
Abstract
Conditional immortalization of hematopoietic progenitors through lentiviral expression of selected transcription factors in hematopoietic stem and progenitor cells provides a promising tool to study stem cell and leukemia biology. In this study, to generate conditionally immortalized lymphoid progenitor (ciLP) cell lines, murine hematopoietic progenitor cells were transduced with an inducible lentiviral “all-in-one” vector expressing LMO2 under doxycycline (DOX) stimulation and the reverse tetracycline-regulated transactivator (rtTA3). For selection of LMO2-expressing ciLPs (LMO2-ciLPs) and longitudinal manipulation in T cell differentiation lymphoid conditions, we developed a robust approach based on coculture with OP9-DL1 stromal cells and improved cytokine conditions allowing a controlled balance between cell proliferation and differentiation in vitro. LMO2-ciLP cell lines with the highest proliferation, vector copy number, and similar insertion pattern were selected for LMO2 “on/off” in vitro study. LMO2 expression under DOX induction resulted in a double negative (DN) 2 differentiation arrest and a propagation of CD44+CD25− myeloid cell population characterized by lymphoid and myeloid phenotypes, respectively. Both DN2 and CD44+CD25− myeloid cell subpopulations expressed c-KIT, suggesting that LMO2-ciLPs were similar to uncommitted progenitors under DOX supplementation. DOX removal resulted in cessation of ectopic LMO2 expression and LMO2-ciLPs continued T cell lymphoid differentiation accompanied by c-KIT downregulation and interleukin 7 receptor expression. Switching off LMO2 expression was accompanied by increased Notch signaling and significant reduction of the CD44+CD25− myeloid cell population under T cell differentiation lymphoid conditions. Although vector insertions in cooperation with LMO2 expression could influence the fate of LMO2-ciLPs and additional experiments are required to evaluate it, our approach provides a promising tool to investigate mechanisms underlying stem cell, leukemia, and lymphocyte biology, leading to novel approaches for disease modeling and therapy evaluation.
Introduction
Generation of hematopoietic cell lines with lymphoid and/or myeloid lineage differentiation potential in vivo is an ambitious goal in regenerative medicine. 1,2 Different approaches to generate and to maintain hematopoietic progenitor cell (HPC) lines in vitro have been tested. One of the most successful strategies not involving genetic manipulation was the establishment of cultured common lymphoid progenitor (CLP) cell lines with a lymphoid restricted uncommitted phenotype and long-term proliferating characteristics in vitro. 2 These cultured CLPs retained the potential to differentiate in vitro into T and B lymphoid cells and to myeloid, but not erythroid and megakaryocytic cells. After transplantation into sublethally irradiated recipient mice, these cultured CLPs transiently repopulated the host and differentiated to all lymphoid subsets and to myeloid cells. 2
Other approaches to generate hematopoietic cell lines are based on transgenic overexpression of transcription factors (TFs) in hematopoietic stem cells (HSCs) or HPCs. Since TFs play a vital role in the regulation of hematopoietic cell fate decision, HSC maintenance, and self-renewal, their overexpression (e.g., Hoxb8, ID3) leads to a cell differentiation arrest at the early progenitor stages, resulting in sustained proliferation in appropriate culture conditions. 3,4 Another example is the combinatory overexpression of eight selected TFs (Run1t1, Hlf, Lmo2, Prdm5, Pbx1, Mycn, Meis1, and Zfp37) to reprogram murine committed HPCs into so-called induced HSCs that demonstrate multilineage differentiation and self-renewal potential in vitro and in vivo. 5
Of note, all TFs that are master regulators of hematopoiesis and influence HSC differentiation and self-renewal in health are commonly overexpressed in cancer and are associated with hematologic malignancies. 6 Therefore, genetic modification of human hematopoietic stem and progenitor cells is very challenging and requires appropriate preclinical models. 7
LIM-domain-only 2 (LMO2) is an example of such a master regulator. LMO2 is a part of a multibinding protein complex and is expressed in hematopoietic cells during normal primitive and definitive hematopoiesis. 8 Lmo2 has important physiological roles and, in the case of its absence, failure of primitive erythropoiesis or early stage of definitive hematopoiesis occurs. 9,10
During normal T cell development, CLPs migrate from the bone marrow to enter the thymus and differentiate from the double negative (DN) stages expressing neither CD4 nor CD8 to the single/double positive stages (SP4: CD4+, SP8: CD8+, DP: CD4+CD8+). Lmo2 expression is required at the DN1 and DN2a stages, when uncommitted lymphoid progenitors retain myeloid, dendritic cell, and natural killer cell potential, whereas at the DN2b stage, the cells are already committed to the T cell fate. 11,12 Starting from the DN3 stage, the Lmo2 gene is downregulated and T cells mature toward the final stages. 12
However, LMO2 overexpression can result in block of thymocyte maturation at the DN2 stage followed by malignancy development 13 and was described for chromosomal translocations frequently found in T cell acute lymphoblastic leukemias (T-ALLs). 14,15 Ectopic LMO2 overexpression can cause severe abnormalities in T cell development. 16 Moreover, enforced Lmo2 expression in the thymus might reactivate an HSC-specific transcriptional program and promote the self-renewal of preleukemic thymocytes followed by leukemia development in mice. 17
Consistently, using a transgenic mouse model, it was shown that Lmo2 overexpression led to an accumulation of immature DN2/DN3 thymocytes within the thymus. 18 A differentiation block at the DN and immature SP stages was observed in a humanized mouse model transplanted with LMO2-expressing hCD34+ cells. 19 However, the median age at which LMO2-expressing mice developed T cell leukemias was 51 weeks. 20 Thus, LMO2 expression alone is not sufficient to cause clonal T-ALL and secondary mutations are required to develop hematologic malignancy. 8 Nevertheless, Lmo2 is able to activate stem cell-like transcriptional programs and mechanisms by which committed T cells can accumulate additional genetic mutations required for leukemic transformation. 17,21
Moreover, upregulation of the LMO2 gene, caused by retroviral insertion, was observed in T cell leukemia cases that occurred within the X-linked severe combined immunodeficiency (X-SCID) and Wiskott–Aldrich syndrome trials. 22 –25 Thus, LMO2 became one of the central genes in the development of surrogate assays addressing the biosafety of gene therapy vectors transducing early murine thymic precursors. 26 It was shown that although Lmo2 vector insertion sites (VISs) occurred in many DN2-blocked samples, gammaretroviral vectors containing full viral long-terminal repeats were more prone to cause DN2 arrest and Lmo2 pathway activation in comparison with lentiviral vectors (LVs) containing the same elements. 26
In this study, we generated conditionally immortalized lymphoid progenitor (ciLP) cell lines that express LMO2 in the presence of doxycycline (DOX). To establish these cell lines, we genetically modified murine HPCs using improved T cell differentiation lymphoid conditions and an inducible lentiviral “all-in-one” vector expressing both LMO2 and the reverse tetracycline-regulated transactivator (rtTA3). 27
The advantages of the drug-controlled system allow us to investigate not only a cell fate under “on/off” DOX conditions (presence/absence of LMO2 expression), but also when LMO2 is expressed at different levels. Of note, it was demonstrated before that level of oncogene expression can define the cell fate. 28 LMO2-expressing ciLP (LMO2-ciLP) cell lines with the highest proliferation, vector copy number (VCN), and similar insertion pattern (Hnrnpul1, Parp8, and Nrip1 gene loci) were selected for LMO2 “on/off” in vitro fate tracking. We observed an LMO2 dose-dependent differentiation arrest at the DN2 stage and a propagation of a CD44+CD25− myeloid cell population accompanied by the features of uncommitted progenitors in LMO2-ciLPs under DOX supplementation.
After the removal of DOX, differentiation toward the DP/SP T cell stages and interleukin 7 receptor (IL7R) expression were detected. Switching off LMO2 expression was accompanied by increased Notch signaling and significant reduction of the CD44+CD25− myeloid cell population under T cell differentiation lymphoid conditions. Our study suggests that investigated LMO2-ciLPs may have originated from a common source and thus insertions in cooperation with LMO2 expression might influence the immunophenotype and fate of LMO2-ciLP cell lines. Although additional experiments are required to evaluate it, modeling of conditionally immortalized hematopoietic progenitors creates an important platform to investigate mechanisms underlying stem cell, leukemia, and lymphocyte biology.
Materials and Methods
Vector construction
To enable constitutive expression, the coding cDNA of human LMO2 (NM_001142315) was cloned into a third-generation lentiviral self-inactivating (SIN) vector containing an internal spleen focus-forming virus promoter (SF), the post-transcriptional regulatory element (PRE), and the enhanced green fluorescent protein (EGFP). 29,30
To enable conditional expression, the coding cDNA of human LMO2 was cloned into a lentiviral SIN vector 29 under the control of the tetracycline-regulated T11 promoter. 31 As was described before, cDNA of the rtTA3 was codon optimized (rtTA3co) (Genscript Corporation, Piscatway, NJ) to remove cryptic splice sites and instability motifs and cloned into the same “all-in-one” vector under the control of the human phosphoglycerate kinase (PGK) promoter (the resulting protein coding sequence, GenBank: CVH74067.1). 27 To track human LMO2 expression, EGFP was linked through an internal ribosomal entry sequence derived from encephalomyocarditis virus. Cloning details are available on request.
Cell lines and vector production
Human embryonic kidney 293T and murine fibroblasts SC1 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM high glucose; Biochrom, Berlin, Germany) as previously described. 32 Vector production and titration were performed as described before. 32 –34 In short, 293T cells were transfected with 10 μg of LV, 10 μg of pcDNA3.GP.4xCTE (gag/pol), 5 μg of pRSV-Rev, and 2 μg of pMD.G (VSVg) using the calcium phosphate method. Virus supernatant was concentrated by ultracentrifugation and titrated using the SC1 cell line. 33,34
For SC1 transduction, cells were seeded the day before onto 24-well plates in the presence of 1.0 μg/mL DOX hyclate (Sigma-Aldrich, St. Louis, MO). The following day, DMEM was replaced with fresh medium supplemented with 4 μg/mL of protamine sulfate (Sigma, Seelze, Germany) and 1.0 μg/mL of DOX before addition of viral supernatants. Spinoculation was performed at 2,000 rpm (700 g) (Multifuge 3S-R; Heraeus, Berlin, Germany) at 32°C for 1 h.
Mice housing and transplantation conditions
C57BL/6J (CD45.2) and B6.SJL-Ptprca-Pep3b /BoyJ (CD45.1) mice were obtained from the central Hannover Medical School (MHH) animal facility and housed in microisolators under pathogen-free conditions (Hannover, Germany). Animal experiments were approved by regulatory authorities and performed according to national and international guidelines. Female CD45.2 mice were used for bone marrow isolation followed by lineage depletion and generation of LMO2-ciLP cell lines. Male CD45.1 mice were used as a source of T cell depleted bone marrow cells (TCDBMs) 35 cotransplanted for radioprotection with LMO2-ciLPs. Female CD45.1 recipient mice (8–12 weeks old) were conditioned by myeloablative irradiation (10.5 Gy) and transplanted with 3 × 106 CD45.1+ TCDBMs and 8 × 106 CD45.2+ LMO2-ciLPs. 36
Recipient mice received 5 mL/L Cotrim K (Ratiopharm, Ulm, Germany) in the drinking water for the first 3 weeks after transplantation for antimicrobial protection. Recipient mice also received standard dry food supplemented with 625 mg/kg DOX hyclate (Ssniff, Soest, Germany) 1 week before and 1 week after transplantation. 37 Peripheral blood cells were acquired by retro-orbital bleeding and measured using an automatic analyzer (ABC Counter; Scil, Vieheim, Germany). Mice were euthanized 2–4 weeks after transplantation and macroscopically examined for pathological abnormalities. Cells from the bone marrow, spleen, and thymus were subjected to cytospin analysis as described before. 33,34 Cell morphology was assessed by light microscope examination (Axiostart plus; Zeiss, Germany) with 60 × objectives.
OP9-DL1 stromal cells
OP9 stromal cells expressing the Notch ligand Delta-like 1 (OP9-DL1) 38,39 were cultured either in ready-to-use alpha minimum essential medium (α-MEM GlutaMAX™; Gibco, Paisley, Scotland, United Kingdom) supplemented with 20% fetal bovine serum Brazil One (FBS; Pan Biotech, Aidenbach, Germany) and 1% penicillin/streptomycin (Pen/Strep; Pan Biotech) or in freshly reconstituted from a powder α-MEM (Gibco) supplemented with 20% FBS, 1% Pen/Strep, and 2.2 g/L sodium bicarbonate (Pan Biotech). 40
OP9-DL1 cells were regularly passaged every 2–3 days and seeded at 1 × 104–1.5 × 104 cells/cm2 in plates 1 day before the coculture assay with hematopoietic cells. Coculture medium is an important component of the assay and freshly reconstituted α-MEM was described to significantly improve T cell differentiation, although not necessarily dramatically changing it. 39,40
Lineage depletion of bone marrow cells, transduction and cultivation of lineage negative cells, and LMO2-ciLPs using OP9-DL1 coculture assay
Lineage negative (Lin−) cells were magnetically isolated from the bone marrow using a Lineage Cell Depletion Kit (Miltenyi Biotech, Bergisch Gladbach, Germany). Lin− cells were prestimulated in serum-free StemSpan medium (Stem Cell Technologies, Vancouver, Canada) supplemented with 1% penicillin/streptomycin, 2 mM
Lin− cells were transduced either with constitutive or tetracycline-regulated LVs expressing LMO2 and/or EGFP (with multiplicities of infection of 33 and 5, respectively) using Retronectin (TaKaRa, Otsu, Japan) coated plates as described before. 34 One day after, transduced Lin− cells were transferred to OP9-DL1 feeder layer in complete α-MEM with cytokines as already described. Hematopoietic cells were transferred to new plates precoated with OP9-DL1 cells every 3–5 days. 39 Casy Cell Counter (Roche Diagnostics, Mannheim, Germany) was used to define cell concentration and viability. The experiments were performed in three biological replicates.
Cultivation of LMO2-ciLPs using myeloid conditions
Cells were cultured in Iscove's modified Dulbecco's medium (Biochrome) supplemented with 15% FBS, 1% penicillin/streptomycin, 2 mM
Flow cytometry
Cells were stained with the following antimouse antibodies: CD44 PE and PE-Cy7 (IM7; Biolegend, San Diego, CA), CD25 APC (PC61; Biolegend) and APC-Cy7 (3C7; Biolegend), CD11b PE (M1/70; eBioscience, San Diego, CA), CD11b APC-Cy7 (M1/70; Biolegend), GR1 APC (RB6-8C5; eBioscience), GR1 Alexa Fluor 700 and PE-Cy7 (RB6-8C5; Biolegend), CD4 Brilliant Violet 605 (RM4-5; Biolegend), CD8a PerCP-Cy5.5 (53–6.7; BD Pharmingen, San Diego, CA), IL7Rα PE (A7R34; eBioscience), c-KIT APC-Cy7 (2B8; Biolegend), c-KIT PE-Cy7 (2B8; Thermo Fisher Scientific, Waltham, MA), CCR9 PE-Cy7 (CW-1.2; Biolegend), CXCR4 APC (L276F12; Biolegend), CD45.2 APC, PE, and Alexa Fluor 700 (104; Biolegend).
Peripheral blood leukocytes were isolated through the BD PharM Lyse™ reagent (BD Biosciences, San Jose, CA) according to the manufacturer's instructions. Bone marrow, spleen, and thymus cell suspensions were prepared for flow cytometry analysis as described before. 37 Sample acquisition was performed using CytoFLEX S (Beckman Coulter Life Sciences, Brea, CA) or FACSCalibur (Becton Dickinson, Heidelberg, Germany) flow cytometers. DAPI (4′,6-diamidine-2′-phenylindole dihydrochloride; Sigma-Aldrich) or PI (propidium iodide; Sigma) were used to label dead cells (for gating strategy see Supplementary Fig. S1). The data were analyzed with FlowJo software (Tree Star, Ashland, OR).
VCN determination
VCN was determined through quantitative PCR (qPCR) as described before. 33 In brief, a plasmid standard containing the sequences of woodchuck hepatitis virus post-transcriptional regulatory element (wPRE) and fetal liver kinase 1 (Flk1) gene as internal reference control was used for quantification with the following primers sets: wPRE_forward, 5′-gaggagttgtggcccgttgt-3′; wPRE_reverse, 5′-tgacaggtggtggcaatgcc-3′; Flk1_forward, 5′-ggtttcaatgtcccgtatcctt-3′; Flk1_reverse, 5′-ctttgccccagtcccagtta-3′. qPCR efficiencies were determined for both sets of primers. 41 qPCR analysis was performed on an Applied Biosystems StepOnePlus System (Foster City, CA) in triplicates using the Quantitect SYBR Green Kit (Qiagen, Hilden, Germany).
Real-time qPCR
Real-time qPCR (RT-qPCR) was performed as described before.
33
In brief, total RNA was extracted using the RNeasy Mini kit (Qiagen) and reverse transcribed into cDNA with Quantiscript Reverse Transcriptase (QuantiTect Reverse Transcription Kit; Qiagen) using random hexamer primers. Real-time qPCR analysis was performed on an Applied Biosystems StepOnePlus System in triplicates using the Quantitect SYBR Green Kit (Qiagen). Real-time qPCR was performed with cDNA in 15 μL using 96-well plates. Relative quantification of a target gene transcript in comparison with a reference β-actin transcript
33
was performed using the method described by Pfaffl.
41
The program
Sequences of primers used: LMO2_forward: 5′-atcgaaaggaagagcctgg-3′; LMO2_reverse: 5′-caatgttctgctggcagc-3′; Gata2_forward: 5′-gcttcacccctaagcagaga-3′; Gata2_reverse: 5′-tggcaccacagttgacaca-3′; Notch1_forward: 5′ ggatgctgactgcatggat-3′; Notch1_reverse: 5′-aatcatgaggggtgtgaagc-3′; Hes1_forward: 5′ acaccggacaaaccaaagac-3′; Hes1_reverse: 5′-cgcctcttctccatgatagg-3′; Jag1_forward: 5′ tggccgaggtcctacactt-3′; Jag1_reverse: 5′-gccttttcaattatgctatcagg-3′; Actb_forward: 5′-cctccctggagaagagcta-3′; Actb_reverse: 5′-tccatacccaagaaggaagg-3′.
Identification of VISs
Analysis of VISs was performed by ligation-mediated PCR (LM-PCR) as reported before.
37
Genomic DNA was digested with MluCI restriction enzyme (New England BioLabs, Frankfurt/Main, Germany). PCR products were extracted after gel electrophoresis using QIA quick Gel Extraction Kit (Qiagen), sequenced directly and analyzed using BLAST/BLAT searches at
Statistical analysis
Results are presented as the mean of individual parameters obtained from different biological replicates (mock, SF.LMO2, SF.EGFP, T11.LMO2, T11.EGFP: n = 3) or LMO2-ciLP cell lines (1B8, 3C1, 10C1, 10H4: n = 4). Statistical analyses and graph plotting were performed using Matplotlib 3.1.0
Results
Improved conditions for T-lymphoid differentiation of murine hematopoietic progenitors in vitro
T cell lymphoid differentiation of hematopoietic progenitors in vitro requires balanced composition of cytokines such as mSCF, hFLT3L, and hIL7 and an activated Notch signaling pathway provided by coculture with OP9-DL1 feeder cells. 39,44 In this study, we took advantage of published protocols and investigated T cell lymphoid differentiation using three different combinations of cytokine concentrations.
Nontransduced Lin− cells were cocultured for 9 days with OP9-DL1 feeder cells in freshly reconstituted α-MEM supplemented with 20 ng/mL of mSCF, 5 ng/mL of hFLT3L, and 1 ng/mL of hIL7 (Fig. 1a). To determine DN stages of T cell maturation on day 9, we investigated the expression of CD44 and CD25 markers (DN1: CD44+CD25−, DN2: CD44+CD25+, DN3: CD44−CD25+, DN4: CD44−CD25−) (Fig. 1b). 38,39 We demonstrated that percentage of cells in DN stages for this time point (day 9) corresponds to previously described in vitro assays: DN1, 13.0% ± 0.6%; DN2, 33.5% ± 1.9%; DN3, 48.8% ± 4.9%; and DN4, 1.4% ± 0.3% (mean ± SD, n = 3) (Fig. 1c). At the same time, as expected, no CD4+ and/or CD8+ T cells were observed (Fig. 1b).
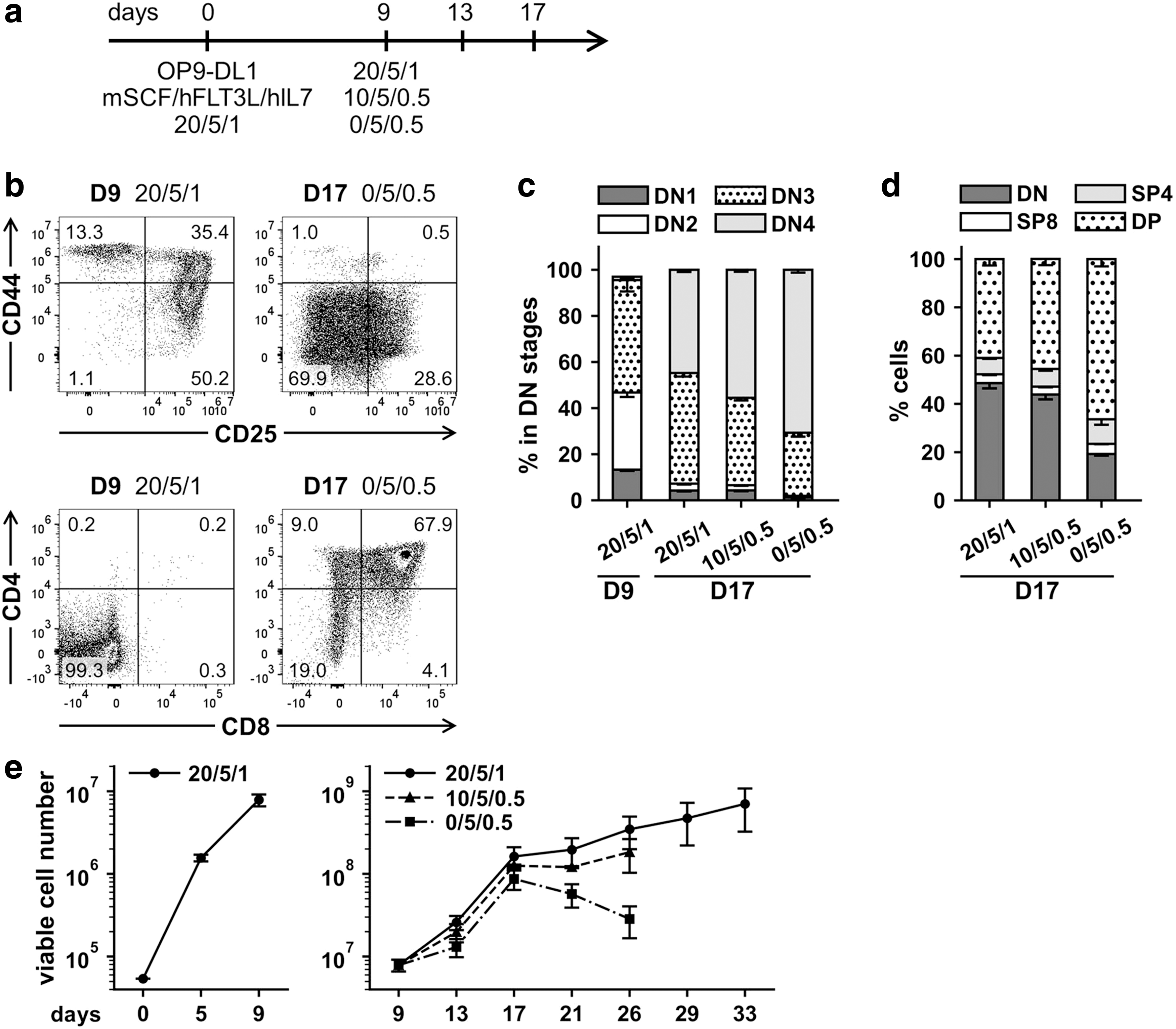
Choice of improved cytokine conditions for T-lymphoid differentiation of murine hematopoietic progenitors in vitro.
To promote T cell differentiation into DP and SP stages, which are characterized by the expression of CD4 and CD8 markers (DP: CD4+CD8+, SP: CD4+ or CD8+), 38,39 the concentrations of mSCF and hIL7 were decreased to 10 or 0 ng/mL and 0.5 ng/mL, respectively, on day 9 (Fig. 1a). The hFLT3L concentration was kept constant at 5 ng/mL. On day 17, CD4+ and/or CD8+ T cells were observed for all cytokine conditions and the maximum percentage of DP differentiated cells (66.3% ± 3.0%, mean ± SD, n = 3) was achieved with cytokine concentrations of 0 ng/mL mSCF, 5 ng/mL hFLT3L, and 0.5 ng/mL hIL7 (Fig. 1b, d).
Considering that T-lymphoid differentiation experiments in vitro require long-term cultivation, the balance between proliferation and differentiation is important. Although differentiating cells under all investigated cytokine concentrations demonstrated comparably high proliferative capacities up to day 17, cell proliferation decreased markedly at later time points in the absence of mSCF (Fig. 1e). At the same time, increased percentages of DP differentiated T cells were revealed for lowest hIL7 concentration (Fig. 1d).
In view of limited proliferating capacity for terminally differentiated T cells, but the requirement for sufficient number of cells, we hypothesized that hIL7 concentrations could be varied at low levels, but the mSCF and hFLT3L concentrations should be kept constant. Because the highest proliferative capacity was shown for 20 ng/mL of mSCF and 5 ng/mL of hFLT3L, these concentrations were kept constant for all further experiments to support sufficient continuous proliferation (Fig. 1e).
Interestingly, on day 9 we determined 10.0% ± 0.4% (mean ± SD, n = 3) of CD11b+ and/or GR1+ cells; however on day 17, the percentage of CD11b+ and/or GR1+ cells decreased to 0 − 3% for all tested cytokine conditions (Supplementary Fig. S2).
Constitutive LMO2 expression in murine HPCs under T-lymphoid conditions in vitro led to arrest at the DN1/DN2 stages
Murine and human LMO2 proteins demonstrate a high degree of homology (99.37%) (Supplementary Fig. S3a). To develop vectors with potential applicability in both murine and human cells, human LMO2 cDNA was chosen for ectopic expression (please, see Materials and Methods section). To study how LMO2 expression will influence T cell lymphoid differentiation of murine HPCs in vitro, Lin− cells were transduced with LVs SF.LMO2.EGFP or SF.EGFP constitutively expressing LMO2/EGFP or EGFP only and cocultured with OP9-DL1 feeder cells in ready-to-use α-MEM supplemented with 20 ng/mL of mSCF, 5 ng/mL of hFLT3L, and 5 ng/mL of hIL7 (Fig. 2a).
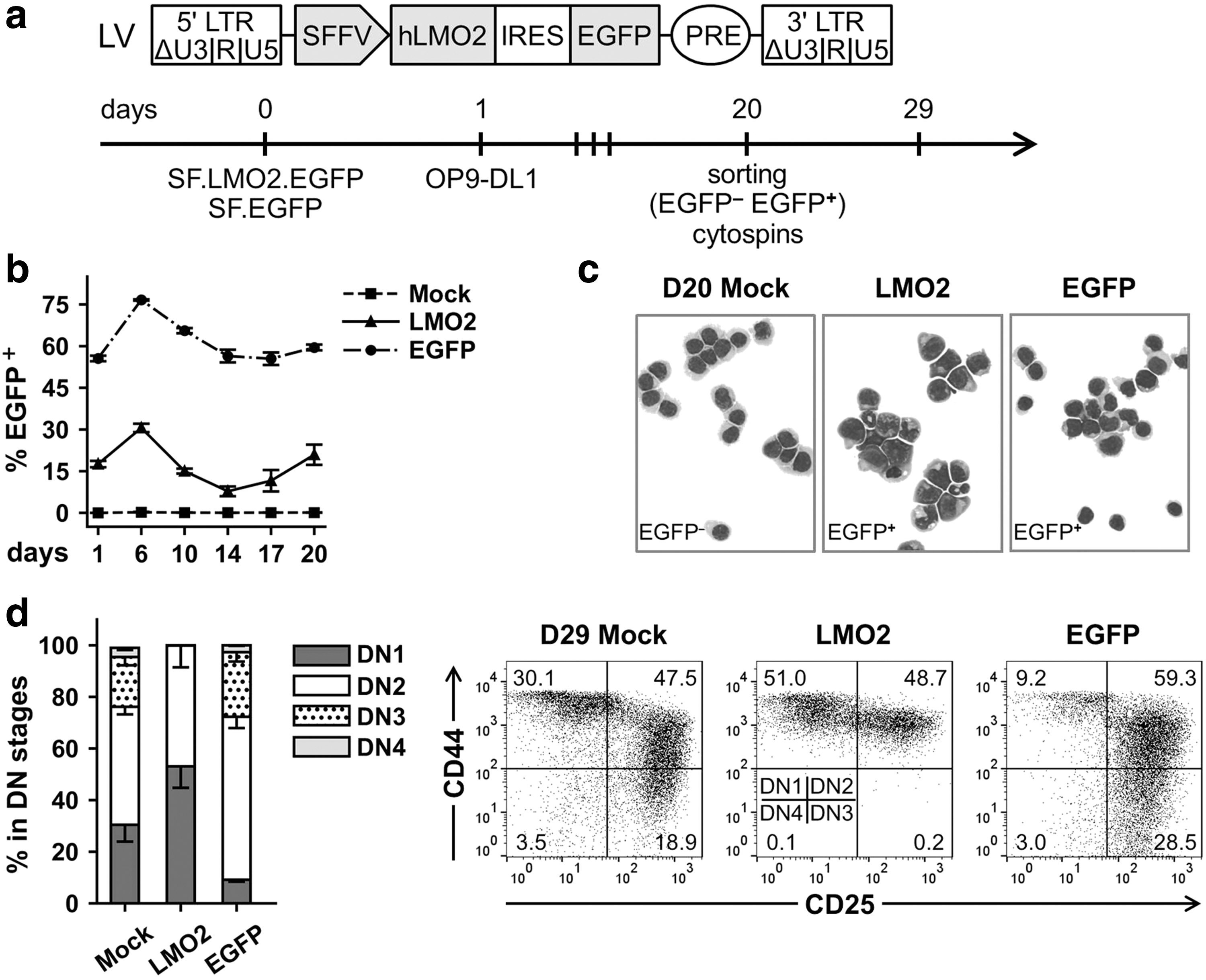
Constitutive overexpression of LMO2 in murine hematopoietic progenitors in T-lymphoid conditions in vitro.
Gene transfer level was tracked over time, and on day 20 post-transduction (PT), we sorted for EGFP+ and EGFP− cell populations (Fig. 2a, b). At the same time point, we investigated morphology of the samples by cytospin analysis and found that nontransduced control Lin− cells (mock) and Lin− cells transduced with SF.EGFP control vector (sorted EGFP+ cells) mostly demonstrated a lymphoid phenotype (Fig. 2c). On day 29 PT (day 9 after the sorting), mock and sorted EGFP+ cultures contained cells of all DN subpopulations (Fig. 2d), indicating efficient lymphoid differentiation.
Surprisingly, LMO2/EGFP+ sorted samples contained cells with both lymphoid and myeloid morphology with different degrees of maturation (Fig. 2c). The level of myeloid marker expression (CD11b+ and/or GR1+), for example, on day 29 PT in LMO2/EGFP+ cell populations, was elevated as compared with mock and EGFP samples (23.0% ± 4.7% vs. 10.8% ± 7.6% and 0.5% ± 0.3%, respectively; mean ± SD, n = 3) (Supplementary Fig. S3b). At the same time point, LMO2/EGFP+ samples demonstrated a block of differentiation at the DN1/DN2 stages, and a corresponding lack of DN3 and DN4 subpopulations (Fig. 2d).
Interestingly, the balance between DN1 and DN2 subpopulations of LMO2/EGFP+ samples shifted toward the DN1 stage over several passages from days 24 to 34 PT: percentage of the DN1 subpopulation increased from 25.0% ± 5.6% on day 24 PT to 71.4% ± 7.0% on day 34 PT (mean ± SD, n = 3) (Supplementary Fig. S3c). The level of myeloid marker expression in LMO2/EGFP+ samples remained relatively constant from day 29 PT to day 34 PT, with 27.7% ± 1.2% (mean ± SD, n = 3) of cells positive for CD11b and/or GR1 markers on day 34 PT (Supplementary Fig. S3d). In contrast, mock and control EGFP+ samples showed only 8.4% ± 4.8% and 2.7% ± 1.0% (mean ± SD, n = 3) of CD11b+ and/or GR1+ cells on day 34 PT, respectively, which is consistent with lymphoid differentiation in these samples (Supplementary Fig. S3d).
Thus, constitutive LMO2 expression in murine HPCs cultured under T cell lymphoid differentiation conditions resulted in a block of differentiation at the DN1/DN2 stages characterized by expression of both T cell lymphoid and myeloid markers.
Generation of ciLP cell lines expressing LMO2
To develop a robust approach to investigate the role of LMO2 in the differentiation arrest, human LMO2 cDNA was cloned into the lentiviral tetracycline(tet)-regulated “all-in-one” vector T11.EGFP.hPGK.rtTA3co (Fig. 3a). This previously described vector expresses EGFP under the control of the tetracycline-regulated T11 promoter 31,37 and codon-optimized rtTA3 under the control of the hPGK promoter. 27 The T11 promoter has an improved dynamic range of expression and when cloned into the “all-in-one” vector has a low basal activity in the absence of DOX, which does not depend significantly on transduction rate and VCN. 27,31,45
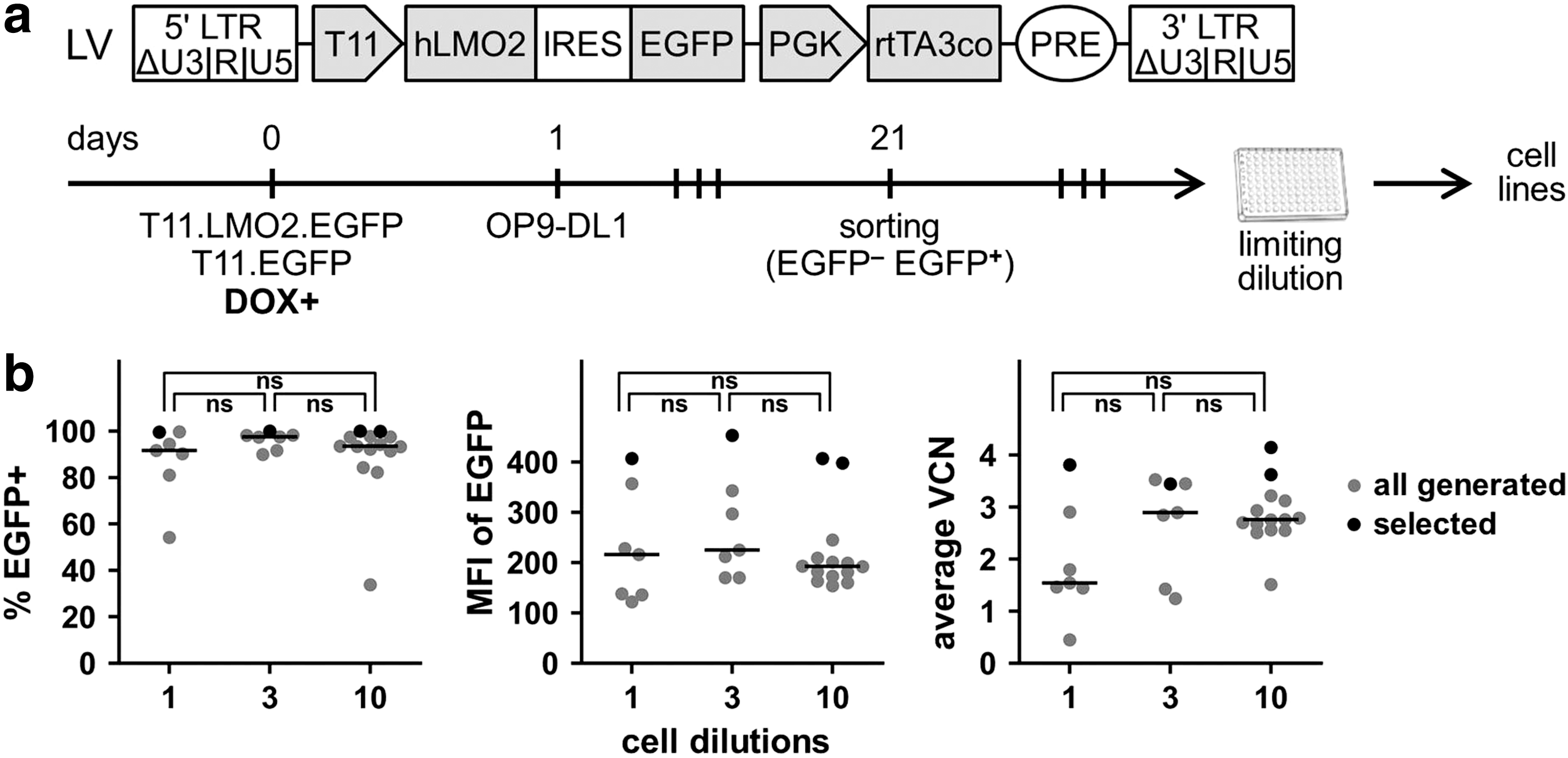
Generation of ciLPs expressing LMO2 in the presence of DOX.
Lin− cells were transduced with T11.LMO2.EGFP.hPGK.rtTA3co (T11.LMO2) or with control T11.EGFP.hPGK.rtTA3co (T11.EGFP) LVs in the presence of 1 μg/mL DOX and cocultured with OP9-DL1 feeder cells in freshly reconstituted α-MEM supplemented with 20 ng/mL of mSCF, 5 ng/mL of hFLT3L, and 1 ng/mL of hIL7 (Fig. 3a). Sorting for EGFP+ cell populations was done on day 21 PT. Gene transfer level after sorting was stable (75–100%) for Lin− cells transduced with the T11.LMO2 vector and the T11.EGFP control vector (Supplementary Fig. S4a).
On day 69 PT, three replicates expressing LMO2 were pooled together and a limiting dilution assay was performed (Fig. 3a and Supplementary Fig. S4b). Using different cell dilutions (1, 3, or 10 cells per well of a 96-well plate), 28 potential individually picked cell lines were obtained (Fig. 3b and Supplementary Fig. S4c). Interestingly, the number of resulting cell lines was the same for experiments in which 1 and 3 cells were seeded per well (n = 7), but 2 times higher when 10 cells were seeded per well (n = 14), revealing a difference in the frequency of immortalization only at the highest seeding density (Fig. 3b and Supplementary Fig. S4c). Obtained cell lines were subjected to a selection process based on the highest gene transfer (percentage of EGFP+ cells), expression levels (mean of fluorescence intensity [MFI] of EGFP), and VCNs (Fig. 3b).
Thus, four LMO2-ciLP cell lines (1B8, 3C1, 10C1, and 10H4) demonstrated the highest LMO2/EGFP expression under DOX supplementation (99.9% ± 0.2%, mean ± SD, n = 4) and similarly high average VCNs as determined by qPCR (3.8 ± 0.3, mean ± SD, n = 4) (Fig. 3b). Insertional analysis revealed that these four cell lines have identical VISs into Hnrnpul1, Parp8, and Nrip1 gene loci (Supplementary Fig. S4d and Supplementary Table 1), suggesting that they may have originated from a common source. Although the clonal composition of these cell lines (e.g., monoclonal or oligoclonal) remains to be fully investigated, all four cell lines (1B8, 3C1, 10C1, and 10H4, n = 4) were selected for further LMO2 “on/off” in vitro fate tracking.
Fate of ciLPs depended on LMO2 expression under T-lymphoid conditions in vitro
We characterized the ciLP cell lines generated previously (1B8, 3C1, 10C1, and 10H4, n = 4) in the presence and absence of DOX under T cell differentiation conditions in vitro. As already described, in the presence of 1.0 μg/mL DOX, LMO2-ciLPs were practically 100% LMO2/EGFP+ (Fig. 4a, b and Supplementary Fig. S5a). However, on day 4 after DOX removal, the number of LMO2/EGFP+ cells decreased to ∼20% and after day 7 after DOX removal, almost no LMO2/EGFP+ cells were detected (Fig. 4a, b and Supplementary Fig. S5a).
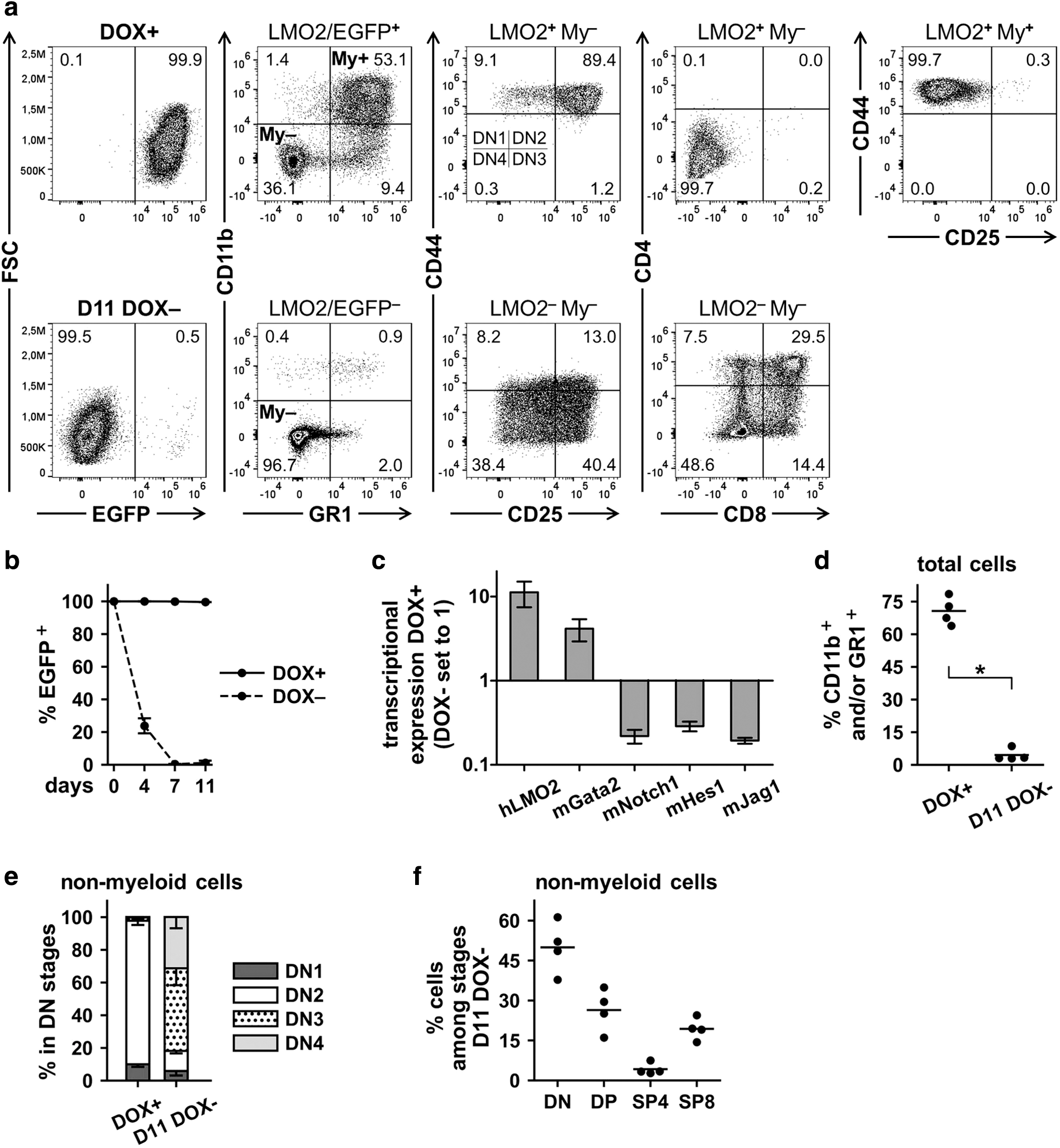
Immunophenotype and fate of LMO2-expressing ciLP (LMO2-ciLP) cell lines in the presence of DOX (1.0 μg/mL) and after its removal.
Relative transcriptional expression of LMO2 measured by RT-qPCR was 11.2 ± 3.8 (mean ± SD, n = 8) times higher in 1.0 μg/mL DOX-treated samples than in the samples cultured without DOX for 7 days (Fig. 4c). As the LMO2 protein can only bind DNA as part of a multiprotein complex together with other TFs, such as the GATA-binding factor 2, we measured Gata2 expression in LMO2-ciLPs and also found this to be upregulated in 1.0 μg/mL DOX+ conditions (Fig. 4c). 8 Interestingly, genes involved in Notch signaling 46 were expressed at lower levels in DOX-treated samples as compared with samples cultured without DOX for 7 days, suggesting increased Notch signaling after LMO2 switching off (Fig. 4c).
From data described previously for constitutive expression of LMO2 in murine HPCs, a differentiation block at the DN1/DN2 stages was induced by LMO2 expression (Fig. 2d and Supplementary Fig. S3). To investigate the detailed phenotype of the DN1 and DN2 subpopulations, eight-color staining was performed (Material and Methods section). Interestingly, LMO2/EGFP+ cells expressing myeloid (My) markers CD11b and GR1 (LMO2+My+) were CD44+CD25− (Fig. 4a, d and Supplementary Fig. S5b). At the same time, ciLPs expressing LMO2 but no myeloid markers (LMO2+My−) revealed the majority of the cells were in the DN2 stage and were negative for CD4 and CD8 markers (Fig. 4a).
Morphological analysis of four investigated ciLP cell lines (1B8, 3C1, 10C1, and 10H4) cultivated with DOX confirmed the presence of both lymphoid and myeloid cells in all samples (Supplementary Fig. S5c). Interestingly, both immature and more differentiated myeloid cells (e.g., neutrophils) were mostly detected in all LMO2-ciLPs, but not in the corresponding mock nontransduced controls (Supplementary Fig. S5c).
After DOX withdrawal (DOX−), LMO2 expression was switched off, which released the differentiation arrest (Fig. 4a–c, e). The cells differentiated from the DN1/DN2 stages toward the DN3/DN4 and further to the DP CD4+CD8+ and SP CD4+ (SP4) or SP CD8+ (SP8) stages (Fig. 4a, f). The differentiation process reached a peak on day 11 after DOX removal and we observed 50.1% ± 9.7% (mean ± SD, n = 4) of cells to be distributed among DP and SP stages (Fig. 4a, f). Importantly, the percentage of cells expressing myeloid markers decreased significantly (4.5% ± 2.8%, mean ± SD, n = 4) at the final stages of this tetracycline-regulated T-lymphoid differentiation (Fig. 4d).
Morphological analysis on day 11 after DOX removal revealed lymphoid cells but lack of neutrophils in LMO2-ciLP cell lines and presence of myeloid cells with increased size and morphological changes resembling differentiation (Supplementary Fig. S5c). Of note, neutrophils have a very short life span. 47,48 Although increased number of viable cells were observed over time for all four LMO2-ciLP cell lines when cultured with DOX, cells ceased to proliferate after DOX removal (Supplementary Fig. S5d). These findings are consistent with our observation that switching off LMO2 expression resulted in terminal T-lymphoid differentiation, accompanied by increased Notch signaling (Fig. 4c).
Interestingly, tracking the CD44+CD25− and CD11b+GR1+ cells in a second independent LMO2 “on/off” experiment (performed for LMO2-ciLPs 1B8, 3C1, 10C1, and 10H4, n = 4) demonstrated that these two populations had a similar fate and were greatly decreased on day 11 after DOX removal and switching off LMO2 expression (Supplementary Fig. S6). At the same time, the T-lymphoid differentiation process again reached the maximum at day 11 after DOX removal (Supplementary Fig. S6b–d).
To better understand the fate of the CD44+CD25−/CD11b+GR1+ cell population, LMO2-ciLPs (1B8, 3C1, 10C1, and 10H4, n = 4) were treated with different DOX concentrations: 0.5, 1.0, 2.0, and 3.0 μg/mL (Supplementary Fig. S7). Although the percentages of LMO2/EGFP+ cells were ∼100% for all DOX groups, significant differences were detected in expression levels (MFI of EGFP) and number of viable cells when LMO2-ciLPs treated with high DOX concentrations (1.0, 2.0, and 3.0 μg/mL) were compared with LMO2-ciLPs treated with 0.5 μg/mL of DOX (Supplementary Fig. S7a, b). Interestingly, decreased levels of LMO2/EGFP expression and proliferative capacities of LMO2-ciLPs treated with 0.5 μg/mL of DOX were associated with decreased percentages of DN2 cells (Supplementary Fig. S7c).
Since the experiment with the different DOX concentrations did not reveal significant differences in MFIs of LMO2/EGFP or the number of viable cells for high DOX concentrations (1.0, 2.0, and 3.0 μg/mL) (Supplementary Fig. S7a, b), to ensure sufficient proliferative capacities of LMO2-ciLPs, further experiments were performed in the presence of 2.0 μg/mL DOX.
We investigated the fate of LMO2-ciLP cell lines (1B8, 3C1, 10C1, and 10H4) under myeloid cytokine conditions (mSCF, hIL6, and mIL3) 34 and 2.0 μg/mL DOX treatment (Supplementary Fig. S8). From days 0 to 8, significant expansion of LMO2-ciLPs cultured in myeloid conditions was observed in comparison with parallel experiments with standard T-lymphoid conditions (Supplementary Fig. S8a). Morphological analysis for myeloid conditions on day 4 revealed immature myeloid cells. However, these cells differentiated predominantly into neutrophils on day 8 (Supplementary Fig. S8b) and demonstrated a myeloid phenotype (CD11b+GR1+, 97.7% ± 1.2%) and nearly no CD44+CD25+ (DN2) (0.7% ± 0.5%, mean ± SD, n = 4) cells (Supplementary Fig. S8c).
On day 12, cytospin analysis showed a lack of neutrophils, 47,48 but the presence of cells with increased size and morphological changes resembling myeloid differentiation (Supplementary Fig. S8b). Although the percentages of LMO2/EGFP+ cells were 100% throughout the experiment accomplished in both cytokine conditions, on day 12 we observed a significant decrease in the viability and number of cells of LMO2-ciLPs cultured in myeloid conditions, indicating a limited survival after myeloid differentiation (Supplementary Fig. S8d).
Thus, ciLPs expressing LMO2 at higher level in DOX+ (1.0–3.0 μg/mL) T-lymphoid conditions revealed the balance between DN2 differentiation arrest and a propagation of CD44+CD25− myeloid cell population characterized by expression of T-lymphoid and myeloid markers, respectively. DOX removal and cessation of ectopic LMO2 expression resulted in T-lymphoid differentiation and a significant decrease of CD44+CD25− myeloid cells. At the same time, under 0.5 μg/mL DOX T-lymphoid conditions, 100% of LMO2-ciLPs retained LMO2/EGFP expression at lower level, which resulted in a CD44+CD25− myeloid shift and decrease of the DN2 population on day 10 (4.7% ± 2.7%, mean ± SD, n = 4). These data indicate that the levels of LMO2 expression could define the fate of LMO2-ciLPs.
Differentiation status of LMO2-ciLPs in the presence and absence of LMO2 expression under T-lymphoid conditions in vitro
To estimate the differentiation status of LMO2-ciLPs, we first investigated the expression level of c-KIT in four LMO2-ciLP cell lines (1B8, 3C1, 10C1, and 10H4, n = 4). As already described, LMO2 upregulation in the DOX+ (1.0 μg/mL) conditions resulted in a differentiation block of LMO2-ciLPs at the DN2 stage and a propagation of CD44+CD25− myeloid cell population (Fig. 4a). We observed that both CD44+CD25− and CD44+CD25+ (DN2) fractions of LMO2-ciLPs expressed c-KIT although at different percentages and levels (Fig. 5a, b). Interestingly, the DN2 subpopulation was ∼100% positive for c-KIT, whereas the CD44+CD25− cells were only 15.9% ± 1.6% (mean ± SD, n = 4) c-KIT+ (Fig. 5b). In this study, we compared MFI of c-KIT and demonstrated that c-KIT expression is significantly higher in the DN2 subpopulation than in the CD44+CD25− fraction, corresponding to c-KIThigh and c-KITlow populations (Fig. 5b). Importantly, c-KIT expression decreased dramatically already 4 days after DOX removal, indicating initiation of the commitment process (Fig. 5a, b).
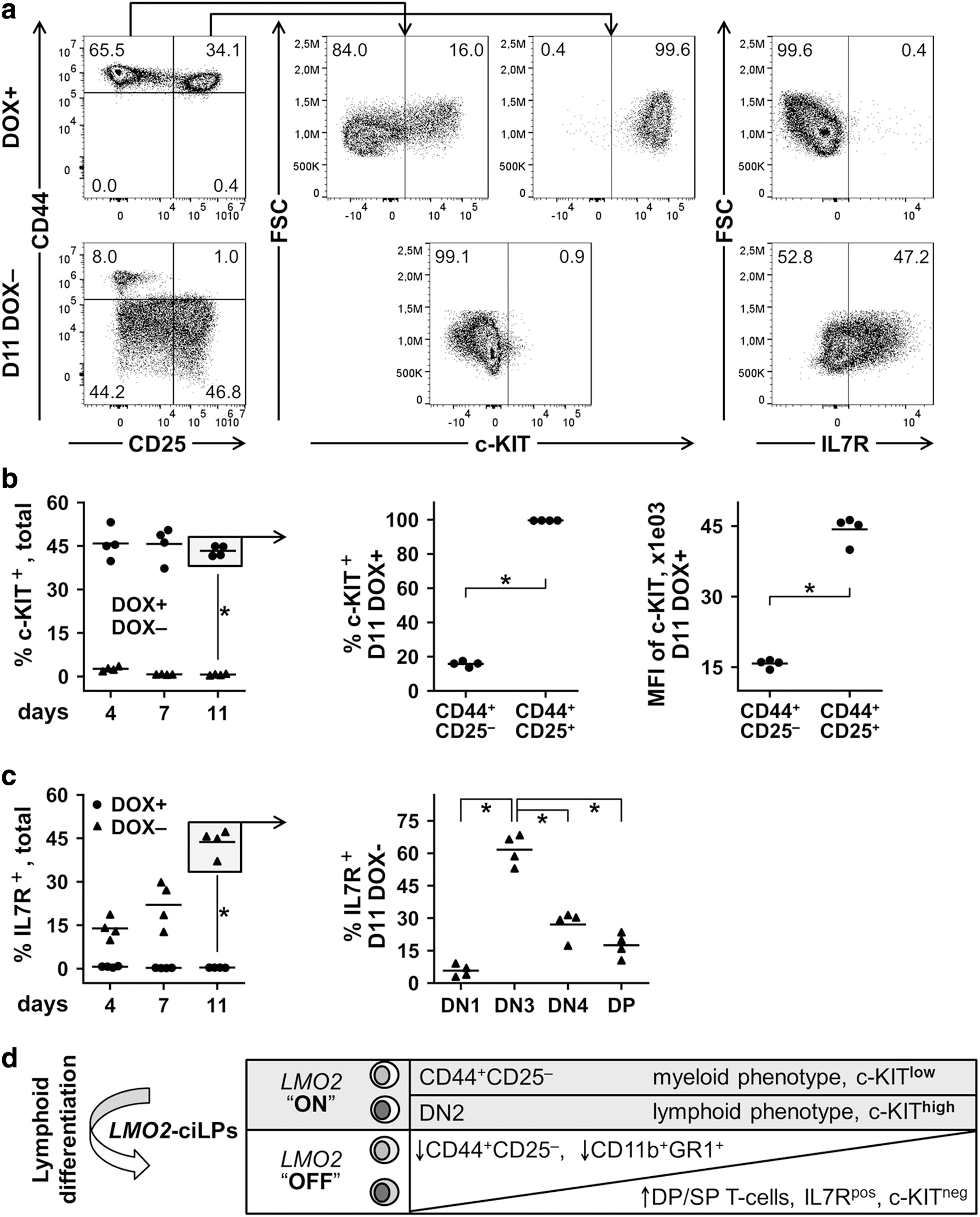
Differentiation status of LMO2-ciLP cell lines in the presence and absence of LMO2 expression in vitro.
Since IL7 is one of the key cytokines for in vitro T cell lymphoid differentiation of murine HPCs and the IL7 signaling pathway plays a vital role in the normal T cell differentiation in vivo by governing IL7R expression on thymocytes during the DN stages, we analyzed expression of the IL7R.
When LMO2 was upregulated (1.0 μg/mL DOX+ conditions), IL7R was not expressed (Fig. 5a, c) in the total population of LMO2-ciLPs. In contrast, after DOX withdrawal, when LMO2 expression was switched off over time from days 4 to 11, IL7R expression increased from 13.9% ± 3.7% to 43.7% ± 4.5% (mean ± SD, n = 4), respectively (Fig. 5a, c). On day 11, the DN3 subpopulation exhibited the highest level of IL7R expression, which was diminished in the following DN4/DP stages, similar to IL7R signaling during normal T cell development (Fig. 5c). 49
Finally, we investigated expression of CCR9 (C-C chemokine receptor type 9) and CXCR4 (C-X-C chemokine receptor type 4) that mediate migration of early hematopoietic progenitors to the thymus and within the thymus. 50,51 In the presence of DOX, LMO2-ciLPs partially expressed CCR9 and CXCR4, however, at low levels according to MFIs (Supplementary Fig. S9). Eleven days after DOX removal, when LMO2-ciLPs progressed to the DN3/DN4 stages, we observed a significant increase in the number of CCR9+ and CXCR4+ cells and, moreover, these CCR9+ and CXCR4+ cells demonstrated much higher MFIs (Supplementary Fig. S9).
To summarize, the LMO2-ciLPs established here expressed LMO2 in the presence of 1.0 μg/mL DOX (“ON”) resulting in a differentiation block at the DN2 stage and a propagation of CD44+CD25− myeloid cell population when cultivated in T-lymphoid conditions in vitro. The CD44+CD25− fraction was characterized by a myeloid phenotype and low expression of c-KIT, whereas the DN2 subpopulation had a T-lymphoid phenotype and enhanced c-KIT expression. DOX removal (“OFF”) resulted in switching off LMO2 expression followed by CD44+CD25− fraction decreasing and T cell differentiation characterized by DP/SP stages and enhanced IL7R expression (Fig. 5d).
To investigate if the established in vitro LMO2-ciLPs are transplantable in vivo, we performed murine transplantation using a congenic mouse model (Supplementary Fig. S10a). Following our hypothesis that better engraftment of LMO2-ciLPs requires DOX+ conditions, DOX supplementation in vivo was performed through dry food starting 7 days before transplantation. 37 Lethally irradiated CD45.1 recipients received LMO2-ciLPs (1B8 cell line, CD45.2+) and freshly isolated TCDBMs (CD45.1+). 36
On day 7 post-transplantation, the DOX food was exchanged with normal chow to cause switching off LMO2/EGFP expression. Red blood cell, platelet, and white blood cell levels were within normal ranges, on days 14, 21, and 28 post-transplantation (Supplementary Fig. S10b). However, we did not observe donor-derived cells in the peripheral blood and hematopoietic organs on days 14 and 28 post-transplantation (Supplementary Fig. S10c, d), indicating that another strategy and additional experiments are required.
Discussion
Simple and efficient systems that allow generation and controlled differentiation of transplantable hematopoietic progenitors will facilitate the study of hematopoietic cell development and create the new platforms for disease modeling. In this study, we present the drug-controlled model based on tetracycline-regulated expression of TF LMO2 in murine HPCs to establish LMO2-ciLP cell lines arrested at early differentiation stages. Drug removal resulted in switching off LMO2 expression, release of the differentiation block, and T cell lymphoid commitment of LMO2-ciLPs (Fig. 5d).
To generate this system, we adapted a T cell lymphoid differentiation in vitro assay. 39,40,44 As was well described previously by Zúñiga-Pflücker and colleagues, T cell differentiation of murine Lin− cells in vitro requires Notch signaling that could be provided by coculture with OP9-DL1 feeder cells and specific cytokines, such as IL7 and FLT3L. 39 Importantly, the differentiation assay terminates when the cells reach DP/SP T cell stages since proliferation capacity is lost at these stages. To find an equilibrium between progenitor proliferation and differentiation, we investigated several cytokine conditions to allow T-lymphoid differentiation of murine HPCs in coculture with OP9-DL1 cells. 39,40,44
We varied concentrations of mSCF and hIL7, but kept hFLT3L concentration constant, and demonstrated that the absence of mSCF and low concentration of hIL7 increased the yield of DP/SP T cells and thus created differentiation conditions appropriate for short-term experiments. However, mSCF addition enabled sufficient long-term HPC proliferation, which is in agreement with previously published data on cord blood-derived CD34+ progenitors during OP9-DL1 cocultivation. 40 Therefore, we chose the cytokine cocktail composed of 20 ng/mL of mSCF, 5 ng/mL of hFLT3L, and 1 ng/mL of hIL7 to keep proliferation and differentiation capacities of LMO2-ciLP cell lines at required levels.
Using improved T-lymphoid culture conditions, 28 LMO2-ciLP cell lines were established in the presence of 1 μg/mL DOX. Although LMO2/EGFP expression was between 80% and 100% for most LMO2-ciLPs, MFIs were more variable, suggesting a role of the LMO2 expression level for the selection process and discrimination between transient and long-term immortalization. Four LMO2-ciLP cell lines (1B8, 3C1, 10C1, and 10H4) demonstrated high proliferation capacity, revealed VCN 3.8 ± 0.3 (mean ± SD, n = 4), and thus were selected for further experiments. Importantly, when these cell lines were treated with different DOX concentrations, decreased levels of LMO2/EGFP expression were associated with decreased proliferative capacities of LMO2-ciLPs treated with 0.5 μg/mL of DOX.
LM-PCR analysis indicated identical insertions in all four investigated LMO2-ciLP cell lines, suggesting that these cell lines may have arisen from a common ancestor(s). Of note, one insertion in the Hnrnpul1 gene is located also in the vicinity of the receptor tyrosine kinase Axl and transforming growth factor Tgfb1. 52,53 Interestingly, the Tgfb1 gene was described to be involved in the regulation of lymphocyte proliferation, differentiation, and survival. 53,54 Nevertheless, both constitutive LMO2 expression in murine HPCs (mass culture) and DOX-dependent (1 μg/mL DOX) LMO2 expression in LMO2-ciLP cell lines caused a differentiation arrest at the DN2 stage and a propagation of CD44+CD25− population. Interestingly, this was observed for both ready-to-use and freshly reconstituted α-MEM used.
LMO2 is a well-known oncogene, the upregulation of which alters T cell development in mice and humans and leads to an accumulation of immature thymocytes. 17 –20 Thymocytes from CD2-Lmo2 transgenic mice demonstrated self-renewal and repopulating capacities in vivo. 17 It was shown that coculture of fetal liver LSK (Lineage−c-KIT+ SCA-1+) cells from CD2-Lmo2 transgenic mice with OP9-DL1 stromal cells resulted in an accumulation of progenitors at the DN2 stage. 18 In our system, LMO2-ciLPs demonstrated both myeloid and lymphoid (DN2-specific) phenotypes: the CD44+CD25− fraction expressed myeloid markers, whereas the DN2 subpopulation did not.
Interestingly, in a previously described early T cell precursor acute lymphoblastic leukemia (ETP-ALL) model induced by Il7r mutant, a similar phenomenon was observed. 55 Thymocytes transduced with vectors expressing the Il7r mutants exhibited a DN2 differentiation block and an accumulation of endogenous Lmo2 mRNA during 20 days of coculture with OP9-DL1. Consistent with our findings with LMO2-ciLPs, ETP-ALL leukemic cells exhibited both lymphoid and myeloid features. Moreover, pharmacological inhibition of the signaling from Il7r mutants resulted in decreased expression of Lmo2. 55 In our study, enforced LMO2 expression led to the loss of IL7R expression, indicating a possible link between related pathways.
Beyond its role in leukemogenesis, LMO2 plays an important role during physiologic hematopoiesis and is expressed by different hematopoietic lineages. 8 During T cell development, Lmo2 is more abundant at the DN1/DN2 stages, but Lmo2 levels decrease from the DN3 stage onward. 12 Under the 1 μg/mL DOX treatment, LMO2 overexpression not only blocked differentiation of LMO2-ciLPs at the DN2 stage but also resulted in a propagation of a myeloid cell population. Interestingly, DOX withdrawal caused switching off LMO2 expression, differentiation arrest release, and commitment of LMO2-ciLPs to the DN3/DN4, DP, and SP T cell stages.
Our study indicates that the level of LMO2 expression is important to support cells with both myeloid and lymphoid phenotypes. Thus, sufficient LMO2 expression levels secured by 1.0–3.0 μg/mL of DOX provide the balance between DN2 differentiation arrest and propagation of a CD44+CD25− myeloid cell population characterized by expression of T-lymphoid and myeloid markers, respectively. Interestingly, it was shown before that primitive DN (CD4−, CD8−) thymic progenitors possess both T and myeloid potential and are able to reconstitute both myeloid and lymphoid lineages. 56
Although we cannot directly explain why the percentages of the CD44+CD25− myeloid cell population decrease after DOX removal, we suggest that switching off LMO2 expression and increased Notch signaling lead to differentiation of CD44+CD25− myeloid cells, loss of proliferative capacity in T-lymphoid conditions, and probably eventual cell death. 47,48 It was previously demonstrated that activation of Notch signaling could induce cell cycle arrest, differentiation and apoptosis of acute myeloid leukemia initiating cells. 57 In this study, increased Notch signaling after DOX removal and switching off LMO2 expression could play a role in the loss of cells with a myeloid phenotype.
Interestingly, culturing LMO2-ciLP cell lines under myeloid cytokine conditions resulted in a myeloid shift characterized by CD11b+GR1+ (97.7% ± 1.2%) and CD44+CD25− (99.3% ± 0.5%) immunophenotype and subsequent myeloid differentiation. Thus, although the percentage of LMO2/EGFP+ cells was 100% over the time course of the experiment in myeloid conditions, it did not lead to a block of myeloid differentiation.
T cell commitment is orchestrated by multiple factors including expression of the IL7R and c-KIT ligand. 11,49 The DN2 stage of T cell development consists of a DN2a and a DN2b substage. DN2a is considered as the last uncommitted stage of T cell differentiation when the pre-T cells retain the potential to develop into myeloid, natural killer, and dendritic cells. DN2a T cells are highly positive for c-KIT, whereas DN2b T cells lose c-KIT expression and become restricted to the T cell fate. 11 LMO2-ciLPs expressed c-KIT in the presence of DOX and moreover, the DN2 subpopulation was ∼100% c-KIThigh positive. After DOX removal, c-KIT expression was abrogated, indicating loss of lineage potential and cell differentiation.
IL7R, another marker of T-lymphoid differentiation, was expressed in an opposite manner. When LMO2 was upregulated, IL7R expression was not observed. However, after switching off LMO2 expression upon drug removal, IL7R was expressed and reached peak expression levels at the DN3 stage before significantly decreasing again at the DN4/DP stages.
Immortalized T cell progenitors derived from LSK cells of CD2-Lmo2 transgenic mice did not require Notch1 signaling to proliferate in culture; however, these cells also failed to engraft in recipient mice. 18 LMO2-expressing hCD34+ cells transplanted into sublethally irradiated humanized mice 2 days after transduction were found in the thymus, peripheral blood, spleen, and bone marrow, but levels of LMO2 overexpression within the total bone marrow cells were very low. 19
We performed a murine transplantation assay of LMO2-ciLPs but were unable to find donor-derived cell populations within the bone marrow or thymus. It is possible that migration of LMO2-ciLPs to the thymus might be impaired due to the absence of chemokine receptor expression required for homing. 58 We demonstrated the expression of CCR9 and CXCR4 in vitro. However, it was shown that CCR7 (C-C chemokine receptor type 7) also plays an important role for the early T cell progenitor homing to the thymus. 59 Normally, CCR7 is downregulated during the DN and DP stages, but it is significantly upregulated during the DN1-2 stage (CD44highCD25int). 60
Therefore, another possibility to achieve engraftment of LMO2-ciLPs into the thymus might be to enhance Notch signaling that regulates Ccr7 and Ccr9 gene expression. 58,61 Transplantation of LMO2-ciLPs in the DOX− conditions should also not be underestimated since we observed upregulation of CCR9 and CXCR4 markers in vitro upon DOX withdrawal.
Moreover, vector insertions in cooperation with LMO2 expression might influence the immunophenotype and fate of LMO2-ciLPs in vitro and in vivo. Therefore, additional experiments are required to evaluate our hypotheses.
Thus, the model of conditional immortalization presented here opens a new chapter in the development of novel tools that allow ex vivo generation and drug-controlled differentiation of hematopoietic progenitors with the potential to investigate mechanisms underlying stem cell, leukemia, and lymphocyte biology, leading to novel approaches for disease modeling and therapy evaluation.
Footnotes
Acknowledgments
We thank Boris Fehse, Michael Heuser, Frank Staal, and Dirk Hoffmann for useful discussions. We acknowledge the assistance of the Cell Sorting Core Facility of the Hannover Medical School supported, in part, by Braukmann-Wittenberg-Herz-Stiftung and Deutsche Forschungsgemeinschaft. We thank Andreas Krueger and Juan Carlos Zúñiga-Pflücker for providing OP9-DL1 cells. We thank Andreas Krueger and Bala Sai Sundarasetty for suggestions regarding the cell differentiation media. We thank Ivan Takmakov for help with graph plotting using Matplotlib. We thank Jessica Herbst and staff of the animal facility of Hannover Medical School for the technical assistance with the animal experiments. We thank Michael Morgan for English proofreading the final draft.
Authors' Contributions
E.K. acquired, analyzed, and interpreted data, and wrote the article; M.St. acquired and analyzed data; L.L. provided critical reagents and interpreted data; M.Sa. designed animal experiments and acquired and interpreted data; O.K. was involved in conception and design, acquisition, analysis, and interpretation of data, and wrote the article; A.S. was involved in conception and design, data analysis and interpretation, and financial support, and wrote the article. All authors reviewed the draft and approved the final version of the article.
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported by the German Academic Exchange Service (DAAD), German Research Foundation (DFG; REBIRTH Exc 62/2; SFB738), and the European Union FP7 integrated project CELL-PID (HEALTH-2010-261387). This project received funding from the European Union's Horizon 2020 research and innovation program under grant agreement Nos. 755170 and 666908.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.