
Abstract
In the last decade, the gene therapy (GT) field experienced a renaissance, thanks to crucial understandings and innovations in vector design, stem cell manipulation, conditioning protocols, and cell/vector delivery. These efforts were successfully coupled with unprecedented clinical results of the trials employing the newly developed technology and with the novel establishment of academic–industrial partnerships. A renewed and strengthened interest is rising in the development of gene-based approaches for inherited neurometabolic disorders with severe neurological involvement. Inherited metabolic disorders are monogenetic diseases caused by enzymatic or structural deficiencies affecting the lysosomal or peroxisomal metabolic activity. The metabolic defect can primarily affect the central nervous system, leading to neuronal death, microglial activation, inflammatory demyelination, and axonal degeneration. This review provides an overview of the GT strategies currently under clinical investigation for neurometabolic lysosomal and peroxisomal storage diseases, such as adrenoleukodystrophy and metachromatic leukodystrophy, as well as novel emerging indications such as mucopolysaccharidoses, gangliosidoses, and neuronal ceroid lipofuscinoses, with a comprehensive elucidation of the main features and mechanisms at the basis of a successful GT approach for these devastating diseases.
Introduction
Inherited metabolic disorders are monogenetic diseases caused by mutations in one or more genes codifying for an enzyme or protein involved in lipid or protein metabolism. The inability to degrade or synthesize a particular substrate leads to an excess of undegraded metabolites and a downstream lack of essential molecular components that would eventually accumulate, resulting in cytotoxicity and cell death. This could affect multiple body tissues, often including the central nervous system (CNS), and results in a wide range of clinical manifestations. CNS involvement can be severe and substantially contribute to the patient's unfortunate prognosis. It ranges from microglial activation, inflammatory demyelination, and axonal degeneration to final neuronal death.
Gene-based approaches for inherited neurometabolic disorders have mainly focused on lysosomal and peroxisomal storage disorders (LSDs and PDs, respectively) with severe neurological involvement. Peroxisomes are cellular organelles highly involved in the degradation (β-oxidation) of very long chain fatty acids (VLCFAs), hydrogen peroxide detoxification, and synthesis of cholesterol, bile acids, and plasmalogens. A typical disease caused by impairment of peroxisomal activity is X-linked adrenoleukodystrophy (X-ALD). In X-ALD, extensive VLCFA accumulation occurs in the oligodendrocytes, where the cholesterol metabolic pathway is essential and extremely active to produce the myelin, a phospholipid derived from this molecule. Impairment in myelin production in turn results in areas of massive demyelination in the CNS and neurodegeneration.
Lysosomes are organelles deputed to the digestion of proteins, nucleic acids, carbohydrates, and some lipids. LSDs are caused by the deficiency of an acidic lysosomal hydrolase, lysosomal membrane proteins, or non-enzymatic soluble lysosomal proteins. 1 The molecular defect finally leads to the initial accumulation of undegraded metabolites in the lysosomes, and later in the whole cell compartments, altering the physiological composition of the plasmatic membrane. 2 Inappropriate sequestration of membrane-associated proteins in storage structures prevents them from normal trafficking and function, leading to a severe alteration of cell homeostasis and cell signaling in neurons and glial cells. 2
Neurodegenerative PDs and LSDs are characterized by undegraded substrate accumulation, oxidative stress, and neuroinflammation, which represent the principal mechanism of neurodegeneration and/or demyelination. It typically occurs in response to a significant activation of the resident microglial cells and astrocytes and can be characterized by the recruitment of peripheral macrophages infiltrating the brain in the late stages of the neurodegenerative pathology. 3
The incidence of PDs and LSDs worldwide is 1:20,0004 and 1:7,0005 live births, respectively, representing a significant fraction of the inherited metabolic diseases. In most of the cases, they share similar clinical manifestations characterized by a progressive neurological impairment. In particular, LSDs present variable clinical manifestations, mostly based on the tissue distribution of the storage material. In the most severe forms, patients appear normal at birth and develop pathological symptoms shortly after, including mental retardation, behavioral abnormalities, progressive deterioration of cognitive and motor functions, and whole-body abnormalities.
Overall, LSDs represent the first cause of pediatric neurodegenerative diseases, characterized by unrelenting neuropathology that is usually the cause of death early in infancy. 5 To date, there are no efficacious treatments for PD and LSD patients. This unmet medical need and their monogenic nature make these diseases very good candidates for cell- and gene-based therapies.
Cell-based therapy, by means of allogeneic hematopoietic stem/progenitor cell transplantation (HCT) from healthy compatible donors, has been applied over the past 30 years to halt or curb the progression of PDs and neurodegenerative LSDs. 6 The rationale is that allogeneic, metabolically competent myeloid cells repopulate patient's tissues, including the CNS, and restore the defective metabolic activity as well as a normal scavenging function, contributing to the rescue of the storage and mitigating the tissue damage. In the case of LSDs caused by enzymatic deficiencies, the allogeneic myeloid cells may release functional lysosomal enzymes in the CNS, which may be taken up by the recipient's enzyme-deficient cells via mannose 6-phosphate and other mannose receptors present on the plasma membrane in a mechanism commonly known as cross-correction (Fig. 1).

The concept of cross-correction. Allogeneic or autologous enzyme-competent cells produce and release a therapeutic lysosomal or peroxisomal enzyme (red dots) in the extracellular milieu, which is subsequently uptaken by enzyme-deficient cells via the mannose-6-phosphate receptor (in black). A genetically modified enzyme-competent cell is indicated by the presence of therapeutic viral vectors in the nucleus (an episomal rAAV or an integrated lentiviral vector, in red). rAAV, recombinant adeno-associated vector.
Despite this rationale, the clinical efficacy of HCT in PD and LSD patients is controversial, and only a limited number of conditions show an overt benefit as a consequence of the treatment. 7 Among these, the childhood cerebral form of X-ALD (ccALD) and mucopolysaccharidosis type I (MPS I) are the conditions for which therapeutic efficacy of HCT is greater, and therefore, transplant indication is stronger. In the other conditions, inconsistent and poor results have been reported, particularly for patients transplanted after disease onset. 5
In a few indications, such as globoid cell leukodystrophy (GLD, also known as Krabbe disease) and metachromatic leukodystrophy (MLD), a milder disease was described in selected cases after HCT, particularly in patients transplanted before onset of the symptoms or very early in the disease course. 5 However, increasing evidences demonstrated lack of clinical benefit in these conditions when the transplant is administered in advanced stages of the disease. 5
Notably, allogeneic HCT remains associated with a significant risk of mortality 6 and morbidity, mostly due to graft-versus-host disease (GVHD). Even in the best scenario of allogeneic HCT performed with human leukocyte antigen (HLA)-fully matched donor or cord blood, the mortality risk remains close to 10–15% in children and slightly higher in adults, in fully myeloablative conditioning regimens. 8 Moreover, the availability of HLA-matched cell donor is usually limited, despite the increasing number of cord blood banks, and requires a too long wait.
Ex vivo gene therapy (GT), schematized in Fig. 2, is based on the autologous transplantation of genetically modified hematopoietic stem/progenitor cells (HSPCs). It was proposed and extensively explored as a safer and more efficacious alternative to allogeneic HCT in several monogenic conditions, including PDs and LSDs. Ex vivo GT overcomes the most severe immunological limitation of allogeneic HCT represented by GVHD and eliminates the need for lifelong immunosuppression in the recipient. Lentiviral gene transfer is nowadays the elective procedure to deliver multiple copies of a therapeutic gene into human HSPCs in ex vivo GT preclinical and clinical studies. Viral transduction of HSPCs allows overexpressing a functional lysosomal enzyme in the HSPC-derived cells in the whole-body tissues, including the CNS, resulting in a robust cross-correction of neurons and glial cells, as observed in multiple preclinical studies and a pivotal clinical trial for neurodegenerative MLD 9,10 that will be discussed in the next section. Lentiviral vectors (LVs) are currently considered by far the safest and most efficient tool for gene transfer in human HSPCs for clinical applications. 11 Ex vivo GT procedures are usually performed in a setting of a partial or full myeloablative pretransplant conditioning to “create space” in the marrow niche to facilitate the engraftment of the transplanted HSPCs. 12 Multiple preclinical studies demonstrated that administration of a myeloablative regimen with CNS-penetrating agents, as busulfan, before cell transplantation, is instrumental to the achievement of a critical myeloid engraftment in the brain, essential for the HSPC-derived myeloid cells to exert a therapeutic effect in PDs and LSDs. 9,13,14
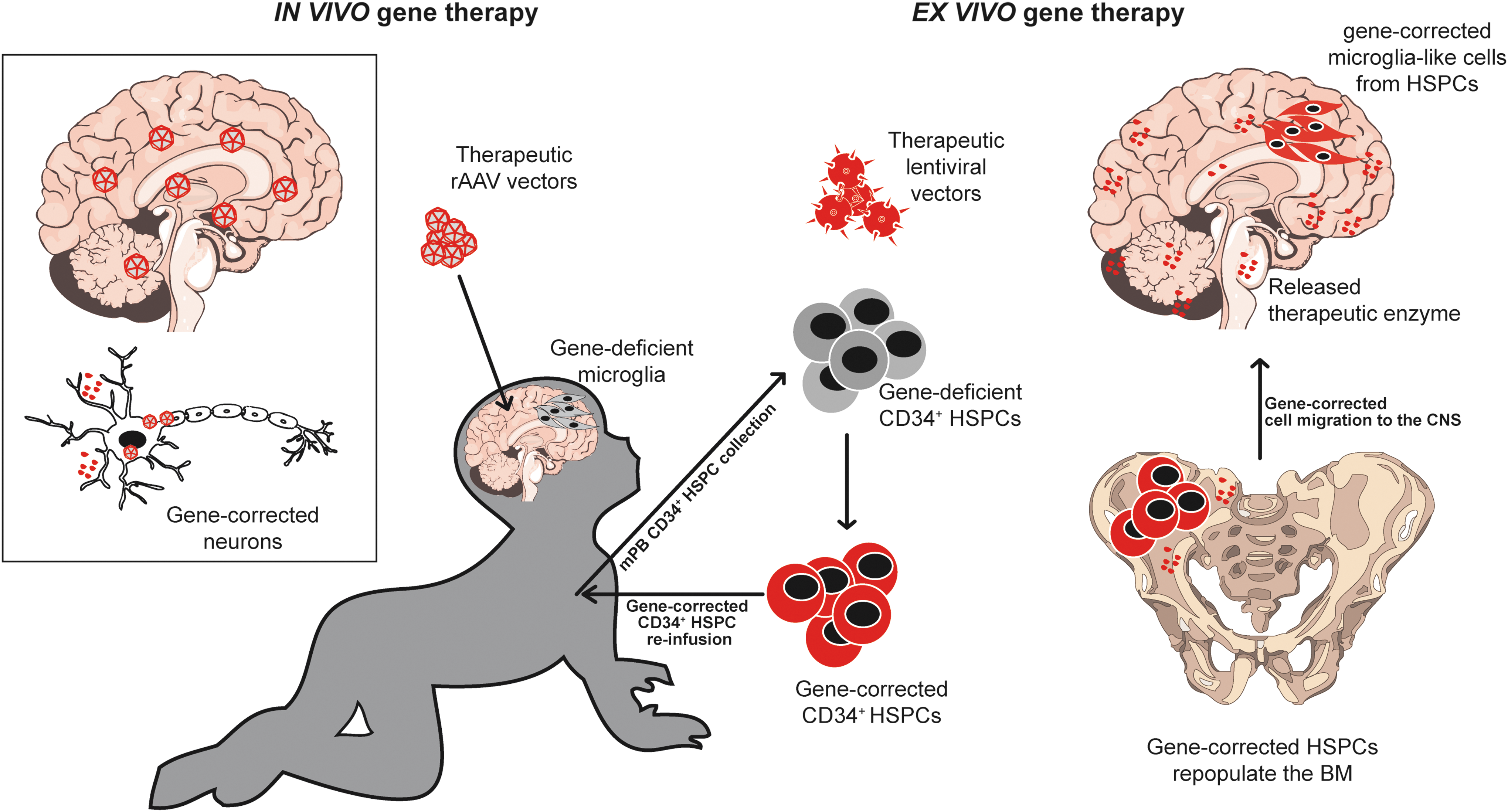
In vivo and ex vivo GT approach. In the in vivo GT setting (left), the therapeutic vector is usually represented by a rAAV, directly delivered into the CNS. The target cells are mostly neurons, which produce and release the therapeutic enzyme (red) into the CSF leading to cross-correction of nontransduced cells. In the ex vivo GT (right), HSPCs are mobilized or collected from the bone marrow of the patients, purified from the blood, in vitro genetically modified by lentiviral transduction, and retransplanted into the preconditioned patient. Once transplanted, HSPCs repopulate the bone marrow and the peripheral hematopoietic organs and reconstitute the whole hematopoietic system. A fraction of these corrected HSPCs repopulates the myeloablated CNS and gives rise to enzyme-competent myeloid cells with microglial phenotype producing and releasing the therapeutic enzyme into the CSF. Enzyme-competent circulating hematopoietic cells will produce and release the therapeutic enzyme systemically. CNS, central nervous system; CSF, cerebrospinal fluid; GT, gene therapy; HSPC, hematopoietic stem/progenitor cell.
LVs are derived from lentiviruses, whose most famous representative is the human immunodeficiency virus (HIV). In self-inactivating configuration, all the original viral genes and regulatory elements are removed, and transgene expression is driven by an exogenous promoter, usually of human origin, placed in internal position. LVs are usually produced as single-stranded RNA particles pseudotyped with the envelope protein-g of the vesicular stomatitis virus to reprogram the original HIV tropism to a very broad range of human cell types, including HSPCs.
Lentiviral genome is stably integrated in the host cell genome after transduction, allowing a long-term and stable transgene expression overtime and in the differentiated progeny of the original transduced HSPCs. Transgene expression may be regulated by a cell-specific or constitutive, strong or weak promoter, based on the particular therapeutic needs. With an appropriate vector design, lentiviral transduction of HSPCs results in a stable transgene expression in the whole committed and differentiated myeloid and lymphoid HSPC progeny, including the HSPC-derived myeloid cells repopulating the CNS after ex vivo GT.
Globally, lentiviral-based gene transfer was demonstrated safe and efficient in multiple authoritative clinical studies to date, for immunodeficiencies, hemoglobinopathies, metabolic diseases, and chimeric antigen receptor T cell without any evidence of vector-related side effects. 11 After the occurrence of severe side effects due to γ-retroviral vectors genotoxicity in the early 2000, an extensive effort was carried out to design LVs as safe and effective tools for ex vivo GT. 15
While γ-retroviral integration occurs preferentially in active regulatory elements, that is, promoters and enhancers, of genes with key regulatory functions in cell-commitment and cell-identity, LVs have a completely different integration pattern, excluding regulatory elements and occurring mostly in the introns of a limited and substantially reproducible cell-specific gene subset, not enriched for oncogenes. 15
Independent of the innate vector integration bias, the peculiar vector design is a crucial factor for avoiding genotoxicity 16 : the usage of promoters of human origin, characterized by a short-range activity, and driving physiological transcriptional levels are strongly recommended for GT applications, rather than strong and long-range trans-activating viral promoters, as the spleen focus-forming virus promoter.
In vivo GT is a valuable alternative gene-based approach for neurodegenerative metabolic diseases (Fig. 2) and is based on direct administration of recombinant adeno-associated vectors (rAAVs) systemically in the bloodstream or locally in the tissue of interest. rAAVs are derived from small, nonenveloped, and nonintegrating AAVs. For their remarkable transduction efficiency and safety profile, rAAVs have been used for direct gene transfer into the CNS in a large number of animal models and multiple clinical trials to date. 17 rAAVs biodistribution, cell tropism, and transgene expression depend on the specific viral capsid serotype, route of administration, and vector design.
The most used rAAV serotypes for gene transfer in the CNS are traditionally the 1, 2, 5, 8, 9, and the recombinant human (rh)10. Usually, rAAV-mediated gene transfer in the brain efficiently targets neurons and scarcely astrocytes, oligodendrocytes, and microglia. 17 For application in LSDs, these vectors are commonly injected in the brain by intraparenchymal injections or into the cerebrospinal fluid (CSF) by intracerebroventricular or cisternal or lumbar-intrathecal administrations. 17
Specific rAAV serotypes, including AAV9 18 and rh10, 17 have the ability to cross the blood–brain barrier allowing transduction of the CNS after systemical administration.
Because of leakage of the system, local rAAV administration into the CNS results in transduction of nontarget tissues and peripheral transgene expression, particularly in the liver. 17 This may trigger an immune response that possibly reduces or abolishes the therapeutic potential of the in vivo GT strategy in patients. 17
Several strategies were put in place to overcome this limitation, ranging from a transient immunosuppression to tolerance induction. 19 In particular, important results were obtained with strategies inducing liver-mediated tolerance 20 or neonatal AAV-mediated systemic expression of a therapeutic protein before CNS-directed in vivo GT. 21,22
X-Linked Adrenoleukodystrophy
X-ALD is one of the most frequent PDs, caused by mutations in the ABCD1 gene, coding for the adenosine triphosphate binding cassette subfamily D member 1, also called adrenoleukodystrophy protein (ALDP). This protein is located in the peroxisomal membrane and promotes the entrance of VLCFAs into the peroxisome to be degraded. ALDP deficiency results in VLCFA accumulation in the CNS and in all the body tissues, resulting in a wide spectrum of systemic and neurological clinical symptoms.
In the most severe X-ALD form, the disease is characterized by a childhood cerebral onset with rapid and progressive demyelination of the cerebral white matter. Patients appear normal at birth, with an initial regular development, when behavioral manifestations begin at the approximate age of 7, progressing very rapidly from visual, auditory, and motor–cognitive impairment to a vegetative state by the age of 10.
Aubourg et al. first reported the beneficial effect of HCT in a boy with early-stage cerebral ALD in 1990, 23 after the report of a previous patient in advanced stage of the disease in which the therapy had a poor clinical outcome. 24 At present, allogeneic HCT is the standard of care for boys with early-stage cerebral ALD. If performed when the neurological and neuropsychological deficits are still minimal and the demyelination is limited, HCT can arrest the cerebral demyelination and disease progression providing a stable clinical efficacy. 25 In a typical scenario, demyelinating lesions usually continue for ∼12–18 months after cell transplantation before arresting. 25
While HCT in LSDs due to an enzymatic deficiency, as MLD and GLD, results in enzymatic delivery and cross-correction of deficient cells in the brain, the dysfunctional protein in X-ALD is a structural protein. Therefore, the therapeutic effect of HCT in X-ALD is not based on the mechanism of cross-correction but rather on a cell-mediated mechanism. 25
HCT likely induces a correction of the abnormal microglial function in the brain, whether or not directly related to the accumulation of VLCFA, finally resulting in a reduction of neuronal death by means of re-establishment of a normal clearance function, and reduction of gliosis and neuroinflammation in favor of a neuroprotective environment. The delay in the HCT benefit after treatment is possibly due to the slow replacement of the resident microglia from donor-derived cells, as has been extensively demonstrated in preclinical studies. 26
Human CD34+ cells transduced by an LV expressing the human ALDP were xenotransplanted in nonobese diabetic/severe combined immunodeficiency mice, migrated into the mouse brain and differentiated into perivascular myeloid cells expressing human ALDP. 26 Supported by these evidences and by allogeneic HCT results, an ex vivo GT strategy based on an LV coding for the wild-type ABCD1 cDNA (CG1711hALD-LV) was originally developed by Cartier et al. 27
Autologous HSPCs transduced with the therapeutic LV were transplanted in two ccALD boys with progressive demyelination and no HLA-matched or cord blood donors, after the administration of a busulfan-based conditioning in a first ex vivo GT clinical trial. 27,28 Cerebral demyelination arrested after 14 and 16 months, respectively, in the two patients, without signs of disease progression up to the last follow-up of over 3 years. The patients showed a polyclonal hematopoietic reconstitution, with ∼9–14% of multilineage gene marking, and a functional ALDP expression correlating with the disease stabilization. 28
Overall, the clinical benefit of the ex vivo GT resulted comparable or superior to the standard allogeneic HCT.
28
After these encouraging results, a larger, multicenter, phase II/III clinical trial was launched in 201329 (
Preliminary results on 17 boys receiving Lenti-D confirmed the initial findings, with 88% of the patients showing only minimal residual clinical symptoms at a median follow-up of ∼30 months. 29 Ex vivo GT resulted in a polyclonal hematopoietic reconstitution with multilineage gene marking and no evidence of clonal skewing. 29 A patient presenting extremely rapid neurological deterioration died because of the disease progression during the time required for myeloid reconstitution of the brain, 29 a factor that still remains as one of the major clinical challenges in the treatment of rapidly neurodegenerative diseases. These results granted the breakthrough therapy designation to the GT product (Lenti-D) from the American Food and Drug Administration (FDA) for the treatment of patients with ccALD in May 2018.
A following phase III trial has been recently opened (January 2019, NCT03852498). Overall, these studies testify the therapeutic value of HSPC-based ex vivo GT in ccALD and encourage its transferability to other similar neurodegenerative diseases.
The same investigators conducting the Lenti-D clinical trial at Massachusetts General Hospital (Boston, MA) have recently developed an rAAV9-based GT strategy for spinal cord axonopathy adrenomyeloneuropathy (AMN), the adult form of neurodegeration due to ABCD1 deficiency affecting primarily the spinal cord. The major challenge in the design of an efficient GT for AMN is represented by the achievement of a critical level of gene correction in the whole spinal cord, minimizing extra-CNS vector leakage that may result in toxicity. 30
To this aim, the investigators developed an osmotic pump to deliver the therapeutic rAAVs into the lumbar CSF of AMN mice. The procedure achieved an efficient gene transfer in the spinal cord and dorsal root ganglia, resulting in a reduction of 20% of VLCFA content in the spinal cord. 30 The vector transduced mainly astrocytes, endothelial cells, and neurons with a limited systemic biodistribution to peripheral organs, in particular the liver and heart. 30
After reviewing these preliminary data, the FDA is now encouraging the investigators to move forward to the clinical experimentation of this in vivo GT for AMN patients. 31
Metachromatic Leukodystrophy
MLD is a rare autosomal recessive LSD (frequency of 1–1,000,000 newborns) caused by mutations in the ARSA gene coding for the lysosomal enzyme arylsulfatase A (ARSA). In MLD patients, lysosomal accumulation of sulfatides occurs particularly in oligodendrocytes and results in a progressive and unrelenting demyelination, which causes a severe deterioration of motor and neurocognitive functions in the affected children. MLD is clinically classified as late infantile, juvenile, or adult based on the age of disease onset and severity.
The infantile form is the most severe, characterized by the disease onset before 2 years of age and progressing very rapidly, with patients becoming bed-ridden few years later. MLD represents one of the LSDs with the largest unmet medical need as it has no available treatment. Allogeneic HCT was extensively tested in infantile and juvenile MLD in the past two decades with very variable and overall poor results. 6
Ex vivo HSPC GT for infantile MLD was developed with the rationale of restoring and boosting ARSA production and secretion in the CNS by the transplant-derived cell progeny. After a proof-of-concept obtained in the mouse model of the disease,
32
–34
the GT strategy has been translated to a first phase I/II clinical trial for infantile and juvenile presymptomatic MLD patients receiving a myeloablative busulfan conditioning before cell transplantation (
The medicinal product (now known as OTL-200) is constituted by autologous CD34+ HSPCs transduced with the therapeutic LV containing the human ARSA. Interim results on the first 9 of 20 patients indicated a stable and sustained engraftment of genetically modified HSPCs over time, with a median follow-up of 36 months. 10 Sustained ARSA activity was detected in circulating hemopoietic cells and in the CSF, reducing the storage material in peripheral nerve samples in most of the patients. 10
In presymptomatic children, the treatment was able to prevent the disease onset, or halt its progression, preserving the motor performances and development similar to those of age-matched healthy children.
10
This first trial has been rapidly followed by a phase II trial based on cryopreserved instead of fresh cell product (
Clinical benefit obtained in the MLD trial compared with standard allogeneic HCT suggests that a supranormal production of ARSA enzyme is required to achieve and maintain a significant control of the disease manifestations. 10 In turn, it suggests a superior capacity of LV-transduced HSPCs with respect to allogeneic HSPCs to induce this supranormal ARSA activity. 9,10
In vivo GT strategies were also developed for MLD, based on AAV-mediated delivery of human ARSA cDNA into the brain parenchyma cells. The rational of this strategy relies on the mechanism of cross-correction operated by neurons transduced with the therapeutic rAAVs, which may produce and release the therapeutic enzyme to be uptaken by affected oligodendrocytes (Fig. 1). Preclinical studies demonstrated the occurrence of axonal transport of rAAVs and/or ARSA enzyme throughout the brain, extending the viral spread beyond the injection site. 35
Multiple preclinical proof-of-concept and safety studies of in vivo GT were performed in the MLD mouse model and nonhuman primates (NHPs), based on an rAAVrh.10 vector carrying a copy of the human ARSA cDNA (AAVrh10-hARSA). 35 Intracerebral administration of the therapeutic AAVrh10 in NHPs resulted in correction of sulfatide accumulation in oligodendrocytes even at a very advanced stage of the disease, without major safety concerns when administered at the dose planned for patients. 36
Based on these results, Aubourg (Paris, France) and co-workers received the authorization from the French Medicine Agency to start a phase I/II clinical trial for presymptomatic MLD children or at early stage of the disease (
Mucopolysaccharidoses
MPSs are a subgroup of autosomic recessive LSDs (with the exception of X-linked MPS II) representing approximately one third of the diagnosed LSDs, with a collective prevalence of 1:22,500–1:52,000. 37 They are caused by the deficiency of a specific lysosomal enzyme involved in the degradation of sulfated glycosaminoglycans (GAGs), which result in the accumulations of their undegraded metabolites in the whole-body tissues and especially in neurons. Age of disease onset, progression rate, and affected organs can vary greatly. Patients affected by MPS I, II, III, and VII present a severe neurological involvement, with progressive neurodegeration and cognitive decline.
Few therapeutic options exist for MPSs, including allogeneic HCT that is considered the standard of care for MPS I children diagnosed before 30 months of age, 37 and enzyme replacement therapy (ERT) that is available for some MPSs without or with minor CNS involvement.
MPSs were extensively studied as candidate for GT applications. Ex vivo GT was developed for MPS I, which is caused by mutations in the IDUA gene coding for the lysosomal α-L-iduronidase enzyme. Multiple preclinical studies performed in the last decade were based on the transduction of HSPCs with γ-retroviral and LVs carrying the IDUA gene. 37 Some ex vivo GT treatments restored IDUA activity in the visceral organs but failed in altering the neurological pathology, most likely due to an insufficient enzyme production in the brain. 38
In a different ex vivo GT strategy, erythroid cells were exploited as enzyme-producer cells after transducing HSPCs with an LV regulating the IDUA transcription with an erythroid-specific promoter. 39 In this study, genetically modified erythrocytes were able to produce and release the therapeutic enzyme into the bloodstream, but the neurological outcome was not substantially improved. 39
The same strategy used for the ex vivo GT of MLD was tested in the MPS I mouse model, transducing autologous HSPCs with a therapeutic LV carrying the IDUA cDNA under the constitutive phosphoglycerate kinase promoter. 40 The study demonstrated a marked therapeutic benefit with correction of the neurological and systemic manifestations of the disease, exceeding the results observed by the transplantation of wild-type cells. 40 The correction of neurological manifestations resulted strongly dependent on the supranormal enzyme activity in the CNS, 40 proving additional evidences to the link between the supranormal enzymatic activity and clinical benefit in multiple neurological LSDs. 40
Based on these preclinical and clinical data coming from the MLD trial, a phase I/II clinical study for the ex vivo GT of MPS I pediatric patients (Hurler variant) was opened in 2018 (
Preliminary data concerning the first six treated patients were presented at the 22th Annual Meeting of the American Society of Gene and Cell Therapy (April 29–May 2, 2019). The first child, at the longest follow-up of 6 months, showed an IDUA expression of ∼10- and ∼40-fold change above normal in the whole blood cells and in the white blood cell compartment, respectively, and the supranormal IDUA activity of approximately threefold change in the CSF (B. Gentner, TIGET, Milan, Italy, oral communication). In this patient, the GT largely exceeded the averaged efficacy of allogeneic HCT in reducing urinary GAGs (∼10-fold change).
A similar ex vivo GT strategy was developed for MPS IIIA (also called Sanfilippo syndrome A), a severe, progressive neurodegenerative LSD caused by loss-of-function mutations in SGSH gene coding for the lysosomal the N-sulfoglucosamine sulfohydrolase. It is based on an LV carrying the human SGSH gene under the control of the CD11b promoter to drive cell-specific gene expression in the myeloid cells repopulating the brain after cell transplantation. 42
The preclinical study performed in busulfan-conditioned MPS IIIA mice resulted in significant disease correction, with normalization of cerebral heparan sulfate accumulation and secondary storage, lysosomal compartment size, neuroinflammation, and finally amelioration of animal hyperactivity. 42 The exploitation of a myeloid-specific promoter resulted in an increased transgene expression in the brain with respect to a constitutive promoter, maintaining the enzyme overexpression in periphery. 42 Based on these encouraging results, safety data concerning this GT strategy and product (OTL-201) were produced 43 leading to the acquisition of the orphan drug designation by the European Medicines Agency (EMA) and FDA.
The same GT approach has been developed by the same investigators for MPS IIIB, caused by the deficiency of the enzyme α-N-acetylglucosaminidase coded by NAGLU gene. 44 An LV expressing the human NAGLU cDNA under the transcriptional control of a myeloid-specific CD11b promoter (LV.NAGLU) has been tested in the MPS IIIB mouse model, in association or not with a supplemental prednisolone-based antiflammatory treatment. 44
The study resulted in supranormal and widespread enzyme expression throughout the brain, normalization of lysosomal storage, correction of astrocytosis and microgliosis, and final behavioral correction. Prednisolone treatment further decreased the overall inflammatory state of the brain. 44 The clinical development of this GT strategy and product (OTL-202) is currently part of collaboration between Manchester University and Orchard Therapeutics.
In vivo GT strategies were also developed for the therapy of MPSs, with promising preclinical data and preliminary clinical results. 37 Multiple in vivo GT clinical studies are currently open in Europe and United States for MPSs based on different rAAV serotypes, vector design, doses, and routes of administration, with the aim of achieving a broad transgene distribution along the CNS, a high transduction level in neurons and glial cells, and an elevated and durable enzymatic activity in the CSF.
Intracerebral administration of therapeutic rAAVs in specific brain areas or intra-CSF was extensively tested in small and large animal models of MPS I, IIIA, IIIB, and VII. 37 Promising results were obtained in the MPS IIIA mouse model receiving intracerebral administration of an AAVrh10 (AAVrh.10-hMPS3A) carrying the human SGSH cDNA and the human sulfatase modifying factor SUMF1 cDNAs, essential and limiting for sulfatase activation. 45 Additional studies in dogs suggested that immunosuppression was required to prevent neuroinflammation and a specific immune response leading to the elimination of both transduced cells and the secreted therapeutic protein. 46
After this extensive preclinical phase, a phase I/II clinical trial was performed in four MPS IIIA patients (
At 1-year follow-up, a moderate improvement in behavior, attention, and sleep was observed in three patients, whereas brain atrophy was stable only in two patients.
47
The invasiveness of the surgical procedure and the limited distribution of the lysosomal enzymes to the injection site represented a limitation of this approach.
47
Recently, a second larger phase II/III trial has been opened based on the same GT strategy and product, called LYS-SAF302 (
Alternative in vivo GT approaches are based on intra-CSF viral delivery. This route of rAAV administration resulted in a widespread CNS transduction and rescue of the clinical phenotype in MPS I, IIIA, IIIB, and VII small and large animal models with different vector serotypes. 37 A clinical trial based on rAAV9 delivery into the lateral ventricles of MPS IIIA patients has been recently opened in Spain (European Clinical Trials Register, 2015-000359-26), whereas clinical trials based on a similar approach are planned for MPS I and II (ESTEVE, Universitat Autònoma de Barcelona, and REGENXBIO).
Finally, an in vivo GT strategy based on AAV8-mediated liver gene transfer resulted efficacious and safe in animal models of MPS VI,
48,49
an LSD caused by arylsulfatase B (ARSB) deficiency. A single systemic administration of a recombinant rAAV2/8 encoding the human ARSB cDNA under the transcriptional control of a liver-specific thyroxine-binding globulin promoter resulted in sustained liver transduction and phenotypic improvement in the mouse and rabbit models of the disease.
48
This extensive preclinical experimentation supported the opening of a phase I/II clinical trial in January 2019 (
Gangliosidoses
GM1 gangliosidosis (OMIM 230500) is an LSD due to the deficiency of the β-galactosidase (β-gal) enzyme, which hydrolyzes the terminal β-galactosyl residues from GM1 ganglioside, glycoproteins, and GAGs. GM1 gangliosidosis is due to mutations in the gene GLB1, and its overall incidence has been estimated to be 1 in 100,000–200,0000 live births, with increased prevalence in Brazil (1:17,000), in the Gypsy population (1:10,000), and in the islands of Malta and Cyprus (1:3,700). 50
GM1 gangliosidosis is characterized by a massive storage of GM1 ganglioside and related glycoconjugates in different tissues and particularly in the CNS. Neuronal cell death and demyelination coupled with severe astrogliosis and microgliosis are typical features of the disease. 50 Neuroinflammatory responses are considered to significantly contribute to the pathogenesis and rapid disease progression. 50
GM1 infants typically appear normal at birth, until their development slows and muscles weaken. They experience developmental regression, seizures, profound intellectual disability, blindness (with typical eye abnormality called cherry-red spot), skeletal and visceral abnormalities, and usually they do not survive after early childhood. Only symptomatic and supportive therapy is available for patients suffering from GM1 gangliosidosis.
However, multiple therapeutic strategies have been explored to date, including allogeneic HCT, GT approaches, and substrate reduction therapy. 50 Naturally occurring GM1 gangliosidosis is observed in cats, dogs, sheep, and calves. 50 Two mouse model lacking a functional GLB1 gene were also generated, closely reproducing the human disease. 50 Several attempts of correction of the enzyme deficiency and glycosphingolipid accumulation were performed in β-gal-knockout (ko) mice following intravenous injection of adenoviral vectors or rAVVs. 50
In particular, the group of M. Sena-Esteves at the University of Massachusetts Medical School (Boston, MA) has recently demonstrated the efficacy of an in vivo GT strategy based on systemic administration of an rAAV9 vector carrying a copy of the specie-specific β-gal gene (AAV9-GLB1) in adult GM1 gangliosidosis mice 51 and in GM1 cats. 52 In treated GM1 mice, biochemical analysis showed high β-gal activity in the liver and serum and moderate β-gal levels in the CNS. 51
Overall, the treatment resulted in a 36–76% reduction in GM1-ganglioside content in the brain and 75–86% in the spinal cord. 51 Histological analyses of the CNS of mice receiving the highest vector dose evidenced the presence of β-gal and clearance of lysosomal storage in the cortex, hippocampus, brainstem, and spinal cord. 51 Storage reduction was accompanied by a marked decrease in astrogliosis, with improved performance in multiple motor function and behavioral test, and increased lifespan. 51 Similar results were obtained in GM1 cats, where treated animals experienced a remarkable preservation of brain architecture and correction of brain metabolites associated with microgliosis, neuroaxonal loss, and demyelination maintaining near-normal functions more than 5 years post-GT. 52
Following this extensive preclinical evaluation, a promising phase I/II clinical trial (
GM2 gangliosidosis is a family of autosomal recessive LSDs characterized by the deficiency of the lysosomal enzyme β-hexosaminidase (Hex), responsible for GM2 ganglioside degradation. The functional Hex enzyme is dimeric and exists in three isozymes, based on the combination of α- and β-subunits. The Hex isozyme A (α/β heterodimer) is the only isozyme that hydrolyzes the GM2 ganglioside. The α- and β-subunits are encoded by separate genes, HEXA and HEXB, respectively. Hex and its cofactor GM2-activator protein catalyze the degradation of the GM2 gangliosides and other gangliosides, which are the main glycolipids of the neuronal plasma membrane. Only the α-subunit is able to hydrolyze GM2 gangliosides because of a key residue and a loop structure, absent in the β-subunit, for the binding of the GM2-activator protein (GM2AP).
TSD (OMIM 272800) occurs when the activity of the α-subunit (HexA) is deficient. TSD heterogeneity in severity of clinical symptoms and age of onset is determined by residual HexA enzymatic activity, with only 10–15% of HexA activity required to prevent GM2 ganglioside accumulation. 53 In SD (OMIM 268800), the causative mutation affects the gene coding for the β-subunit (HexB). The clinical manifestations of these diseases are very similar to GM1 gangliosidosis.
Three animal models are available for GM2 gangliosidosis, the SD mouse, the SD cat, and the TSD sheep. 53 The mouse and cat models have some limitations, whereas the TSD sheep closely reproduce the human genetics and the biochemical pathology, with progressive neurological symptoms characteristic of TSD patients. 53 Intracranial administration of AAVrh8 vectors encoding the α-subunit of Hex (TSD α) or a mixture of two vectors encoding both the α- and β-subunits separately (TSD α+β) was tested in TSD sheep. 54
rAAVs were injected in a single lateral ventricle and into the thalamus bilaterally. 54 The treatment had positive effects on behavior, clinical biomarkers, and neuroinflammation but did not significantly increase the animal lifespan. 54 Histopathological evaluation evidenced a scarce enzyme delivery to the cerebellum and spinal cord. SD cats receiving the same GT demonstrated better results, 55 correlated with a higher Hex activity in the cerebellum and spinal cord, 55 indicating the need for improved enzyme delivery in the cerebellum and spinal cord to reach a clinical benefit.
Following these efficacy studies, the same authors performed a safety study of the strategy in cynomolgus macaques (cm). A single bilateral infusion of AAVrh8 vectors encoding cm-Hex α- and β-subunits (AAVrh8-cmHexα/β) was performed in the thalamus and in the lateral ventricles of the animals. Unexpectedly, the treatment resulted in dyskinesia, ataxia, and loss of dexterity in most of treated monkeys, with animals receiving the highest vector dose eventually becoming apathetic. 56 In these animals, the neurological function declined so rapidly to require a premature euthanasia between 20 and 28 days post-GT. 56
Histopathology in monkeys injected with AAVrh8-cmHexα/β showed severe white and gray matter necrosis along the injection track, reactive vasculature, and abnormal neurons, 56 whereas minimal or no lesions were observed in the control animals. 56 A dramatic increase in Hex activity was measured in the thalamus. 56 The extreme overexpression of Hex protein in a very limited number of thalamic neurons has been imputed by the authors as the main cause of this severe neurotoxicity. 56
Since previous studies of direct intracranial injections of AAVrh8 vectors encoding species-specific Hex α- or β-subunits at 1:1 ratio in mice, cats, and sheep have all indicated safety and widespread enzyme distribution, 54 –56 the occurrence of these severe side effects in monkeys suggests the need for careful safety consideration and extensive multispecies preclinical experimentation of similar in vivo GT approaches before proceeding to the clinical phase.
Neuronal Ceroid Lipofuscinoses
Other very attractive indications for GT are represented by the family of disorders called neuronal ceroid lipofuscinoses (NCLs; Batten disease), a group of clinically overlapping and fatal LSDs with primarily neurodegenerative symptoms based on the deficiency of lysosomal enzymes (CLN1, CLN2, and CLN5) or transmembrane proteins (CLN3, CLN6, CLN7, CLN8, and CLN12). Promising preclinical and clinical data were recently produced for the enzymatic forms, whereas the design of efficacious GTs for the other forms is particularly challenging, since the causative transmembrane proteins are usually poorly characterized. The most advanced therapeutic strategy for these indications to date is represented by in vivo GT for the restoration of the lysosomal enzyme of interest in the CNS.
Relevant preclinical data of in vivo GT has been obtained for CLN1, caused by the deficiency of palmitoyl protein thioesterase 1 (PPT1). Multiple intracranial injections of an rAAV2 vector encoding the human PPT1 in newborn Ppt1-ko mice resulted in detectable enzyme activity at the injection sites, reduction of storage material, reduction of neurodegeneration, and improvement of motor and behavioral functions. 57 However, the treatment failed in reducing the frequency of seizures and in increasing the mice survival. 57 Similar results were obtained by multiple intracranial injections of an rAAV5-PPT1 vector into young Ppt1-ko mice. 58
In this study, the authors showed an increased therapeutic benefit when the rAAV-mediated gene transfer was coupled with bone marrow cell transplantation, whereas bone marrow cell transplantation alone showed no therapeutic effects. 58 Finally, simultaneous injection of rAAV9-PPT1 in the brain and spinal cord of Ppt1-ko mice resulted in a significantly better therapeutic outcome than the direct vector delivery in a single region of the CNS, suggesting the relevance of targeting the whole CNS to achieve a significant therapeutic efficacy in NCLs. 59
For CLN2, caused by the deficiency of the tripeptidyl peptidase 1 enzyme (TPP1), several in vivo GT strategies has been developed and clinically tested. A phase I clinical trial (
Additional phase I/II in vivo GT clinical trials are ongoing for the treatment of CLN3 and CNL6 based on intrathecal injections of rAAV9 expressing the cDNA for the therapeutic enzyme (
Overall, an efficacious therapy for NCL still represents an urgent and challenging unmet medical need, which will hopefully be fulfilled in the next future by novel strategies for the efficient delivery of enzyme-competent cells or therapeutic vectors that could potentially offer a better clinical outcome and an improved quality of life for NCL patients and their families.
Novel Genome-Editing Approaches for LSDs
Among the novel gene-based therapeutic strategies for neurometabolic diseases, very promising approaches are based on site-specific editing of the human genome, to specifically correct the disease-causing gene or add a copy of the therapeutic gene in a specific genomic location. The genome-editing system based on the clustered regularly interspaced short palindromic repeats-associated protein-9 nuclease (CRISPR/Cas9) is a relatively simple and versatile tool to engineer eukaryotic cells. 61 The system is based on the delivery of a Cas9 nuclease and a short guide RNA (sgRNA) to the target cells.
When guided to the target genomic sequence by the sgRNA, the Cas9 nuclease creates a double-stranded DNA break (DSB). In case a donor DNA template is provided, the DSBs induce homologous recombination with the donor DNA molecule that contains the desired genetic modification or the therapeutic gene, flanked by homology arms centered at the break site. 61 This cellular mechanism is commonly indicated as homologous recombination-mediated genome editing and has been extensively used for in vitro gene correction in several primary human cells to date. 61
With this tool, a therapeutic gene can be potentially inserted downstream to its endogenous promoter, in its natural genomic locus. It has the general advantage of a physiological transgene expression, but it may not be sufficient in the context of LSDs to reach a critical level of enzyme expression, necessary to secure the cross-correction of unedited cells. Therefore, the insertion of a complete expression cassette (exogenous promoter-gene of interest) into a genomic region considered nonessential for a particular cell type (or “safe harbor”) may be preferential to achieve gene overexpression and the supranormal enzymatic activity in the brain. Besides, a safe harbor provides a platform that can be easily adaptable to several lysosomal enzymes just changing the donor DNA template and allows for a predictable expression level in a maximum of two copies for genome (biallelic insertion).
The most advanced preclinical studies testing a CRISPR/Cas9 genome editing approach for LSDs are based on the genetic engineering of engraftable human fetal brain–derived neural stem cells (NSCs) to express the lysosomal enzyme galactosylceramidase for the treatment of Krabbe disease 62 and of autologous CD34+ HSPCs to express the lysosomal enzyme ialuronidase in a standard setting of ex vivo GT for MPS I. 63 In both cases, the human target cells have been edited by inserting the therapeutic transgene in a safe harbor locus, represented by the interleukin-2 receptor gamma locus in the NSCs 62 and by the C-C chemokine receptor type 5 locus 63 in HSPCs.
With this approach, the investigators obtained in one case the biochemical cross-correction of patient-derived cells cultured with the edited NSCs, 62 and in the other study, the phenotypical correction of immunodeficient MPS I mice receiving the human edited HSPCs. 63 These exciting results inspire the development of a similar editing-based strategy for other LSDs, possibly exploiting a safe harbor approach that may represent a novel and flexible platform for gene expression in multiple neurodegenerative diseases.
Final Considerations
A successful therapy for neurometabolic diseases is based on the achievement of high levels of gene-corrected cell chimerism in the CNS, coupled with a sustained expression and widespread activity of the therapeutic factor(s). The delivery of multiple copies of the therapeutic gene to the proper target cells and regulating its expression by an appropriate promoter are crucial aspects for the design of an efficacious GT strategy. Although overexpression of the therapeutic enzyme was well tolerated and likely instrumental to achieve clinical benefit in the ex vivo GT clinical trial for MLD, 9,10 an excessively high enzyme production may result in cytotoxicity.
An example is represented by the overexpression of lysosomal galactocerebrosidase enzyme (GALC), defective in GLD, and involved in the maintenance of a functional HSPC niche by contributing to the control of the intracellular content of key bioactive sphingolipids. 64 Either abrogation of GALC activity, as in GLD, or gene transfer and supraphysiological GALC expression resulted in alterations of human HSPC survival, clonogenic potential, and engraftment capability. 64 Interestingly, supraphysiological GALC activity in differentiated hematopoietic cells did not induce any toxicity, 65 suggesting the possibility of cell-specific sensitivity to alterations in the normal lysosomal enzyme activity.
In addition to the potential cell-specific response to a particular enzymatic imbalance, a general mechanism of cytotoxicity triggered by stable protein overexpression is mediated by the chronic unfolded protein response. 66 Activation of this cellular pathway, coupled with a very limited number of extensively transduced cells, has been suggested as the main cause of acute neurotoxicity observed in monkeys receiving intraparenchimal injection of AAVrh8 vectors expressing N-β-acetylhexosaminidase in a recent in vivo GT study for GM2-gangliosidoses. 56
To reduce the vector load, alternatives have to be investigated, as modulating the therapeutic gene expression or engineering the therapeutic enzyme to increase its uptake. 67 Concerning the proper target cells for an efficient gene-based approach to neurometabolic diseases, microglial cells are emerging as a key cell type, not just as producer of therapeutic enzymes but also as principal modulator of neuroinflammation. Neuroinflammation is a common hallmark of neurodegenerative diseases, primarily contributing to neurodegeneration by creating a neurotoxic environment as consequence of a massive release of cytokines, chemokines, and proapoptotic molecules. 3
Activation of microglia, and of astrocytes in a lesser extent, often precedes and predicts regions of neuronal loss. 3 If the endogenous activated microglia has been suggested as the main determinant of neurodegeneration in LSDs and other neurodegenerative diseases, HSPC-derived enzyme-competent microglial cells has been demonstrated as the principal mediator of the therapeutic effect in at least two successful ex vivo GT clinical trials, for ALD and MLD. 9,28 For this crucial role, microglia represent the prime target to achieve a significant therapeutic effect, by stopping the neurodegeneration and mediating other fundamental therapeutic effects, as reverting the CNS environment from neurotoxic to a neurotropic. 3
To achieve this goal, a robust engraftment of HSPC-derived enzyme-competent microglial cells is essential, in properly pretreated recipients. 13 In this setting, genetically modified microglia can be exploited to locally produce additional disease-modulating molecules with neurotrophic and antinflammatory abilities, as well as to restore normal scavenging and immune surveillance functions. 68
This approach can be particularly useful to treat other neurodegenerative diseases with a complex and multifactorial etiology, such as Alzheimer disease, Parkinson disease, Lewy body dementia, frontotemporal dementia, amyotrophic lateral sclerosis, Huntington disease, and prion diseases. Thus, we may envisage the development of novel and efficacious GT strategies for rare neurological LSDs as well as for these devastating neurological pathologies, considering that the dementias are overall responsible for the greatest burden of disease nowadays in the developed countries, affecting more than seven million people in Europe, a number doubling every 20 years as the population ages (EU Joint Programme, Neurodegenerative Disease Research).
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
No funding was received for this article.