
Abstract
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disorder caused by repetitive trauma to the central nervous system (CNS) suffered by soldiers, contact sport athletes, and civilians following accident-related trauma. CTE is a CNS tauopathy, with trauma-induced inflammation leading to accumulation of hyperphosphorylated forms of the microtubule-binding protein Tau (pTau), resulting in neurofibrillary tangles and progressive loss of neurons. At present, there are no therapies to treat CTE. We hypothesized that direct CNS administration of an adeno-associated virus (AAV) vector coding for an anti-pTau antibody would generate sufficient levels of anti-pTau in the CNS to suppress pTau accumulation thus interrupting the pathogenic process. Using a serotype AAVrh.10 gene transfer vector coding for a monoclonal antibody directed against pTau, we demonstrate the feasibility of this strategy in a murine CTE model in which pTau accumulation was elicited by repeated traumatic brain injury (TBI) using a closed cortical impact procedure over 5 days. Direct delivery of AAVrh.10 expression vectors coding for either of the two different anti-pTau antibodies to the hippocampus of these TBI mice significantly reduced pTau levels across the CNS. Using doses that can be safely scaled to humans, the data demonstrate that CNS administration of AAVrh.10anti-pTau is effective, providing a new strategy to interrupt the CTE consequences of TBI.
Introduction
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disease caused by repetitive trauma to the central nervous system (CNS). 1 –4 Clinically, CTE is associated with behavioral and mood changes, including impulsivity, violence, depression, and irritability, with eventual cognitive and memory impairment. 1,2,5 Originally identified in boxers exhibiting “punch drunk syndrome,” 6 CTE occurs in military personnel following traumatic brain injury (TBI), in civilians following accident-related CNS trauma, and a range of contact sport athletes, including football, hockey, soccer, boxing, and rugby players. 7 –12 TBI is a significant, underrecognized public health threat. 13
While professional football players and military personnel have received the most attention, 14 –16 2.5 million people in the United States sustain TBI every year, with related health care costs estimated to be $80 billion annually. 17 –19 There are no accurate estimates of the proportion of TBI cases who develop CTE, but as more attention is focused on the risk, the numbers of identified cases are increasing. 4 CTE is common in military personnel exposed to blast injury. 20 –22 Up to 2 million soldiers (20% of returning service members) deployed to Iraq and Afghanistan have experienced some TBI and are at risk for CTE. 22,23
Biologically, CTE is a CNS tauopathy characterized by pathologic hyperphosphorylation of the microtubule-associated Tau, coded by a single gene on chromosome 17. 1,24 The pathogenesis of CTE starts with repetitive CNS trauma initiating chronic inflammation that, in turn, mediates the development of hyperphosphorylation of protein Tau (pTau), resulting in the generation of neurofibrillary Tau tangles, causing progressive loss of neurons and the clinical symptoms of CTE. 1,5,24 –26 Whereas Aβ pathology is upstream and appears to initiate Tau pathology in the Alzheimer's mouse model, in CTE, Tau pathology appears to be independent of CTE and thus a distinct mechanism from that of age-related neurodegenerative disease. 27 –29 In humans, CTE Tau pathology also has a distinctly different distribution in the brain than that found in Alzheimer's disease. 30
At present, there is no therapy for CTE. 31,32 Systemic anti-pTau antibody therapy has been successful in treating a murine model of CTE, 33,34 but this strategy is unlikely to be successful in humans, as has been found with systemic administration of anti-pTau in clinical studies of Alzheimer's disease 35 –37 ; the blood/brain barrier limits the amount of systemically administered antibodies reaching the brain to <0.5% of the administered dose. 38 To circumvent the blood/brain barrier, we hypothesized that direct CNS administration of an adeno-associated virus (AAV) gene transfer vector coding for an anti-pTau antibody would mediate expression of the anti-pTau antibody within the brain, leading to suppression of the accumulation of pTau following TBI.
To test this hypothesis, we used an AAVrh.10 serotype gene transfer vector because there is low anti-AAVrh.10 seroprevalence in humans, 39 AAVrh.10 mediates high expression of transgenes in neurons in experimental animal models, 40 –43 and AAVrh.10 vectors have been used safely in clinical trials to deliver the therapeutic genes to the CNS of children with mucopolysaccharidosis IIIB and metachromatic leukodystrophy. 44,45 To evaluate an AAVrh.10 anti-Tau-based therapy, we developed a murine repetitive TBI model that is mechanistically unlike the age-related degenerative disease models. The data with this model demonstrate that direct CNS administration of AAVrh.10 vectors coding for anti-pTau antibodies can be used to genetically modify the CNS to persistently secrete an anti-phospho-tau antibody to suppress the progression and spread of pTau, suggesting a novel strategy to treat CTE.
Methods
Mouse model
All animal protocols were reviewed and approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee. C57Bl/6 mice (8–10 weeks old) were used in all CNS impact studies. Immunocompromised NOD-scid IL2γ-null (NSG mice) were used to assess antibody expression in the CNS. P301S Tau mice were used for the positive pTau brain controls for the Western blot analysis; these transgenic mice express the human 0N4R variant form of the microtubule-associated pTau. 46,47
We developed a closed cortical impact procedure resulting in consistent, increased levels of brain pTau. 48 –50 C57Bl/6 mice (males, 8–10 weeks old) were anesthetized using 1–4% isoflurane. The superior aspects of the head were shaved and the mouse positioned within a stereotactic frame, on top of a 2-cm-thick Type E foam padding (Foam to Size, Ashland, VA) to allow for the acceleration–deceleration component of injury. 51,52 The foam pads were changed daily after each day to ensure a similar spring constant for all impacts. The mice were positioned on the foam pads in a prone position and the head held in place with ear bars with rubber end caps adjusted in height to level the skull to maintain positioning in the stereotactic frame during impacts. Head holders were positioned such that lateral movements would not occur when the head was impacted. 53
The impacts were regulated using a stereotaxic impactor actuator (Leica Biosystems, Buffalo Grove, IL) attached to a stereotaxic frame (Stoelting, Wood Dale, IL). Impacts were delivered using a modified controlled device that was adjusted to include a widened tip of vulcanized rubber (9 mm). 51,52 The impactor tip was positioned at an angle of 20°, 3 mm left of the intersection of the sagittal plane and a coronal line drawn halfway between the eye and the ear of the left hemisphere. The impact was zeroed so that it was directly perpendicular to the impact surface and orthogonal to the skull. Using settings that were modified from the Zhang et al. 54 protocol, the impactor actuator was set at a velocity of 3 m/s and the tip driven to 2.2 mm past zero point with a 100 ms dwell time. We used 2 impacts/day (6-h interval) for 5 consecutive days, with each mouse receiving a total of 10 impacts. To prevent Tau activation by hyperthermia following the impacts, 55 the anesthetized mice were placed on top of a heating pad to maintain their body temperature until they regained consciousness and sternal recumbency.
Impacted mice were closely monitored during recovery using the Neurological Severity Score (NSS) adapted by Petraglia et al. 56 Any mouse scoring higher than a 1 (out of 10) was separated from the colony and closely evaluated over the next 24 h; failure to recover by the next day led to euthanasia. Mice were observed for apnea, failure in righting response, signs of sluggish recovery, paresis/paralysis, or other severe adverse events following the impacts. Any such occurrences resulted in euthanasia of that mouse for postmortem evaluation. No mice during this trial using the study, described above, failed the NSS or developed severe complications requiring euthanasia.
AAVrh.10 vectors
All vectors were based on the nonhuman primate AAVrh.10 capsid, with an expression cassette that comprised 5′ to 3′ of the AAV2 5′ inverted terminal repeat (ITR), packaging signal (Ψ), CMV enhancer, chicken β-actin promoter with splice donor and rabbit β-globin intron with splice acceptor, the cDNA for the different antibodies followed by the rabbit β-globin polyA signal, and the AAV2 3′ ITR. Five vectors were generated with different coding sequences, including AAVrh.10PHF1 coding for the anti-pTau monoclonal PHF147; AAVrh.102B6 coding for the anti-pTau monoclonal 2B657; AAVrh.10IPN007 coding for the anti-pTau monoclonal IPN00758; AAVrh.10CisTau coding for the anti-Tau monoclonal CisTau 33,59 ; and AAVrh.10Null used as a control, identical to the other vectors but with no gene coding sequence. 41
PHF1 hybridoma cell cDNA (gift from Peter Davies, Albert Einstein College of Medicine) was amplified and synthesized in two independent reactions using primer annealing conserved regions of the constant heavy and light chains. The cDNA containing the mouse heavy chain sequence from IgG1 and the mouse Kappa light chain sequence was amplified using nested primers, cloned into a TOPO vector (Thermo Fisher Scientific, Inc., Carlsbad, CA) and sequenced. The full antibody sequence was cloned into a pAAV plasmid with the heavy and light chains separated by the Tav2A sequence downstream of a furin 2A cleavage recognition site.
The cDNA sequences corresponding to a variable region of the antibodies 2B6, IPN007, and CisTau were obtained from the publicly available patent documents (WO2013151762, WO2015081085, and WO2014152457, respectively). Each sequence was synthetized and cloned in a pUC57 vector (GenScript Biotech, Piscataway, NJ). The cassette coding variable region of the antibodies and constant regions of human IgG4 heavy chain and human Ig Kappa were similarly subcloned into a pAAV plasmid for vector production.
All vectors were produced using 293T cells as described previously. 40 –43,60 Briefly, the expression plasmid and the AAVrh.10 packaging-Ad helper hybrid plasmid pPAK-MArh.10 were cotransfected into 293T cells using the PEI transfection reagent (Polysciences, Warrington, PA). At 72 h post-transfection, cells were harvested and lysate prepared using five cycles of freeze/thaw. The cell lysate containing the virus was clarified by centrifugation at 3,500 rpm for 15 min. The vectors were purified from the crude viral lysate by iodixanol gradient and QHP anion exchange chromatography (GE Healthcare, Piscataway, NJ). The purified vector was concentrated using a Biomax 100K membrane concentrator (Millipore, Billerica, MA) and stored in phosphate-buffered saline, pH 7.4 at −80°C.
Vector genome titers were determined by TaqMan qPCR using a CMV–chicken β-actin promoter (CAG)-specific primer–probe set, forward primer: GTCAATGGGTGGAGTATTT-ACGG and reverse primer: AGGTCATGTACTGGGCATAATGC, designed using Primer Express software (Applied Biosystems, Foster City, CA). The purified AAVrh.10 vectors were digested with proteinase K in the presence of 0.5% sodium dodecyl sulfate, 25 mM ethylenediaminetetraacetate at 70°C, 1 h followed by inactivation of the protease at 95°C for 15 min. The vector was then used as a template for TaqMan analysis using an AAV plasmid DNA standard of known copy number to generate a standard curve. 40
The capacity of all four anti-pTau antibodies to bind pTau was assessed in vitro by Western blot analysis. HEK293T cells were transfected individually with the four different vectors described above coding for anti-pTau antibodies. After 72 h, the cell culture supernatants were collected and the antibodies purified using the NAb™ Spin Kits for antibody purification (Thermo Fisher Scientific, Inc.). In each case, the antibody concentration was measured using the NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.) with readings at 280 nm. To test the ability of the produced antibodies to recognize pTau, SDS-PAGE gels containing samples from brain homogenates from the transgenic mouse P301S 46,47 were assessed using the purified antibodies as detection antibody and, as secondary, a goat anti-murine IgG1- or a goat anti-human IgG-specific antibody conjugated to horseradish peroxidase (HRP).
Administration of AAVrh.10 anti-pTau vectors
Following TBI, mice were administered anti-pTau AAV vectors directly into the hippocampus, bilaterally, 3 weeks following impacts. The mice were anesthetized by inhalation of isoflurane (2% to 3%) and prepped for surgery with povidone/iodine scrub (Betadine scrub, Stamford, CT) and 70% isopropyl alcohol. Local anesthetic, 0.1 mL bupivacaine (0.25% solution), was administered subcutaneously into the tissue adjacent to the intended incision line, and an injection of buprenorphine (0.5 mg/kg; SQ) given for pre-emptive analgesia immediately after the animal was anesthetized. The head was secured with ear bars and clamped to a stereotaxic frame mouse stereotaxic instrument (Stoelting) and anesthesia maintained using a nose cone. A small skin incision (∼2 cm) made along the cranial midline from just behind the eyes to the beginning of the neck. The periosteum was removed and the skull area cleaned with H2O2.
A high-speed Dremel drill was used to make a small burr hole in the skull corresponding to the stereotactic locations of the target structure, with carbide bits 0.5 mm in diameter and with drill stops to prevent brain penetration. The bregma was identified and coordinates recorded. Mice were injected intracranially into the hippocampal region, bilaterally. Stereotactic coordinates used for the hippocampus (for both hemispheres) in adult mice were as follows: −1.7 mm anteroposterior (AP) from bregma; ±1.2 mm mediolateral from bregma, and −1.7 mm dorsoventral below the dura. A Hamilton 710 series syringe (1–5 μL) with neuroadapter and a 33-gauge needle was secured to the syringe pump of the stereotactic apparatus, and the appropriate injection volume and rate set. The syringe needle was slowly lowered to the appropriate target structure and left in place 1 min before starting the injection. The vector was administered bilaterally via 2 stereotactic injections into the hippocampal region of the mouse brain: at 2 μL per site, 4 μL total volume/mouse, using a syringe pump at a rate of 0.5 μL/min.
After each injection, the needle was left in place for 2 min to minimize backflow and then slowly withdrawn. Following the injection, the skin over the skulls was closed with a tissue adhesive (Vetbond, 3M, St. Paul, MN). After surgery, animals were observed until they could maintain sternal recumbency. The mouse was observed for neurologic complications following recovery from anesthesia.
The mice were administered either a control vector (AAVrh.10Null, expressing no transgene, 1011 gc) or AAVrh.10anti-Tau (PHF1, 2B6, IPN007, or CisTau, all 1011 gc) 3 weeks after the last day of impacts. After a period of 6 weeks to 6 months, mice were sacrificed via CO2 gas asphyxiation, transcardially perfused, and brains were harvested and sectioned into 6 coronal sections (2 mm thickness) and hemisected for 12 brain samples/mouse. Samples were immediately frozen at −80°C for Western blot analysis or were fixed in 4% paraformaldehyde for immunohistochemistry.
Immunohistochemistry
For detection of pTau in situ in the CNS, mouse brains were excised during necropsy, sectioned using a mouse brain block (Kent Scientific, Torrington, CT) into six, 2 mm coronal sections (Fig. 1A), and then fixed immediately in 4% paraformaldehyde (diluted in 1 × PBS). Brain specimens were processed and preserved as paraffin blocks, and sectioned at 5 μm (Histoserv, Inc., Germantown, MD). Brain sections containing different coronal sections were deparaffinized, treated with IHC Antigen Retrieval Solution (Thermo Fisher Scientific, Inc.) at 88°C, 20 min, and then blocked using the SuperBlock Buffer (Thermo Fisher Scientific, Inc.) overnight in a humid chamber at 4°C. The sections were incubated at 23°C, 2 h with an AT8 biotin-labeled anti-pTau antibody (Thermo Fisher Scientific, Inc.) diluted 1:100 in SuperBlock Buffer.
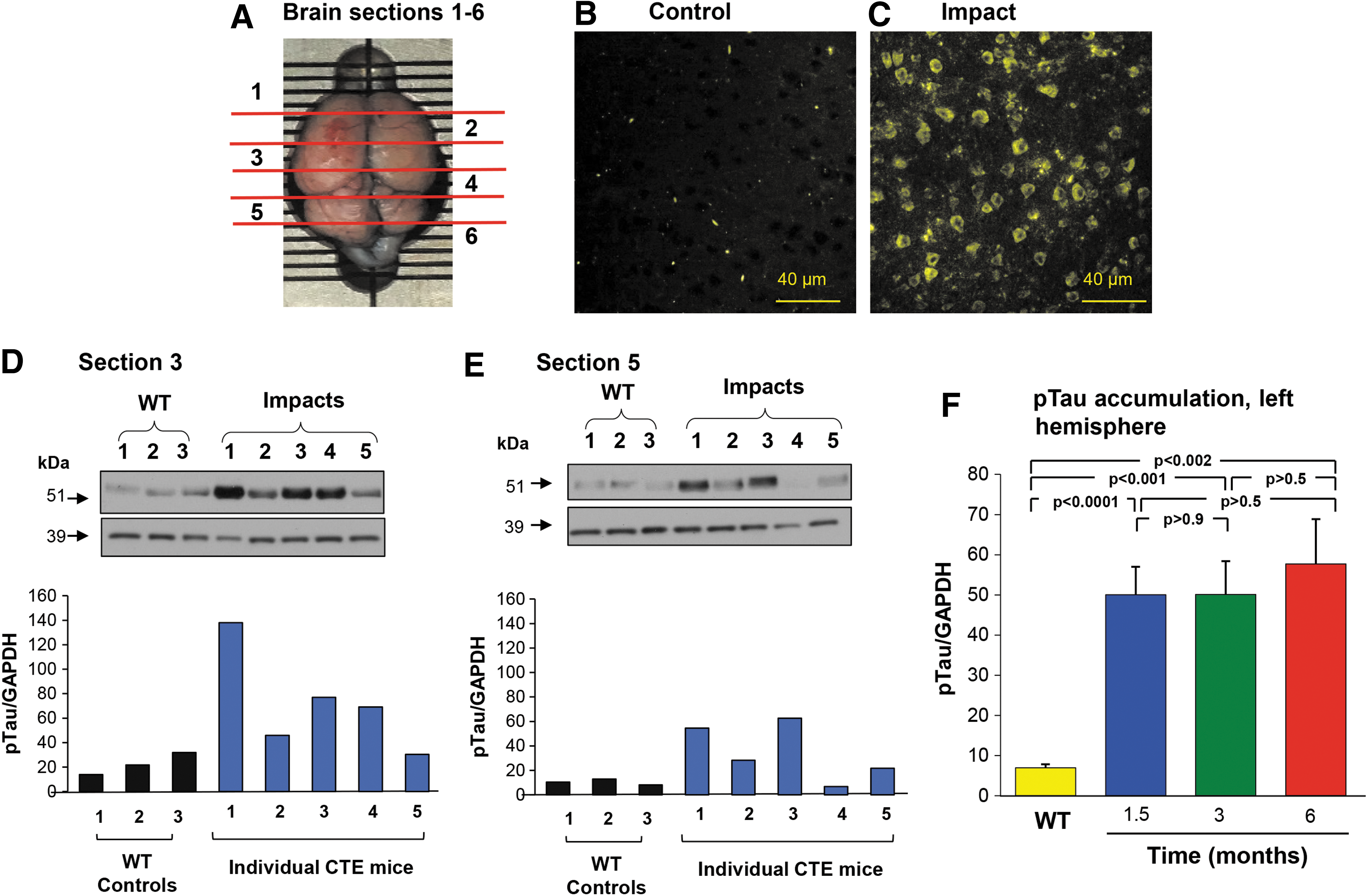
CTE repeat trauma brain impact murine model. C57Bl/6 mice (8–10 weeks old) were impacted twice daily for five consecutive days. The mice were sacrificed at various time points 3 weeks to 6 months after last impact, and CNS prepared for either immunohistochemistry (3 weeks post) or Western blot analysis (6 weeks, 3 and 6 months postimpacts).
After incubation with the primary antibody, the sections were washed 4 × in TBS-T and incubated for 30 min with HRP conjugated to streptavidin (Jackson ImmunoResearch Laboratories, West Grove, PA), diluted 1:1,000 in 20% SuperBlock in TBS. The immunoreactivity was detected using the ImmPACT DAB (3,3′-diaminobenzidine) peroxidase substrate (Vector Laboratories, Burlingame, CA) and counterstained using hematoxylin QS (Vector Laboratories). The slides were covered with coverslips using the EMS-Mount Mounting Medium (Electron Microscopy Science, Hatfield, PA) and imaged using a Nikon Eclipse 80i microscope.
For immunofluorescent microscopy, after incubation with the primary antibody AT8-biotin, sections were washed 4 × in TBS-T and incubated for 30 min with alkaline phosphatase conjugated to streptavidin (Vector Laboratories), diluted 1:1,000 in 20% SuperBlock in TBS. The immunoreactivity was detected using the substrate ImmPACT vector red alkaline phosphatase substrate (Vector Laboratories) and counterstaining incubation in 4′,6-diamidino-2-phenylindole (DAPI) solution for 2 min. The slides were covered with coverslips using the EMS-Mount Mounting Medium (Electron Microscopy Science). A Leica TCS SP5 confocal imaging system (Leica Microsystems, Buffalo Grove, IL) with Ar/He/Ne lasers and 63 × oil immersion lens was used to detect the pTau in the CNS samples. Visualization of the AP substrate was made with excitation at 514 nm and emission at 560 nm and images obtained were analyzed using ImageJ (ImageJ/Fiji software version 1.52i—National Institutes of Health, Bethesda, MD).
Quantification of pTau by immunofluorescent microscopy was done in ImageJ by converting stained 10 × images to 8-bit grayscale and using a fixed threshold of 170. The percentage of positive pixels was measured per coronal section and normalized to the negative control of the corresponding coronal section, accounting for background and nonspecific staining. For each coronal section, 5 images were taken at 10 × magnification at specific localized regions for both the left and right hemispheres of the cortex, for a total of 10 images per coronal section, resulting in 60 images per animal. With five animals per group, this led to analysis of 300 images per group. Four images were taken at 10 × magnification in coronal section 4, for quantification of the hippocampal staining. The quantification data are expressed as percentage of total area stained compared with the total area measured.
Western blot analysis
For Western blot analysis of pTau levels in the CNS, brain samples were homogenized with cold homogenization buffer (20 mM Tris, pH 7.5/150 mM NaCl/1 mM ethylenediaminetetraacetic acid (EDTA)/1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA)/1% Triton/2.5 mM sodium pyrophosphate/1 mM β-glycerophosphate/1 mM Na3VO4/1 μg/mL leupeptin) supplemented with a protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Inc., Danvers, MA) in 2 mL tubes using 5 mm stainless-steel beads in a TissueLyser LT (Qiagen, Valencia, CA) for 10 min at an oscillation of 50/s. The homogenates, after an incubation at 35°C for 10 min, were centrifuged at 16,000 g for 30 min, 4°C, and the supernatants were stored in aliquots at −80°C until use.
To standardize the Western blot analysis, the amount of total protein from each sample was measured using the BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.) before gel loading. The samples were then diluted in LDS Sample Buffer (Thermo Fisher Scientific, Inc.) and 15 μg of total protein loaded per lane in a 4% to 12% Bis-Tris Mid Gel (Thermo Fisher Scientific, Inc.) separated by electrophoresis and blotted to polyvinylidene difluoride membranes (Thermo Fisher Scientific, Inc.). The membranes were blocked overnight at 4°C with 20 mM Tris-buffered saline containing 0.1% (vol/vol) Tween (TBS-T; Cell Signaling Technology, Inc.) and 10% skim milk powder (Santa Cruz Biotechnologies, Inc., Dallas, TX).
On the next day, the blots were incubated for 2 h, 23°C with the anti-pTau CP13 antibody 61 (gift from Peter Davies), diluted 1:100 in TBS-T+5% skim milk. After four washes with TBST +5% milk, the membranes were incubated for 1 h, 23°C, with a HRP-conjugated secondary antibody specific to the mouse light chain (Millipore Sigma, Burlington, MA) diluted 1:10,000 in TBS-T+5% milk. The membranes were then incubated with the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Inc.) and the signals detected using the CL-XPosure Film (Thermo Fisher Scientific, Inc.) and the Medical Film Processor SRX-101A (Konica Minolta Medical, Wayne, NJ).
Protein loading was controlled by reprobing the membranes with a rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:10,000; Cell Signaling Technology, Inc.) and a HRP-conjugated secondary antibody (goat anti-rabbit IgG-HRP, 1:10,000; Cell Signaling Technology, Inc.). For quantification, the autoradiograms were scanned at 600 DPI using the Epson Perfection V600 Photoscanner and the band intensity measured with NIH ImageJ software. All Western blot analysis results were normalized to GAPDH as the control protein.
Statistics
All statistical analyses were executed using GraphPad statistical software (Prism v8.0.1, GraphPad, San Diego, CA). Analyses comparing the different mouse vector cohorts with nontreated controls for pTau levels in the CNS were performed by a two-tailed unpaired t-test and by nested analysis of variance at various time points to identify significant differences due to treatment. Statistical comparisons were considered to be significant if p < 0.05.
Results
CTE mouse model
No mice involved in the study developed severe complications following the TBI procedures. To demonstrate increased expression of pTau in impacted brains, mouse brains were subdivided into 12 sections (6 coronal 2 mm sections, bisected by hemisphere) to allow monitoring of the pTau levels on the ipsilateral (impacted) and contralateral hemispheres (Fig. 1A). The impact zone was centered in coronal section 3 (left hemisphere). The resulting pTau pathology was readily detectable in the cerebral outer cortex area 3 weeks after impacts (Fig. 1B, C), near the impact site and in multiple areas across the CNS (Fig. 1B, C).
Quantification of pTau in brain coronal sections demonstrated the increased pTau in the CNS of the impacted mice. However, as expected, there was variable increase in pTau in different regions of the CNS (Fig. 1D, E). For this reason, subsequent studies were carried out by pooling the pTau data from CNS samples from several or all regions as indicated. The coronal section data from the left hemisphere were combined into one set (sections 2, 3, 4, and 5). The increase in pTau was observed at 6 weeks after impact, with similar elevated levels of pTau at 3 and 6 months (Fig. 1F, n = 3 mice/time point, p < 0.002, all comparisons with wild-type mice with no impact). There was no difference in the amount of pTau observed 6 weeks to 6 months after impact (p > 0.5, all comparisons).
Expression of anti-pTau monoclonal antibody in the CNS
To assess the ability of the AAVrh.10 vector to direct CNS expression of anti-pTau, murine NSG mice (n = 3) were administered 1011 genome copies (gc) of AAVrh.10PHF1 or AAVrh.10Null. NSG mice were required for assessing PHF1 expression due to confounding high levels of endogenous mouse IgG in C57Bl/6, used in all other studies here. The vectors were injected by the intrahippocampal route and the analysis was carried out 3 weeks postadministration. Localized production of AAVrh.10PHF1 was readily detectable across the mouse brain observed by immunohistochemistry and Western blot analysis (Fig. 2). In contrast, no antibody expression was observed in the negative control AAVrh.10Null-treated mice.

Expression PHF1 in the CNS following administration of AAVrh.10PHF1.
Efficacy of AAV vectors in suppressing TBI-induced CNS pTau accumulation
To assess for efficacy of AAVrh.10 vectors in a murine model of CTE, a treatment model was established analogous to relevant human scenarios of postimpact therapy, in which impacted mice were administered an AAVrh.10 vector following impact. Direct CNS administration was performed 3 weeks after impacts to allow the mice a recovery period before surgical administration of the AAV treatments. The efficacy of AAV-expressed PHF1 to reduce pTau in the CTE mice was compared with that of 3 other anti-pTau antibodies [2B6, IPN007 (referred subsequently as “IPN”), and CisTau]. 33,57 –59 All four AAV vectors expressed anti-pTau antibodies that bound to pTau in brain homogenates from P301 transgenic (tau) mice (Supplementary Fig. S1).
C57Bl/6 mice were delivered controlled repeated impacts 2 × day for 5 days. Three weeks later, the mice were randomly assigned to cohort groups (n = 10 mice/group), and then 1011 gc (5 × 1010 gc in 2 μL/hemisphere) of the anti-pTau vectors (AAVrh.102B6, AAVrh.10-IPN, AAVrh.10-CisTau, or AAVrh.10PHF1) or the control vector (AAVrh.10Null) was administered to the hippocampus (Supplementary Table S1). Additional controls included a baseline impact-only nonsurgical control group (“Impacts, no AAV”; n = 10) and a control nonimpact, nonsurgical wild-type group (“Negative, no impacts”; n = 10). The animals were euthanized 6 weeks postvector administration, and the pTau levels in the brain coronal sections were assessed by immunohistochemistry and Western blot analysis.
Both left and right hemisphere coronal sections were analyzed separately to determine if there was a hemispheric preference for pTau accumulation and reduction resulting from the unilateral impacts on the left side. Although the repeated impacts were delivered to the same location on the left hemisphere, the elevated pTau levels were observed in both the left and right hemispheres (Fig. 3A). The non-AAV treated and AAVrh.10Null-impacted control mice (green and yellow bars, respectively) showed significant increases in the pTau levels in both the left and right hemispheres, (p > 0.4, p > 0.6, respectively), demonstrating the broad distribution of elevated levels of pTau across the CNS on the ipsilateral and contralateral sides. Mice treated with AAVrh.10PHF1 (blue bars) and AAVrh.10-IPN (orange bars) all demonstrated marked suppression of the accumulation of pTau in the CNS in both hemispheres (Fig. 3B; p > 0.9 and p > 0.7, respectively, compared with untreated controls).
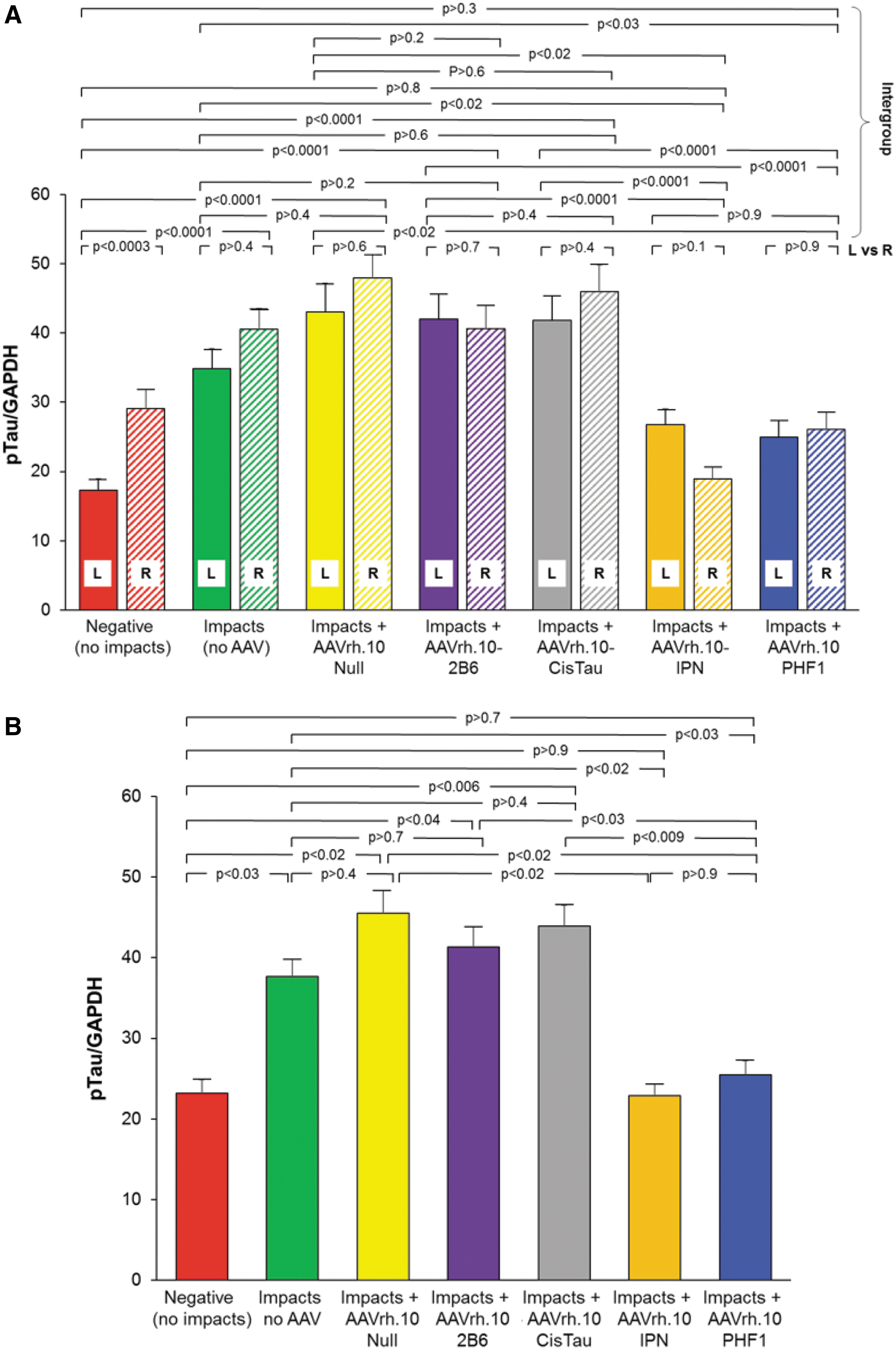
Quantification of CNS pTau in mice treated with AAVrh.10 viral vectors expressing various anti-tau antibodies (2B6, CisTau, IPN, and PHF1) following CNS impacts in the CTE murine model. Wild-type C57Bl/6 mice (n = 60) had repeated TBI delivered 2 × /day for five consecutive days. At 3 weeks postimpact, AAVrh.10 vectors expressing 2B6, CisTau, IPN, or PHF1 anti-pTau antibodies were stereotactically injected bilaterally into the hippocampus (5 × 1010 gc per site) of the impacted mice (n = 10/group). Additional impacted mice that did not receive any treatment (“Impacts, no AAV”; n = 10) served as internal controls as did wild-type mice receiving no impacts (“Negative”; n = 10). As a vector control, 10 impacted mice received AAVrh.10Null (no transgene). Six weeks after vector administration, mice (including the controls) were euthanized, and brains were rapidly collected, divided, and processed for protein quantification and Western blot analysis. Detection of pTau was carried out using anti-pTau CP13, with HRP-conjugated goat anti-mouse light chain as the secondary antibody. The loading control was GAPDH.
When the left and right hemispheres were combined, expression of the antibodies PHF1 and IPN significantly reduced the pTau levels after impacts (IPN vs. untreated group [impacts, no AAV], p < 0.02; and PHF1 vs. untreated group [impacts, no AAV], p < 0.03). In contrast, the vectors coding for antibodies 2B6 and CisTau failed to significantly reduce the pTau levels (p > 0.7 and p > 0.4, respectively; Fig. 3B). The AAVrh.10Null control showed no difference in pTau level reduction (p > 0.4). Since the AAVrh.10PHF1 and AAVrh.10IPN vectors were similarly efficacious, all subsequent studies were carried out with one therapeutic vector (AAVrh.10PHF1).
Assessment of the brains from the AAVrh.10PHF1 versus controls using immunofluorescent microscopy demonstrated that the impacted AAVrh.10PHF1-treated mice substantially reduced the levels of pTau in the mouse cortex (Fig. 4). In contrast, the impacted mice treated with the Null vector control or no treatment showed abundant detection of pTau deposits throughout the cortex. Analysis of the mouse CNS using immunohistochemical detection of pTau deposits in the cortex and hippocampus of impacted mice receiving the AAVrh.10PHF1 versus controls demonstrated elevation of pTau in the cortex with impacts (no AAV) and AAVrh.10Null control (Fig. 5A–F). Images from the cortex area of impacted mice revealed many areas with pTau accumulation around small blood vessels (example in Fig. 5C, arrow) after impacts, consistent with the diagnosis of early-stage CTE. 62
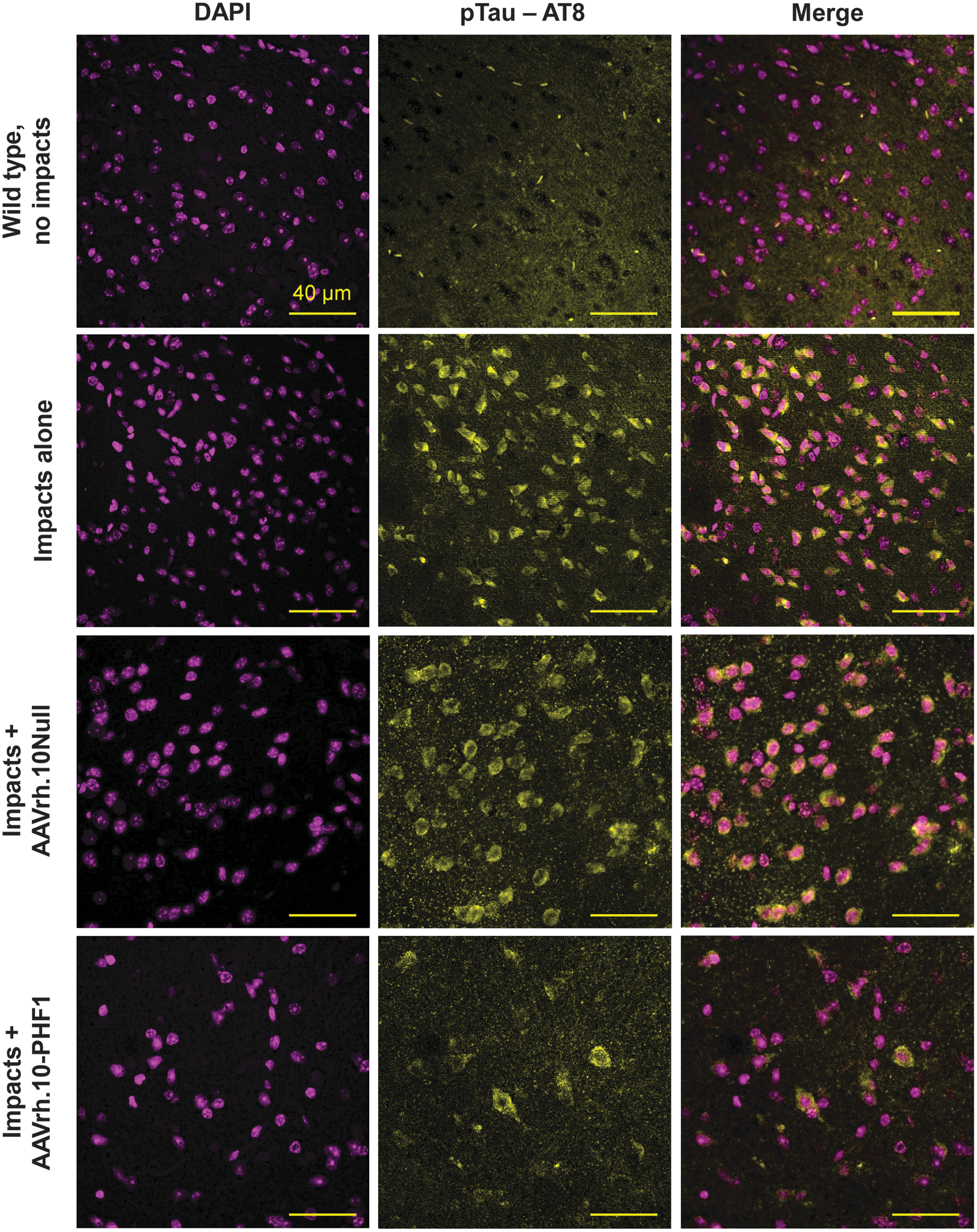
Immunofluorescent analysis of brain pTau in the cortex region of impacted mice treated with AAVrh.10PHF1 compared with the control vector AAVrh.10Null. C57Bl/6 mice (n = 15) had repeated TBI delivered 2 × /day during 5 consecutive days. At 3 weeks postimpact, AAVrh.10 vectors expressing the PHF1 anti-pTau antibody or the control vector AAVrh.10Null were stereotactically injected bilaterally into the hippocampus (5 × 1010 gc per site) of the impacted mice (n = 5 each group). An additional five impacted mice did not receive any treatment (“impacts alone”) and served as internal controls along with wild-type mice (“wild-type, no impacts”; n = 5). Six weeks after vector administration, mice (including the controls) were euthanized for necropsy. Brains were rapidly collected, divided into six coronal sections, and preserved in 4% PFA until processing. Imaging of pTau was with anti-pTau AT8 conjugated to biotin, detected with fluorescent substrate of alkaline phosphatase. Imaging of the brain sections was performed using a laser scanning confocal microscope. Left column panels are the DAPI channel (obtained using UV light originally in blue, modified to magenta using ImageJ for better visualization), the middle column panels are the alkaline phosphatase substrate channel (excitation 514 and emission 560 nm, originally in red, modified to yellow for better visualization), and the right column panels are the merged images from both channels. Scale bars, 40 μm. DAPI, 4′,6-diamidino-2-phenylindole.
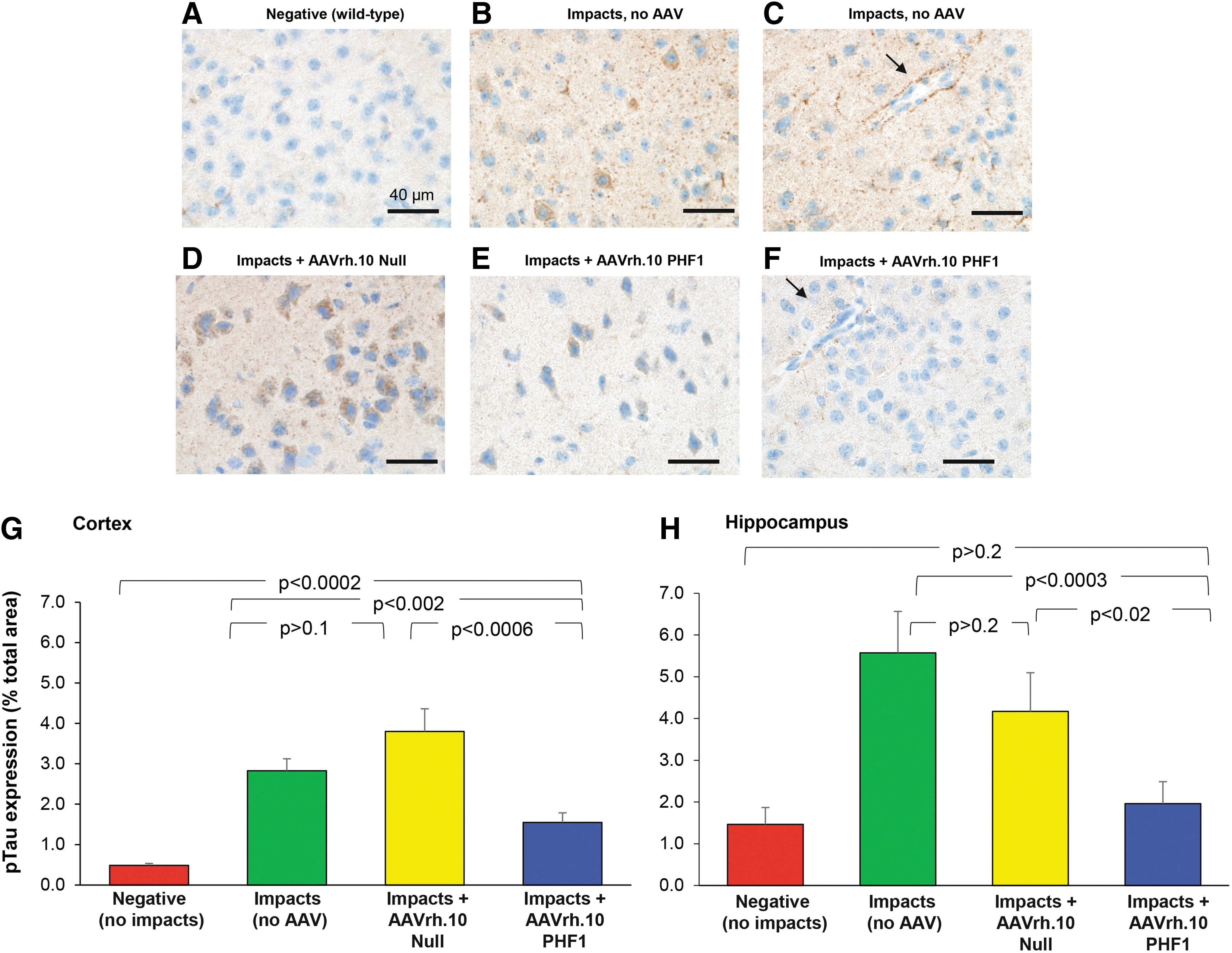
Immunohistochemical analysis of brain pTau in impacted mice treated with AAVrh.10 viral vector expressing anti-pTau antibody PHF1 and the control vector AAVrh.10Null. The mice were impacted and treated with AAV vectors 3 weeks postimpacts, and sacrificed at 6 weeks after vector administration.
In contrast, the mice receiving impacts and AAVrh.10 PHF1 demonstrate significantly less than that of the impact-only group and the AAVrh.10Null control. When the cortex from each animal within each treatment group was assessed for pTau in situ quantification, the levels of accumulation of pTau were significantly less in the impact+AAVrh.10PHF1 group compared with both the impact-only group and vector control impact+AAVrh.10Null group (p < 0.04 and p < 0.04, respectively; Fig. 5G). There was no significant difference between the impact-only and the control vector group (p > 0.8). Similarly, images taken from the hippocampal region demonstrated the similar efficacy of PHF1 treatment, with pTau levels significantly less in the impact+AAVrh.10PHF1 group compared with both the impact-only group and vector control impact+AAVrh.10Null group (p < 0.02 and p < 0.03, respectively; Fig. 5H).
Discussion
Repetitive trauma to the CNS leads to CTE, a debilitating, progressive neurodegenerative disease. 1 –4 At present, there are no approved therapeutics to treat CTE. 31,32 Based on the knowledge that CTE is a tauopathy, characterized by elevated levels of pTau in the CNS, we hypothesized that gene transfer-mediated expression of an anti-pTau monoclonal in the CNS would function to clear pTau from the CNS, thus eliminating a central pathobiologic process in the pathogenesis of CTE. Using direct CNS administration of an rh.10 serotype AAV to transfer an anti-pTau monoclonal coding sequence to the CNS, the data demonstrate effective clearance of pTau, suggesting a possible therapy for this disorder. This strategy circumvents the challenge of the limitation of systemically administered anti-pTau antibodies from reaching the brain because of the blood/brain barrier. 38,63
Strategies to treat CTE
The current concept of the pathogenesis of CTE suggests that repetitive trauma induces chronic inflammation, which in turn mediates abnormal phosphorylation of Tau, resulting in Tau tangles and progressive loss of neurons. 1,5,24 –26,64,65 Several strategies have been investigated to treat CTE, including the following: (1) interruption of pTau accumulation by prevention of its phosphorylation using inhibiting kinases 28,66 ; (2) use of pharmaceuticals to block elements responsible for the inflammation and inflammation-induced downstream signaling 67 ; (3) increasing neuronal microtubular stabilization 68 ; (4) antioxidants to confer neuroprotection after a TBI event 69 ; (5) inducing CNS repair with stem cells 70 ; and (6) use of hyperbaric oxygen therapy to increase tissue oxygenation in the brain. 71
Albayram et al. 34 have demonstrated that systemic administration of an anti-pTau monoclonal antibody in a murine model of CTE suppressed the elevated pTau levels in the CNS. While these are promising data, it is likely that in the humans, where only <0.5% of a systemically administered antibody will reach the CNS, 38 as has been found with attempts to treat Alzheimer's disease with systemic anti-tau monoclonals, systemic administration of an anti-pTau antibody to treat CTE will be challenging. 35,37
Rationale for CNS AAVrh.10 anti-pTau therapy
When considering a strategy to interrupt the pathogenic process in the CNS associated with the development of CTE, it might be possible to target the persistent CNS inflammation associated with repetitive CNS trauma. 64,65 However, the inflammatory process is complex, and it is not clear which component of inflammation is dominating. 64 Our focus for CTE therapy is to target the step resulting from chronic inflammation, the accumulation of pTau, a process that is responsible for the development of Tau tangles, which leads to neuron loss and eventual cognitive impairment. 1,5,24,25,72,73 Importantly, since inflammation in CTE is chronic, persisting long after the trauma, 64,65 effective therapy will require persistent therapy. AAV-based gene therapy provides persistent expression of a transgene and thus a useful approach for anti-pTau therapeutics, although it may not address the upstream inflammatory responses.
In our murine model of CTE, the neuronal accumulation of pTau following repetitive trauma was detected as early as 3 weeks and remained at high levels throughout the cortical regions up to 6 months after implant, the longest time point assessed. Using this CTE mouse model, we tested the delivery of four different anti-pTau monoclonal antibodies directly to the CNS mediated via gene transfer, using AAV serotype rh.10 vectors. Our findings demonstrated that expression of anti-tau antibodies mediated by AAVrh.10PHF1 and AAVrh.10IPN significantly reduced the accumulated pTau resulting from TBI. PHF1 is a mouse IgG1 antibody that targets pTau phospho-serines 396 and 404, 74 whereas IPN is a humanized IgG4 antibody that works by targeting a small number of residues at the N-terminus of tau. 58 Thus, at least for a murine model of TBI, neither the specific antibody target site on the phosphorylated tau protein nor the isotype of the antibody is the determining factor for efficacy. However, since the IPN anti-Tau antibody recognizes an N-terminal epitope (within residues tau 17–28) 75 and there is no requirement for phosphorylation, we decided to proceed with the pTau-specific PHF1 antibody for future studies. The data demonstrated that direct bilateral hippocampal administration of AAVrh.10PHF1 at a total dose of 1011 gc, a dose that could be safely scaled to humans, 76 demonstrated marked suppression of the accumulation of pTau in the CNS of the impact animals.
The AAVrh.10PHF1 therapy has the potential to treat patients suffering from the early effects of CTE and reverse the neurologic pathology. While efficacy studies in the murine CTE model were carried out with direct hippocampal administration of the anti-pTau therapy vectors, there are data that suggest that other CNS routes of delivery would be equally efficacious. 43 Since patients with CTE have already suffered brain trauma, surgical intraparenchymal delivery of the AAV vectors could carry additional and unnecessary risk. Based on our studies of AAV-based gene therapy to the CNS, AAVrh.10 vectors could be administered via the intracisternal route safely and with broad expression throughout the CNS. 41 This provides an opportunity for clinical development, which would include the standard product development steps, including safety and toxicology studies along with a more formal evaluation of efficacy.
Footnotes
Acknowledgments
We acknowledge Veronica Mitchell, Elizabeth Fisher, and Eleni Papadopoulos for assisting with the development of the CTE animal model; we thank N. Mohamed for helping in preparing this article.
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported, in part, by Weill Cornell Medicine's Daedalus Fund for Innovation and Department of Genetic Medicine funds.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.