
Abstract
Lung gene therapy requires efficient transduction of slow-replicating epithelia and stable expression of delivered transgenes in the respiratory tract. Lentiviral (LV) vectors have the ideal coding, expression, and transducing capacity required for gene therapy. A modified envelope glycoprotein from the Jaagsiekte Sheep Retrovirus, termed Jenv, is well suited for LV-mediated lung gene therapy due to its inherent lung tropism. Here, two novel Jenv-pseudotyped LVs that effectively transduce lung tissue and yield titers similar to the gold standard, vesicular stomatitis virus glycoprotein (VSVg)-pseudotyped LVs, were generated. As the concentration efficiency of LVs was found to depend on envelope pseudotype, a large-scale production method tailored for Jenv-pseudotyped LVs was developed and the most appropriate method of concentration was determined. In contrast to VSVg and Ebola virus glycoprotein-pseudotyped LVs, ultracentrifugation through a sucrose cushion drastically reduced the yield of Jenv LVs, whereas polyethylene glycol precipitation and tangential flow filtration (TFF) proved to be more suitable methods for concentrating Jenv LVs. Importantly, pressure during TFF was found to be crucial for increasing LV recovery. Finally, a unique mouse model was developed to test the suitability of these novel Jenv-pseudotyped LVs for use in lung gene therapy applications.
Introduction
Lentiviral vectors (LVs) remain attractive tools for gene delivery due to their ability to encode larger transgenes, than for example adeno-associated virus (AAV) vectors, and insert these transgenes permanently into the genome of transduced cells. 1,2 Lentiviruses are capable of infecting both dividing and non-dividing cells 3 and it is this ability to target quiescent cells that is of special interest for lung gene therapy applications, where the epithelia generally have low cell division rates. 4 LVs have been modified to mitigate the risk of oncogenesis-linked genome integration, 5 which has led to increased demand for LVs in gene therapy applications. 1,2 Other viral vectors being developed for lung gene therapy include those based on adenovirus and AAV. However, the immunogenic potential of adenovirus vectors and the small coding capacity of AAV vectors hinder their potential use. 4,6 In contrast, LVs have relatively low immunogenicity and relatively large coding capacity. 7,8 Despite these advantages, LVs continue to lag behind adenovirus and AAV vectors for use in lung gene therapy trials.
For LVs to be effective for use in lung gene therapy applications, it is critical to identify envelope glycoproteins that when used for pseudotyping promote efficient lung gene transfer in vivo. Vesicular stomatitis virus glycoprotein (VSVg)-pseudotyped LVs efficiently transduce a wide range of cell and tissue types both in vitro and in vivo 9 and are, thus, considered the “gold standard.” However, studies have shown that VSVg cannot readily access the lung epithelia from the apical surface due to the basolateral location of the low-density lipoprotein receptor. 10,11 Thus, chelating and surfactant agents are being developed and utilized to transiently disrupt the tight junctions in the lung epithelium and facilitate VSVg-LV entry. 12,13 Though this approach may be suitable for some genetic diseases of the lung, the use of chelators might not be appropriate for others, such as Cystic Fibrosis (CF). 7,14 Cumulative disruption of tight junctions could lead to inflammation or bacterial translocation, thereby enhancing the pathology of CF. 7,15 Efforts to find alternatives to VSVg have identified envelope glycoproteins from a range of viruses, including baculovirus, Ebola virus (EBOV), and influenza virus, that can efficiently pseudotype LVs and facilitate transduction both in vitro and in vivo. 16 –19
Recently, the glycoprotein of Jaagsiekte Sheep Retrovirus (JSRV) was identified as a promising envelope for pseudotyping LVs. 20,21 JSRV is the only retrovirus known to target the respiratory tract. 21 Due to its natural tropism for the lung, the JSRV envelope glycoprotein (Jenv) represents an ideal envelope with which to pseudotype LVs for use in lung gene therapy. Indeed, Liu et al. established that Jenv could efficiently pseudotype LVs. 22 In 2005, Sinn et al. demonstrated that flanking Jenv with promoter regions from the JSRV LTR increased expression of the envelope and, consequently, production of LVs pseudotyped with Jenv 20,21 ; they termed their new Jenv construct delta gag pol Jenv (ΔGP Jenv). Further, in 2012, Davey et al. showed that the Jenv-pseudotyed LV was capable of efficiently transducing the fetal ovine airway, in contrast to EBOV glycoprotein-pseudotyped LV, baculovirus GP64 LV, and AAV2/6.2. 21 Despite the demonstrated ability of ΔGP Jenv to transduce the lung, it has remained difficult to produce high titer vector, at levels similar to that of VSVg-LV, leading to reduced interest in Jenv as a pseudotyping protein.
In this study, we optimized a scalable method for purifying Jenv-pseudotyped LVs suitable for use in preclinical studies. By mutating amino acid residues N592 and M593 in the cytoplasmic tail (CT) of Jenv, which are known to enhance protein expression and reduce toxicity, 23 respectively, we were able to dramatically increase Jenv protein production levels in human embryonic kidney (HEK) 293T cells and this correlated with a significant increase in LV titers, similar to that of VSVg LV. By employing AAV-mediated pulmonary delivery of the human homologue of hyaluronoglucosaminidase-2, the cellular receptor for JSRV, we developed a novel mouse model that allowed us to evaluate the transducing properties of Jenv-pseudotyped LVs in ex vivo lung slices. Finally, we demonstrate that these novel mutant Jenv-pseudotyped LVs can efficiently transduce both murine and ovine lung tissue. The results of this study have important implications for optimized gene delivery to lung epithelial cells.
Materials and Methods
Cell lines
HEK 293T cells were cultured and maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated Cosmic Calf Serum (ThermoFisher Scientific, Mississauga, Ontario, Canada), 2 mM L-glutamine 100 U/mL penicillin, and 100 μg/mL streptomycin (P/S) in a humidified 5% CO2 incubator at 37°C.
Animals
All animal experiments were conducted in accordance with the Canadian Council on Animal Care guidelines and approved by the Animal Care Committee of the University of Guelph. Four 6-week-old BALB/c female mice were purchased from Charles River Laboratories (St. Constant, Quebec, Canada). The mice were acclimated to the environment for 7 days before intranasal administration of 1 × 1011 viral genomes (vg) of AAV6.2FF-FTHyal2 (n = 3), as previously described. 24,25 Control mice were administered phosphate buffered saline (PBS; n = 1). One specific-pathogen free Cornell Star neonate lamb was obtained from Ponsonby Research Station (University of Guelph) and euthanized via an intravenous injection of pentobarbital (Euthansol, Merck Animal Health, Quebec, Canada) before harvesting tissues.
Lung tissue slices
Lung slices were prepared and maintained as previously described. 26 Briefly, mouse and lamb lungs were excised immediately after euthanasia. Each murine lung lobe and intact lamb lungs were perfused via the trachea with 2% low melting point agarose. A Microslicer DTK-3000W (PALL Corporation, Mississauga, Ontario, Canada) generated 300-μm-thick tissue slices. These were maintained in culture for 3 days before transduction and an additional 2 days post-transduction for fluorescence microscopy imaging or alkaline phosphatase (AP) staining.
DNA constructs
Lentivirus vector particles were generated with the use of psPAX2 (second-generation packaging plasmid devoid of vif, vpr, vpu, and nef genes) (Addgene plasmid no. 12260, Cambridge, MA), pSinGFP or pLV-CASI-hPLAP (backbone constructs with reporter gene), and envelope glycoprotein-encoding vectors pCI-Neo-VSVg, pCMV-EBOV GP, or pCMV3JS21ΔGP for VSV, Zaire EBOV, and JSRV glycoproteins, respectively. Development of the AAV6.2FF vector has already been described. 24 For the cloning of pLV-CASI-hPLAP, ΔGP Jenv N592T and M593E, and pACAG-PPT-FT-hHyal2 please refer to Supplementary Data.
LV production
LV particles were produced in HEK 293T cells. Two million cells were seeded per 10-cm dish for a total of fifteen 10-cm dishes. One day post-seeding, cells were transfected by using the following per dish: 5 μg of genome plasmid (pSinGFP or pLV-CASI-hPLAP), 3.25 μg of helper plasmid (psPAX2), and 1.75 μg of envelope plasmid (pCI-Neo-VSVg, pCMV-EBOV GP, pCM3JS21ΔGP, pCMV3JS21ΔGP N592T, or pCMV3JS21ΔGP M593E) plus 67.5 μL of 1 mg/mL polyethylenimine transfection polymer (Polysciences, Inc., Warrington, PA), and 450 μL of DMEM per dish. Two hours post-transfection, the medium was replaced with basal DMEM supplemented with 2 mM L-glutamine and P/S. Every 24 h, for a total of 72 h, the supernatant was collected and replenished with fresh basal DMEM. Once collected, the supernatant was spun down in a swinging-bucket centrifuge at 500 g for 5 min to pellet cellular debris. The clarified supernatant was passed through a 0.45-μm polyethersulfone syringe-tip filter (VWR, Mississauga, Ontario, Canada) and stored at 4°C until all three LV collections were procured. Ten milliliters of the neat supernatant were set aside and maintained at 4°C for LV titrations.
AAV6.2FF-FT-Hyal2 production
Production of AAV6.2FF was previously described. 24 The AAV6.2FF-FT-Hyal2 vector was produced in the same fashion.
Lentivector purification and concentration
Ultracentrifugation
Concentration of LVs by ultracentrifugation has been extensively described. 27 Briefly, LV-containing clarified supernatant was layered on top of a 6-mL 20% sucrose cushion in thin-walled ultra-centrifuge tubes (Beckman Coulter 360743, Mississauga, Ontario, Canada) (30 mL per tube) suited for the SW32Ti rotor (Beckman Coulter 369650, Mississauga, Ontario, Canada) and centrifuged at 60,000 g for 2 h at 4°C. Immediately after centrifugation, the LV pellet suspended in 100 μL of Tris-NaCl-EDTA (TNE) buffer, pH 7.4 (100 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA), 28 yielding a total of 600 μL of concentrated LV.
Polyethylene glycol-8000 precipitation
This study employed similar techniques, as has been previously detailed, with a few exceptions. 29 In brief, LV-containing supernatant was distributed into five 50-mL conical, high-speed centrifuge tubes and polyethylene glycol (PEG)-8000 (Sigma-Aldrich, Oakville, Ontario, Canada) was added in a 1:2 PEG to supernatant ratio. The tubes were then gently rocked overnight for 16 h at 4°C. The next day, the LV-PEG mixture was subjected to swinging-bucket centrifugation at 4,000 g for 15 min. Each tube was then decanted into a waste bottle and set upside down on top of paper towels to remove any residual media. Finally, the LV-containing white pellet was re-suspended in TNE buffer at 1/100th the initial supernatant volume.
Tangential flow filtration
Three different sized tangential flow filtration (TFF) membranes were used in this study: 50K, 100K, and 300K. Other than the different membranes, the Centramate Cassette (PALL Corporation) was assembled (Fig. 1) as specified by the manufacturer and as previously described. 30 The TFF process was run with two different differential pressures: 5 and 0 psi, or 15 psi and 10 psi for inlet and outlet pressures, respectively. A buffer exchange with PBS was performed. Elutions were collected in TNE buffer. Neat (input) virus, elutions, and waste aliquots were collected for titration by p24 enzyme-linked immunosorbent assay (ELISA) and flow cytometry (transducing units [TU]/mL).

Graphical representation of the TFF setup. TFF, tangential flow filtration. Color images are available online.
Downstream TFF purification and PEG-20,000 concentration
The production of lentivirus particles via a sucrose gradient and PEG-20,000 concentration has been previously described. 30 Briefly, ultra-clear centrifuge tubes (344058; Beckman Coulter, Brea, CA) were filled with 10 mL of 60% sucrose, followed by 10 mL of 20% sucrose and finally, by 20 mL of pooled TFF elutions. The gradients were centrifuged at 51,000 g for 2 h at 4°C. Immediately after centrifugation, the tubes were clamped to a retort stand and a syringe was used to remove ∼3 mL of the band between the 20% and 60% sucrose cushions, as previously described. 31 The LV was then dialyzed and precipitated by using PEG-20,000 as described. 30
Lentivector titration
An ELISA kit to detect the p24 capsid protein of HIV-based lentivirus particles was used (Takara Clontech, Mountain View, CA), according to the manufacturer's specifications. The assay used to determine TU/mL was extensively described earlier, and the same approach was employed here. 32 HEK 293T cells were seeded at 300,000 cells and transduced the next day with serial dilutions of vector particles in six-well plates in the presence of 8 μg/mL polybrene. Twenty-four hours later, the percentage of cells expressing the green fluorescent protein (GFP) reporter protein was measured by flow cytometry (Canto II; BD Biosciences, Mississauga, Ontario, Canada), and it was analyzed by using FlowJo LLC software (version 10; BD Biosciences). The concentration of the vectors was measured as described. 32
Transduction of lung slices
Lung slices (n = 5 for lamb lung slices and n = 9 for murine lung slices) were transduced with 1 × 106 TU per vector in the presence of 8 μg/mL of polybrene. Immediately after transduction, the plate containing the lung slices was placed on a nutating mixer overnight in a humidified 5% CO2 incubator at 37°C. Cells and lung slices transduced with GFP LVs were imaged with a Zeiss AXIO Observer.A1 48 h after transduction. A Zeiss Stereo Microscope CL1500 ECO (Zeiss, Toronto, Ontario, Canada) was used to image the murine lung slices after AP staining, as previously described. 33
Immunohistochemical staining
Lung lobes from PBS-treated and AAV6.2FF-FT-Hyal2-transduced BALB/c mice were harvested for immunohistochemical staining as previously described. 34 Briefly, rabbit α-FLAG (cat. no. Ab21536; Abcam) was used at 1:100 in 3% bovine serum albumin in 1 × PBS-Tween-containing SignalStain Boost IHC Detection Reagent (HRP Rabbit) (Cell Signaling, NEB, Burlington, Ontario, Canada) as recommended by the manufacturer. SIGMAFAST 3,3′-Diaminobenzidine DAB (Sigma-Millipore, Oakville, Ontario, Canada) was used for detection following the manufacturer's instructions. The slides were counterstained with hematoxylin and mounted with Richard-Allan Scientific Cytoseal XYL (ThermoFisher Scientific), before being imaged with a Clinical Olympus BX45 microscope (Olympus, Tokyo, Japan).
Western blotting
HEK 293Ts were transfected with Jenv GFP LV, ΔGP Jenv GFP LV, ΔGP Jenv N592T GFP, and ΔGP Jenv M593E GFP as described in the LV Production section of the Materials and Methods. Cell lysate preparation and western blotting were performed as described. 35
Statistical analysis
All experiments were performed in triplicate on three separate occasions. Standard error was calculated for all figures (error bars) and data tables (standard error of the mean). Unpaired t-tests and two-way analysis of variance were conducted to determine statistical significance where appropriate. All statistical analyses were conducted with Prism7 software (GraphPad, La Jolla, CA).
Results
LVs pseudotyped with the envelope glycoproteins from VSV, EBOV, and JSRV differentially withstand ultracentrifugation and PEG precipitation
It is well known that LVs pseudotyped with envelope glycoproteins from disparate viruses differ with respect to their stability during purification. 36 Initially, we compared two standard methods of concentrating LVs to determine whether they would be suitable for ΔGP Jenv pseudoypted LVs expressing GFP: ultracentrifugation through a 20% sucrose cushion and PEG-8000 precipitation. For comparison, we included the gold standard LV pseudotype, VSVg, and EBOV GP-pseudotyped LV as an additional lung tropic vector recently investigated for use in CF gene therapy. 17,18 Vector titers, in the form of capsid protein in pg/mL, were determined by using a commercial p24 ELISA, which detects capsid protein in HIV-based LVs. TU/mL was measured as the percentage of GFP-positive cells per volume of LV used to transduce cells. Using these two assays, we determined the relationship between p24 levels and infectivity and found that 1 infectious unit corresponded to 100 LV particles (Supplementary Fig. S1).
For VSVg LV, which is known to be a stable vector, PEG-8000 precipitation and ultracentrifugation yielded average recovery rates of 78.0% and 86.4%, respectively (Table 1). Interestingly, in the case of EBOV GP LV, average recovery rates were nearly 100% for both concentration methods. LVs pseudotyped with ΔGP Jenv were stable during PEG-8000 precipitation, yielding 100% average recovery, but they were destroyed during ultracentrifugation such that, on average, 31.3% of LV was recovered.
Titers and percent recovery of VSVg, EBOV GP, and ΔGP Jenv-pseudotyped LVs concentrated by sucrose cushion ultracentrifugation or PEG-8000 precipitation
The values are in mean ± SEM.
TU were determined by using flow cytometry to quantify number of GFP-positive cells.
Experiments were conducted in triplicate on three separate occasions. Average recovery represents infectious titer (TU) recovery.
ΔGP Jenv, delta gag pol Jenv; EBOV, Ebola virus; GFP, green fluorescent protein; PEG, polyethylene glycol; SEM, standard error of the mean; TU, transducing units; VSVg, vesicular stomatitis virus glycoprotein.
Pressure use during TFF increases the rate of recovery of VSVg and ΔGP Jenv LV-pseudotyped vectors
Large-scale scale production of LVs is often required to obtain vector quantities that are sufficient for in vivo applications. Processing large volumes by ultracentrifugation or high-speed centrifugation for PEG-8000 precipitation is cumbersome and time-consuming. However, TFF is a simple method used to concentrate liter-scale volumes of LV-containing supernatant. 37,38 TFF does not appreciably trap LV and allows for buffer exchange, as volumes are considerably reduced. Therefore, we explored the use of TFF as an alternative to centrifugation for concentrating large volumes of LVs. VSVg-pseudotyped LV was used in the optimization process due to its relatively stable nature. 39 Initial efforts to concentrate VSVg LV by TFF involved the use of 5 psi of differential pressure, as this had been previously reported. 38,40 Here, the inlet/input pressure was set to 5 psi and no pressure was maintained on the outlet/output (Fig. 1). We designated this condition as low-pressure TFF. Although there are reports in the literature of close to 100% recovery of LVs when using TFF with 5 psi of differential pressure, 37,38 we were unable to reproduce those results by using a 100K molecular weight cut-off (MWCO) membrane with five and zero psi of inlet and outlet pressure, respectively (Table 2). To ensure this was not due to differences in the MWCO membrane used, we tested three different MWCO membranes (50K, 100K, and 300K) with the same pressure conditions. However, TFF purification of VSVg LV with the 50K, 100K, and 300K membrane all resulted in very poor average recovery rates of 2.5%, 21.0%, and 10.6%, respectively (Table 2: Low-pressure TFF). To introduce additional pressure into the system, the inlet pressure was subsequently increased to 15 psi without changing the outlet pressure; however, this also did not improve yields. Next, inlet pressure was kept at 15 psi and the retentate tubing was clamped to increase the outlet pressure to 10 psi, thereby maintaining a differential pressure of 5 psi. We designated this condition as high-pressure TFF. This alteration greatly increased average recovery yields (Table 2: High-pressure TFF). Further, using the high-pressure TFF conditions, all membranes showed a higher average recovery rate than with low-pressure TFF conditions. The 50K membrane had the highest average recovery yields of all MWCO membranes tested.
Titers and percent recovery of VSVg LV concentrated using TFF under low- and high-pressure conditions
The values are in mean ± SEM.
TU were determined by using flow cytometry to quantify number of GFP-positive cells.
Experiments were conducted in triplicate on three separate occasions. Average recovery represents infectious titer (TU) recovery.
Sucrose gradient ultracentrifugation, dialysis, and PEG-20,000 concentration
TFF, tangential flow filtration.
Vectors recovered directly from TFF, even after PBS buffer exchange, were then used to transduce HEK 293T cells; however, this was somewhat toxic to cells. To resolve this issue, we implemented a post-TFF purification sucrose gradient ultracentrifugation and dialysis step by using the vector recovered from the 50K MWCO membrane as proof of principle. Employing these additional downstream purification steps eliminated toxicity and vector titers increased to >108 TU/mL without compromising yield (99.6% for VSVg LV; Table 2, bottom row). Having optimized the TFF concentration method for VSVg LV, we then applied it to ΔGP Jenv LV. In contrast to ultracentrifugation by sucrose cushioning, TFF using a 50K membrane under high pressure resulted in high ΔGP Jenv LV-pseudotyped vector recovery (94.1%; Table 3). Thus, we determined that a scalable TFF method, which did not affect recovery or rely on multiple rounds of centrifugation, could be used to concentrate large volumes of ΔGP Jenv LV.
Titers and percent recovery of ΔGP Jenv LV using TFF under low- and high-pressure TFF conditions
The values are in mean ± SEM.
Experiments were conducted in triplicate on three separate occasions. Average recovery represents infectious titer (TU) recovery.
Sucrose gradient ultracentrifugation, dialysis, and PEG-20,000 Concentration.
Mutating amino acid residues in the CT of Jenv increases protein expression levels and, in turn, LV titers
We consistently observed that the titer of ΔGP Jenv LV-containing supernatant was an order of magnitude lower than VSVg LV-containing supernatant. We reasoned that this low titer could be due to cytotoxicity induced by the Jenv protein, which in our hands causes HEK 293T cells to lift off the plate, or due to poor Jenv expression levels in producer cells. To address these two possibilities, we introduced M593E or N592T mutations into the CT of ΔGP Jenv to either abolish transforming activity, which results in less frequent cell death in the producer cells, or increase protein expression, respectively. 23 To test whether these mutations would lead to increased Jenv expression, HEK 293T cells were transfected with Jenv, ΔGP Jenv, ΔGP Jenv N592T, or ΔGP Jenv M593E expression vectors, along with a luciferase reporter gene construct to control for transfection efficiency, and cells were harvested 48 h post-transfection. Using an antibody against the surface glycoprotein of Jenv (anti-SU), Western blot analysis on protein lysates normalized for transfection efficiency revealed that both the M593E and N592T mutant versions of ΔGP Jenv were expressed at higher levels than wild-type ΔGP Jenv (Fig. 2A). Densitometry on Western blots performed in triplicate revealed that ΔGP Jenv N592T and ΔGP Jenv M593E expressed significantly more Jenv protein than wild-type ΔGP Jenv (Fig. 2B and Supplementary Fig. S2).

Mutations in the cytoplasmic tail of Jenv increase protein expression levels and lead to production of higher titer LVs.
To investigate whether higher Jenv expression levels resulted in higher titers, equal volumes of supernatant containing lentivectors pseudotyped with VSVg, ΔGP Jenv, ΔGP Jenv N592T, and ΔGP Jenv M593E were titered (Fig. 2C); then concentrated by TFF; and subjected to downstream ultracentrifugation (Fig. 2D) in three separate experiments. Before centrifugation, the mutant ΔGP Jenv N592T and ΔGP Jenv M593E pseudotyped LVs yielded average titers of 3.4 × 108 TU/mL (3.37 × 108 ± 8.659 × 107) and 1.4 × 108 TU/mL (1.44 × 108 ± 4.02 × 107), respectively, which were more than a log higher than ΔGP Jenv LV (4.50 × 106 ± 3.78 × 105 TU/mL) and not significantly different than VSVg LV titers (3.10 × 108 ± 2.484 × 107 TU/mL) (Fig. 2C). After centrifugation, the mutant ΔGP Jenv N592T and ΔGP Jenv M593E pseudotyped LVs yielded average titers of 5 × 109 TU/mL (5.035 × 109 ± 1.418 × 109) and 3 × 109 TU/mL (2.934 × 109 ± 5.490 × 108), respectively, which were more than 10-fold higher than ΔGP Jenv LV (7.75 × 107 ± 2.69 × 107 TU/mL) and in the case of ΔGP Jenv N592T, not significantly different than VSVg LV titers (7.87 × 109 ± 3.577 × 108 TU/mL) (Fig. 2D). Taken together, introducing mutations into the CT of Jenv to enhance protein expression led to increased LV particle production, rather than increased particle stability, with titers similar to those of VSVg-pseudotyped LV.
Development of a mouse model to evaluate Jenv-pseudotyped LV-mediated transduction
We wanted to compare VSVg, EBOV GP, ΔGP Jenv, ΔGP Jenv N592T, and ΔGP Jenv M593E LVs in tissue explants to qualitatively evaluate transduction in both murine and ovine lung tissues. Although ovine and human hyaluronoglucosaminidase-2 (hHyal2) can function as receptors to mediate ΔGP Jenv LV infection, 41 the murine Hyal2 homologue does not. 42 Therefore, to test Jenv-pseudoypted LV vectors in mouse lung, we developed a novel murine lung slice model. Briefly, 1 × 1011 vg of an AAV6.2FF vector 24 expressing a FLAG epitope tagged version of hHyal2 (AAV6.2FF-FT-hHyal2) was administered to 6-week-old BALB/c mice intranasally as described 25 (Fig. 3A). Three weeks later, lungs were harvested and used to generate hHyal2-expressing lung tissue slices as previously described. 26 Immunohistochemical staining for FLAG revealed robust AAV-mediated expression of FT-hHyal2 throughout the lung parenchyma (Fig. 3B, bottom panel). Widespread expression of the cellular receptor for Jenv allowed us to test Jenv-pseudotyped LVs in murine tissues. Lung slices were transduced with 1 × 106 TU LV vectors expressing the human placental alkaline phosphatase (hPLAP) reporter gene pseudotyped with the VSVg, ΔGP Jenv, ΔGP JenvN592T, ΔGP JenvM593E, or EBOV GP envelopes. The ΔGP Jenv LV-pseudotyped vectors were capable of transducing tissue slices prepared from the lungs of mice previously transduced with AAV6.2FF-FT-hHyal2, as evidenced by the purple staining indicative of hPLAP activity (Fig. 3C). Mutant ΔGP Jenv LV pseudotypes (N592T and M593E) were also capable of transducing murine lungs at similar or greater levels than ΔGP Jenv, VSVg, and EBOV LV-pseudotyped vectors, as shown by the higher incidence of dark purple staining observed in mutant ΔGP Jenv LV pseudotypes (N592T and M593E, Fig. 3C right panel) transduced murine lungs expressing FT-hHyal2.
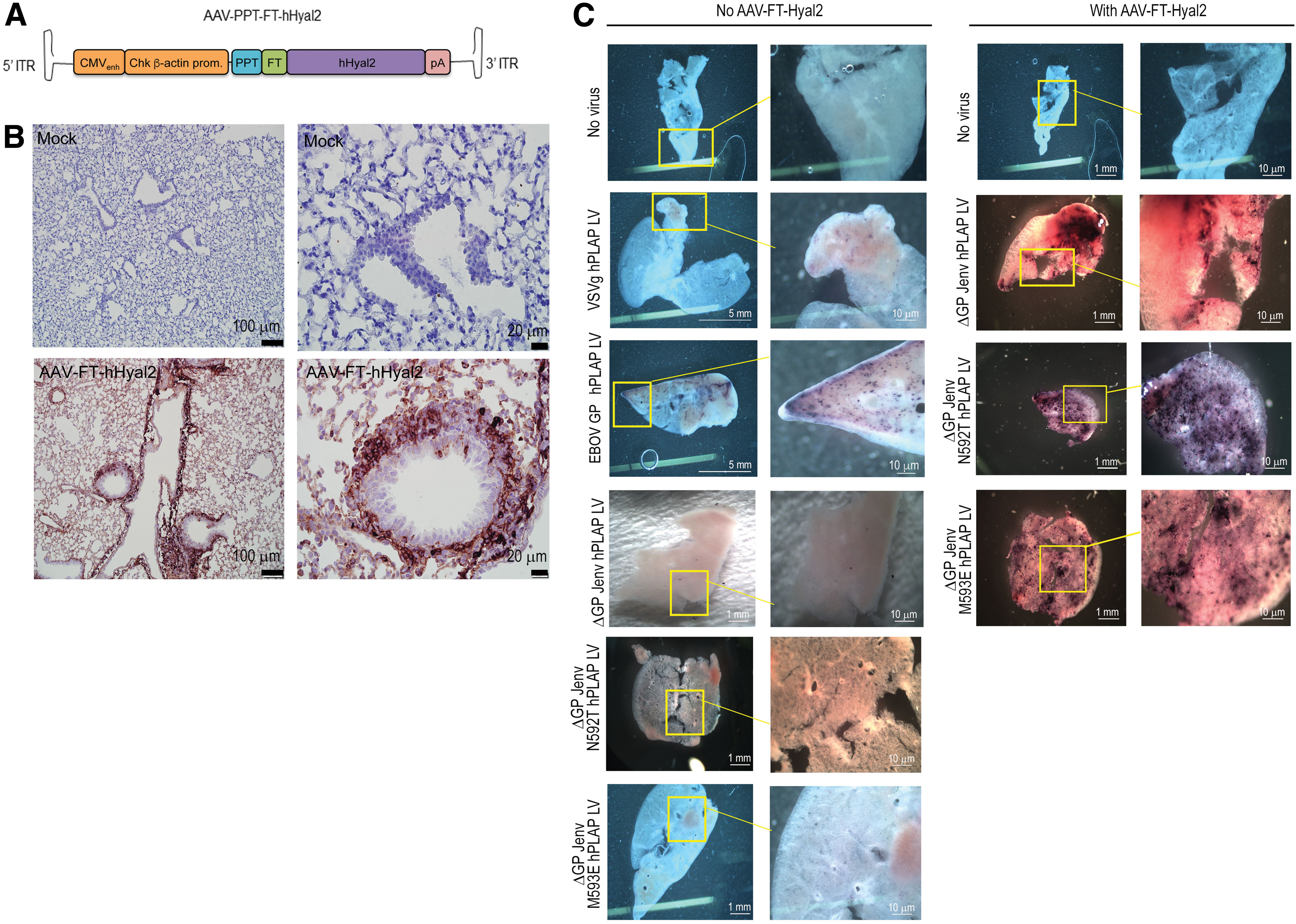
Transduction of murine lung tissue slices with VSVg, EBOV GP, and ΔGP Jenv LVs.
We also examined whether GFP expressing LVs pseudotyped with the various envelopes would transduce ovine lung tissue slices. GFP expression was observed in ovine lung tissue slices transduced with ΔGP Jenv, ΔGP Jenv N592T, ΔGP Jenv M593E, and VSVg LVs, with the latter being less efficient than the Jenv-pseudotyped LVs (Fig. 4). Interestingly, although the EBOV GP LV was able to transduce murine lung slices (Fig. 3C), ovine lung slices were not efficiently transduced by EBOV LV (Fig. 4).

Transduction of ovine lung tissue slices with VSVg, EBOV GP, and ΔGP Jenv LVs.
Discussion
LV-based vectors are attractive gene transfer vehicles for multiple reasons. 43 They efficiently deliver DNA into the nuclei of both dividing and nondividing cells, they integrate into the host genome leading to transgene expression for the lifetime of the cell, and they can be pseudotyped with envelope glycoproteins from a wide range of viruses. Pseudotyping can provide LVs with an expanded ability to target multiple cell types or can restrict LVs to specific cells that are of therapeutic interest. 44 Pseudotyped LVs may also have altered stability and/or interaction with the host immune system that can increase efficacy. 45,46 Finally, some envelope glycoproteins confer LVs the ability to be produced and/or concentrated to higher titers than native viral envelopes. 47
Although VSVg is a commonly used envelope for pseudotyping LVs, VSVg-pseudotyped vectors are comparatively inefficient at transducing airway epithelial cells and require the addition of chemicals to disrupt tight junctions, for example, lysophosphatidylcholine, to permit virus entry into airway cells.
48
–50
Several groups have attempted to improve LV transduction of the airway epithelium by altering the viral envelope proteins. The EBOV glycoprotein and derivatives thereof showed early promise in being able to transduce the large airways and submucosal glands of the murine respiratory tract.
17,18
More recently, LVs pseudotyped with the HA and M2 envelope glycoproteins from fowl plague virus (also known as highly pathogenic avian influenza),
51
GP64 from baculovirus,
8
or the F and HN envelope proteins from Sendai virus
52,53
have been shown to transduce airway epithelia in the nasal cavity. Lentivectors pseudotyped with Jenv are able to transduce human cells in vitro, including the BEAS-2B bronchial epithelial cell line, suggesting that such vectors might be useful for lung-directed gene therapy.
22
Indeed, in utero administration of Jenv-pseudotyped HIV-based LVs to fetal lambs resulted in transduction of >90% of distal airways (with an internal diameter between 20 to 80 μm) with an average epithelial transduction of between 16% and 25%, and airway epithelial transgene expression was maintained for up to 10 weeks in utero.
21
In addition, Jenv-pseudotyped FIV vectors can transduce the distal lung epithelia of rabbits. When tested in vitro, Jenv-FIV vectors transduced primary cultures of rabbit type II alveolar epithelial cells (AEC2) with 100-fold greater efficiency than primary cultures of rabbit tracheal cells, mimicking the cell tropism of wild-type JSRV, which preferentially infects AEC2 both in vitro and in vivo.
54,55
On the other hand, Jenv-FIV vectors were found to transduce primary cultures of human airway epithelia derived from the trachea or upper bronchus poorly in comparison to VSVg-pseudotyped FIV
56
and complementation of primary cultures of human airway epithelia with an adenovirus vector (Ad5) expressing the receptor for JSRV, Hyal2, did not significantly enhance the transduction efficacy.
56
It is important to note, however, that in this study, human airway epithelia grown at the air
Here, we generated two novel LVs pseudotyped with ΔGP Jenv N592T and ΔGP Jenv M593E, mutant versions of the envelope glycoprotein from a lung tropic betaretrovirus, which can produce vector titers equivalent to VSVg LV, and optimized a method for their large-scale purification. Using a lung slice method we previously optimized, 26 we developed a novel murine lung tissue slice model that allowed us to test these pseudotyped LVs ex vivo. We showed that TFF, followed by gradient ultracentrifugation, dialysis, and PEG-20,000 concentration, is a scalable method for producing high-titer ΔGP Jenv-pseudotyped LVs for in vivo use. Further, we demonstrated that murine and ovine lung tissue slices were capable of being transduced by these LVs.
We first evaluated three different LV concentration methods: ultracentrifugation, PEG-8000 precipitation, and TFF. Ultracentrifugation was the most suitable method for concentrating infectious VSVg and EBOV LVs, as previously reported. 39,57,58 However, this was not the case for ΔGP Jenv-pseudotyped LVs. Ultracentrifugation imposes considerable shearing forces that not all envelope glycoproteins are able to sustain. 29,39,59,60 In fact, it has been demonstrated that LV particle stability during ultracentrifugation depends on the particle core stability and the envelope protein used to pseudotype the LV. 39 Studies have evaluated the resilience of the VSVg and found it to be resistant to the shear forces of ultracentrifugation. 39,60 Ultracentrifugation of EBOV LV has also been shown to yield high titers and recovery yield, 17 which we confirmed. Conversely, JSRV envelope-pseudotyped LV has a low recovery yield when ultracentrifuged, compared with LVs pseudotyped by VSVg and EBOV 17,18,58 ; thus, ultracentrifugation is not an ideal method to concentrate ΔGP Jenv-pseudotyped LVs. Although we did not perform a comprehensive analysis of the three different concentration methods for LVs pseudotyped with mutant ΔGP Jenv N592T and ΔGP Jenv M593E envelopes, based on the dramatic loss of ΔGP Jenv-pseudotyped LV (∼70% loss) during ultracentrifugation and the fact that for both VSVg- and ΔGP Jenv-pseudotyped LVs high-pressure TFF resulted in near complete recovery, we believe that high-pressure TFF purification would be equally suitable for LVs pseudotyped with mutant ΔGP Jenv. In addition, although we analyzed both p24 units (pg/mL) and TU/mL, it would be interesting to further characterize the ratio of infectious to non-infectious particles throughout the concentration process.
Unlike sucrose cushion ultracentrifugation and PEG-8000 precipitation, which are acceptable methods for concentrating small volumes of LVs for use in vivo in mice, TFF is a promising technique for concentrating large volumes of LVs and is better suited for large animal gene therapy and human clinical applications due to its scalability. The importance of pressure during TFF purification of LVs has not been clearly defined in the literature. The pressure values reported tend to reflect the differential pressure and do not clearly specify whether or not the outlet pressure is altered. 38,40 We found that increasing the outlet pressure while maintaining the same pressure differential was critical for increasing recovery yields for VSVg and for ΔGP Jenv LV pseudotypes (Tables 2 to 3). The high recovery yields we observed after pressure conditions were adjusted were consistent with previous reports using the 100K and 300K membranes, 37,38,40 however we were unable to replicate the 100% recovery rates as previously observed. 37 Since no LV was observed in the waste fraction for these two membranes (data not shown), suggesting that the pore size of the membrane was appropriate, it is possible that without increased pressure, some LV was retained on the membrane; further pressure modulation might be needed to reach 100% yield. Although the 50K membrane yielded the highest recovery of VSVg LV, it should be noted that the smaller pore size of the 50K membrane resulted in the purification process taking several hours longer than when using the 100K and 300K membranes. Therefore, 100K and 300K membranes might be more favorable if conducting large-scale purification, as they do not severely compromise recovery. Despite a high recovery yield from TFF, it was necessary to include additional purification steps as the end product was found to be somewhat toxic to cells in vitro. Given that TFF can be used to concentrate LVs down to relatively small volumes (<10 mL), gradient ultracentrifugation followed by PEG-20,000 concentration and dialysis would be an appropriate downstream purification method. Indeed, subjecting the TFF eluant to gradient ultracentrifugation, PEG-20,000 concentration, and dialysis yielded increased titers with a high rate of recovery and abolished the toxicity observed with TFF elution fractions. Now that we have identified an optimal concentration method, the next steps are to scale up production for large animal studies and evaluate methods for removal of contaminants such as host cell protein and host cell DNA.
Although VSVg is the standard envelope used for pseudotyping LVs, it is not ideally suited for lung gene therapy. In vivo studies in mice have shown that the VSVg is unable to efficiently transduce the lung and relies on chelating and surfactant agents for its entry into respiratory epithelial cells. 9,12,13,61 Indeed, we also observed that VSVg LV transduced murine lung slices poorly, despite transducing ovine lung tissue slices similarly to ΔGP Jenv and the ΔGP Jenv mutant LVs. Conversely, the EBOV GP-pseudotyped LV has shown promising results in mice. 17,18 We also observed similar results in murine lung slices for EBOV GP LV but were unable to transduce ovine lung tissue slices with this vector. Others have also shown that EBOV GP and GP64 glycoprotein LV pseudotypes were unable to efficiently transduce the ovine airway. 21 Future studies involving in vivo delivery of pseudotyped LVs followed by histologic and single-cell RNAseq analysis will be important to determine the exact cell type transduced by the various pseudotyped LVs.
As a first step toward developing a small animal model for testing ΔGP Jenv LVs, we used the lung tropic AAV6.2FF vector 24 to deliver Hyal2, the receptor for JSRV, to mouse lungs, resulting in high-level expression in the lung parenchyma. This novel murine model also serves as an example of how AAV vectors can be used, in a rather straightforward and rapid manner, to express pathogen-specific receptors in a small animal model, thereby enabling in vivo studies. For example, the murine receptor for the Severe Acute Respiratory Syndrome (SARS) corona virus (CoV) S1 protein differs substantially from the human receptor and thus does not allow SARS-CoV entry. 62,63 To develop an animal model that would allow scientists to study the pathogenesis of SARS, as well as to test various vaccines and therapeutics, transgenic mice encoding the human receptor for SARS-CoV were created. 63 Delivering an AAV vector expressing the human receptor for SARS-CoV offers a rapid alternative to transgenic mice, and this may be particularly relevant in the case of newly emerging infectious diseases.
Using this murine AAV6.2FF-FT-hHyal2 model allowed us to compare VSVg, EBOV, and the two novel ΔGP Jenv mutant LV pseudotypes. Both ΔGP Jenv N592T and ΔGP Jenv M593E LVs were capable of transducing the FT-hHyal2-bearing murine lung tissue slices, which were otherwise uninfectable. In addition, production of the mutant ΔGP Jenv LV pseudotypes yielded titers that were much higher than those observed for EBOV GP LV in this or previous studies, 17,18,21,64 and this generated titers similar to those achieved with VSVg LV. 17,20,21,37,65
It should be noted that lung slices may make certain cell types accessible to vectors that would otherwise not be accessible in an intact animal. A more detailed histologic and molecular analysis of gene transfer would confirm which cell types in the lung are being transduced. Indeed, we intend to perform these studies. Despite this caveat, lung slices provide insight into whether the appropriate receptors are expressed within a target tissue and whether the machinery needed to facilitate vector transduction and transgene expression are present, particularly in species where live animal experimentation is either not possible or too expensive. As more gene therapy vectors are tested in lung slices and then eventually in larger animal species, we will be able to answer the question of how reflective these lung slice models are of what occurs in vivo.
In summary, our study presents a scalable concentration method for two novel lung tropic LV pseudotypes that can yield high titers and emphasizes their suitability as lung gene therapy vectors.
Footnotes
Acknowledgments
The plasmid psPAX2 was a gift from Didier Trono (Addgene plasmid no. 12260). The pL-Hyal2-SN plasmid was a gift from Dusty A. Miller. The construct pCMV3JS21 was kindly provided by Massimo Palmarini. The authors would like to acknowledge Brian McDougal and Pam Hasson for their efforts in donating sheep lungs for this work, Betty-Ann McBey of the Wootton lab and Bruce Cornfield of the Animal Health Laboratory for technical services, and Jodi Morrison for histology help. MCRG is the recipient of an NSERC PGS-D and an Ontario Veterinary College PhD Scholarship. JD is the recipient of the Andrea Leger Dunbar Summer Research Assistantship.
Authors' Contributions
M.C.R.G. and S.K.W. conceptualized the experiments and P.J.K., B.W.B. and T.J.M. contributed to experimental design and editing of the article. M.C.R.G. performed all experiments, with the exception of two flow cytometry experiments completed by J.P.V. and J.D. J.M.D. and L.P.V. produced AAV6.2FF-FT-Hyal2. J.C.I. harvested the lungs from the SPF lamb. L.P.V., D.L.Y., and J.d.J. generated DNA constructs. M.C.R.G. wrote the original draft and all authors reviewed and edited the article.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by NSERC Discovery (grant no. 355661), Lung Association – Ontario (grant no. 41217) and Mason Research Fund (grant no. 053671) grants awarded to SKW and an NSERC Discovery (grant no. 436264) awarded to BWB.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.