppress cervical cancer progression through mitochondrial pathway. 33
Emerging evidence has shown that TGF-β1/Smad signaling regulates liver fibrosis. 23,24 Blocking Smad3 signaling could suppress type I collagen expression and inhibit epithelial–myofibroblast transition. 23 We found that HSC-specific expression of MFN2 resulted in suppressed TGF-β1/Smad signaling and decreased type I, type III, and Type IV collagen expression (Fig. 3). Consistent with our findings, Yutaka Inagaki et al. reported that by using HSC-specific enhancer of the mouse COL1A2 gene, they could suppress collagen gene expression and liver fibrosis through interfering TGF-β/Smad signaling. 34 Intriguingly, although we found that overexpression of MFN2 induced apoptosis with suppressed TGF-β1/Smad signaling, other groups reported that TGF-β/Smad signaling enhanced apoptosis. 35 –37 The discrepancy indicates the complicated regulation networks involved in MFN2-induced apoptosis in HSCs. The function of MFN2 was also evaluated in the classic CCl4-induced liver fibrosis mouse model. Overexpression of MFN2 not only suppressed the liver fibrosis-related gene expression levels, but also reduced the infiltration of immune cells (Figs. 4 and 5). These findings suggest that MFN2 ameliorates the liver injury and liver fibrosis process in a coordinated manner.
In conclusion, we suggest that overexpression of MFN2 promotes HSC apoptosis and ameliorates liver fibrosis through regulating TGF-β1/Smad signaling. Targeting MFN2 could provide a potential efficient therapeutic strategy for liver fibrosis treatment. It should be pointed out that the data would be more convincing if one or two other animal models of liver fibrosis were employed. Future studies could focus on validating these results in different animal models and screening other genes would exhibit similar effects.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This work was funded by Zhejiang Provincial Natural Science Foundation of China (Grant No. LY17H030002), National Natural Science Foundation of China (Grant No. 81160037), and Medicine and Health Science and Technology Plan Projects of Zhejiang Province (Grant No. 2018KY569).