
Abstract
For most lysosomal storage diseases (LSDs), there is no cure. Gene therapy is an attractive tool for treatment of LSDs caused by deficiencies in secretable lysosomal enzymes, in which neither full restoration of normal enzymatic activity nor transduction of all cells of the affected organ is necessary. However, some LSDs, such as mucopolysaccharidosis type III (MPSIII) diseases or Sanfilippo syndrome, represent a difficult challenge because patients suffer severe neurodegeneration with mild somatic alterations. The disease's main target is the central nervous system (CNS) and enzymes do not efficiently cross the blood–brain barrier (BBB) even if present at very high concentration in circulation. No specific treatment has been approved for MPSIII. In this study, we discuss the adeno-associated virus (AAV) vector-mediated gene transfer strategies currently being developed for MPSIII disease. These strategies rely on local delivery of AAV vectors to the CNS either through direct intraparenchymal injection at several sites or through delivery to the cerebrospinal fluid (CSF), which bathes the whole CNS, or exploit the properties of certain AAV serotypes capable of crossing the BBB upon systemic administration. Although studies in small and large animal models of MPSIII diseases have provided evidence supporting the efficacy and safety of all these strategies, there are considerable differences between the different routes of administration in terms of procedure-associated risks, vector dose requirements, sensitivity to the effect of circulating neutralizing antibodies that block AAV transduction, and potential toxicity. Ongoing clinical studies should shed light on which gene transfer strategy leads to highest clinical benefits while minimizing risks. The development of all these strategies opens a new horizon for treatment of not only MPSIII and other LSDs but also of a wide range of neurological diseases.
Introduction
Mucopolysaccharidosis type III disease
Mucopolysaccharidoses (MPSs) are a group of rare, inherited lysosomal storage diseases (LSDs) caused by specific lysosomal enzyme deficiencies that lead to intracellular accumulation of partially degraded glycosaminoglycans (GAGs, formerly called mucopolysaccharides) within cells. 1,2 Depending on the nature of stored material and the deficient enzyme, MPSs have been classified into different types. 3
MPS type III (MPSIII), also known as Sanfilippo syndrome, is an autosomal recessive disease characterized by intralysosomal accumulation of the GAG, heparan sulfate (HS). 1,3,4 It is caused by deficiency in one of the four enzymes involved in lysosomal degradation of HS: deficiency in N-sulfoglucosamine sulfohydrolase or sulfamidase (SGSH, EC 3.10.1.1) causes type IIIA (OMIM No. 252900) 5 ; in α-N-acetylglucosaminidase (NAGLU, EC 3.2.1.50), type IIIB (OMIM No. 252920) 6 ; in acetyl CoA α-glucosaminide acetyltransferase (HGSNAT, EC 2.3.1.78), type IIIC (OMIM No. 252930) 7 ; and in N-acetylglucosamine-6-sulfatase (GNS, EC 3.1.6.14), type IIID (OMIM No. 252940). 8
MPSIII is one of the most common types of MPSs. 4 MPSIIIA is the most frequent subtype of Sanfilippo syndrome found in Northwest Europe, North America, and Australia, 9 –14 and type B has the highest prevalence in Southeast Europe and Brazil. 13,15 –19 MPSIIIC and D are much rarer diseases with few cases reported in the literature. 4,9,16,20 –22
GAGs, including HS, play an important role in the mechanical support of tissues and are also involved in regulating cell growth and development, cell–cell interactions, immunity and defense against viral infections, coagulation, and lipid metabolism. 23 The excess of HS fragments and HS-derived oligosaccharides that accumulate in lysosomes and are released into the extracellular medium in MPSIII disease could interfere with all of these processes. 3,4 However, the exact cascade of pathological events that leads to cell dysfunction and death is not completely understood. In addition to HS, neurons from Sanfilippo patients accumulate GM2 and GM3 gangliosides and, in some cases, also unesterified cholesterol. 24 –30 This secondary accumulation also appears to play an important role in the neurological pathology characteristic of the disease. 31
Clinical manifestations appear once HS accumulation causes cellular damage, after which symptoms progressively worsen. MPSIII is generally characterized by severe, progressive central nervous system (CNS) degeneration. Affected children appear normal at birth, and the first symptoms are detected around 1–4 years, generally under the form of delayed intellectual development. 4,20,22,32,33 The disease progresses to severe neurological manifestations, including hyperactivity, sleeping problems, loss of speech, and epilepsy. 4,20,22,32,33 At later stages of the disease, affected individuals develop profound dementia and progressive loss of motor functions, most patients being wheelchair-bound and fully dependent by the second decade of their lives. 4,20,22,32 –34
In contrast to the majority of other MPS disorders, children with MPSIII have relatively mild somatic symptoms. 4,35 Facial dysmorphisms, although usually slight, are detected in most individuals. 4,20,22,32,36 Patients frequently present visceromegaly, mainly hepatosplenomegaly, and mild skeletal alterations. 20,32,33,36 Recurrent ear, nose, throat, and chest infections are commonly found in young patients, as well as frequent diarrhea. 4,20,32,33,37 Death usually occurs in the mid-late teenage years. 4,22,32,33,36 –38
Treatment of MPSIII disease
Presently, there is no specific treatment approved for MPSIII. Control of the disease is merely symptomatic, aimed at improving the quality of life of patients and their families. 3,39 Several new therapies are being developed to treat Sanfilippo syndrome and some of them have already reached the clinical stage.
Most of these therapies are forms of enzyme replacement therapy (ERT), in which the enzyme is provided periodically as a recombinant protein, or a classic example of gene augmentation gene therapy, in which the enzyme is endogenously synthetized by cells that have received a correct copy of the mutated gene. Underlying all these strategies is the principle of cross-correction, whereby a soluble lysosomal enzyme present in the extracellular compartment can be taken up by endocytosis through binding to mannose-6-phosphate receptors present on the plasmatic membrane of cells. 40 –43 Therefore, the enzyme present in the extracellular milieu after recombinant protein administration or produced by engineered/corrected cells can cross-correct other cells. 41 –43
All these novel therapeutic approaches need, however, to overcome a major challenge in drug development for LSDs with CNS involvement: the existence of the blood–brain barrier (BBB), which limits efficient access of compounds to the CNS after systemic administration. To bypass the BBB and allow for repeated drug administrations, the use of a drug delivery device permanently implanted in the intrathecal space for delivery of recombinant proteins directly to the cerebrospinal fluid (CSF) has been tested for MPSIIIA (
Although technically feasible, permanent implantation of intrathecal devices is associated with complications, such as implant site infections or device malfunction.
44
–46
Despite these shortcomings, clinical studies of intrathecal ERT with recombinant human sulfamidase for MPSIIIA (
An investigational, intrathecal ERT drug for the treatment of MPSIIIB (NCT02754076) consisting in a recombinant chimeric protein in which human NAGLU is fused to a truncated form of human insulin-like growth factor 2 has shown HS reduction in the CSF and stabilization of development quotient. 47
In Vivo Gene Therapy for MPSIII Diseases
To overcome the need for periodic administration of the therapeutic protein of classic ERT approaches, gene therapies for MPSIII are being developed to achieve constant production of active enzymes available to cross-correct cells. A key advantage of gene therapy strategies is that correction of the genetic defect in all cells of an organ is not required since few corrected cells could, in principle, produce sufficient amount of the active enzyme to become available to neighboring cells or to other cells within the target organ or nontargeted organs in the body if the enzyme reaches main fluids, for example, CSF and serum. 48 –58
Since the CNS is the most affected organ in MPSIII, and the CNS is anatomically isolated from the rest of the body, a sufficient degree of gene correction has to be achieved in this organ. CNS-targeted gene therapy is a very active field of research. Recent years have witnessed the publication of numerous preclinical and clinical studies that demonstrate the potential of gene therapy for treating neurological conditions, particularly monogenic hereditary diseases. 59,60
Most of the gene therapy strategies that are being developed for MPSIII are based on in vivo gene transfer mediated by adeno-associated virus (AAV)-derived vectors. These vectors have shown high transduction efficiencies in vivo as well as excellent safety profiles in clinical studies. 61,62 In addition, preclinical studies in large animal models as well as clinical data have provided strong evidence supporting long-term expression in the CNS mediated by AAV vectors in the absence of clinically significant adverse events. 48,49,51,63 –67
This overview will focus on in vivo gene therapies for MPSIII. Nevertheless, ex vivo gene therapies have also been developed for MPSIIIA and MPSIIIB. 68 –71 They are based on the autologous transplantation of hematopoietic stem cells genetically modified using lentiviral vectors to express SGSH or NAGLU for the treatment of MPSIIIA or MPSIIIB, respectively. 68 –70 The progeny of transplanted gene-corrected cells traffics to the brain, bypassing the BBB, to become resident cells that produce the therapeutic protein in the CNS. Proof-of-concept studies demonstrated normalization of brain HS, secondary GM2 storage and neuroinflammation, and improvement of behavioral deficits in MPSIIIA and MPSIIIB mice. 68 –70 A clinical trial is to be initiated soon in MPSIIIA patients. *
Similar ex vivo gene therapy approaches have demonstrated clinical efficacy for the treatment of other neurometabolic storage disorders, such as adreno and metachromatic leukodystrophies, as well as other indications such as MPS type I. 72 –78 These studies are discussed in detail in the review by Poletti and Biffi included in this special issue of Human Gene Therapy. 72
Routes of administration to deliver AAV vectors to the CNS
MPSs are diseases in which the neurodegenerative process affects the whole CNS, although little information is available from human specimens on whether there are structures that are more affected than others. 27,79,80 Several AAV-based strategies for delivery of genes to extensive areas of the CNS have been described.
Initial efforts administered the AAV through multiple direct injections to the brain parenchyma. Other systems followed that exploited the ability of certain AAV serotypes, such as serotype 9 (AAV9), to cross the BBB after intravenous administration, resulting in widespread transduction of the brain as well as of the liver and other peripheral organs through a noninvasive procedure. More recently, in an effort to maximize gene transfer to the most affected organ while minimizing vector dose, direct delivery of AAV vectors to the CSF has been proposed as an alternative for global CNS gene transfer. Additionally, at least in animal models, this approach results in transduction of the peripheral nervous system and the liver, providing for somatic disease correction.
Figure 1 depicts a schematic representation of potential vector and enzyme biodistribution after AAV vector delivery through these different routes of administration. 49,51,54,81 –85 Several clinical trials are currently testing the safety and efficacy of these approaches in MPSIII disease and are listed in Table 1.
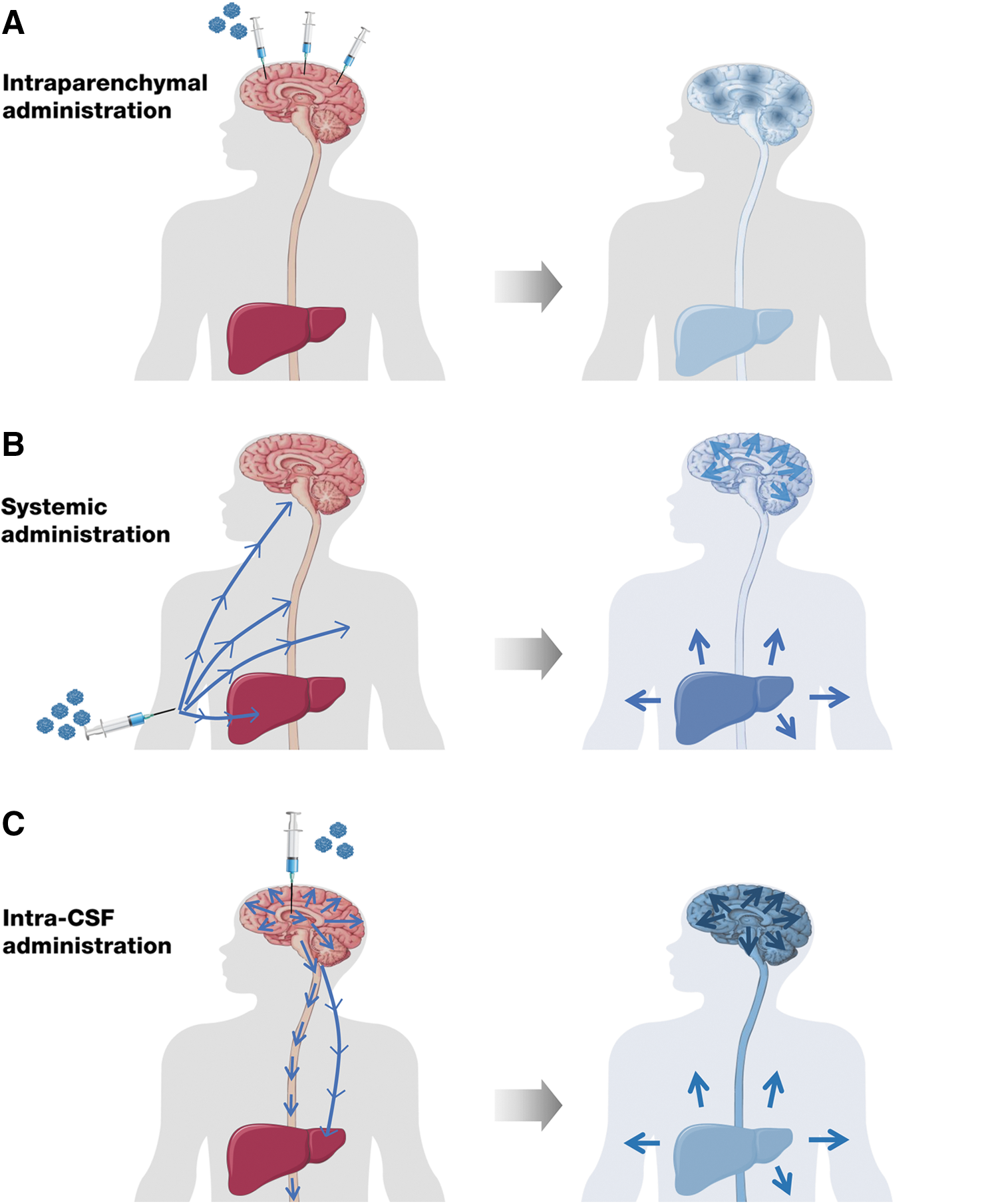
Possible routes of vector administration and subsequent enzyme biodistribution, in vivo, AAV-mediated gene therapy strategies for Sanfilippo syndrome. Based on results obtained in proof-of-concept studies in small and large animal models of MPSIII,
49,51,53,54,56,81
–84,91,96,97,99,102,117
–120,125,126
a schematic representation of the possible biodistribution of the gene therapy product in patients is presented.
In vivo gene therapy clinical trials for Sanfilippo patients
Clinical trial registered in
Clinical trial registered in
Information based on Tordo, 2018.
AAV, adeno-associated virus; CSF, cerebrospinal fluid; LT, long-term follow-up; NA, nonavailable; NAGLU, α-N-acetylglucosaminidase; SGSH, N-sulfoglucosamine sulfohydrolase or sulfamidase; SUMF1, sulfatase-modifying factor 1; HGSNAT, acetyl CoA alpha-glucosaminide acetyltransferase.
Direct administration of AAV vectors to the brain parenchyma
Vectors can be delivered directly to the CNS by intraparenchymal injection, and this approach has been used safely in patients to target the putamen in the context of investigational gene therapies for Parkinson's disease. 65,86 –90 MPSIII, however, affects the whole of the CNS.
Given the limited diffusion of AAV vectors from the site of injection, 67,91 the applicability of this method to global neurodegenerative diseases has required the development of a rather complex surgical procedure by which small volumes of vectors are simultaneously deposited in the brain white matter at different sites. 64,92 –94 There is a limit to the number of injections and to the locations at which these injections can be safely performed. 67,92,94 Moreover, intraparenchymal injection fails to transduce deep CNS structures, a fact that has been considered the culprit of the limited efficacy in previous studies in humans using this approach for other neurodegenerative diseases. 64 In the original procedure, a total of six burr holes were drilled laterally from the midline and the vector was administered at two depths per site. 94
Intraparenchymal administration of AAVs at multiple sites has been tested in the clinic for MPSIIIA (NCT01474343) and MPSIIIB (NCT03300453), using AAVs of serotypes rh10 and 5, respectively. 92,93 In the case of the phase I/II trial for MPSIIIA, the vector encoded for SGSH as well as for the sulfatase-modifying factor 1 (SUMF1) protein. SUMF1 post-translationally generates C-alpha-formylglycine (FGly), the catalytic residue in the active site of eukaryotic sulfatases, from a cysteine. 95 The rationale behind SUMF1 supplementation was to avoid potential toxicity derived from competition for SUMF1 sequestration at sites with very high levels of expression of sulfamidase (e.g., injection sites).
Preliminary results of the 1-year follow-up of this trial reported moderate improvements in behavior, attention, and sleep disturbances. 92 Nevertheless, these patients have remained under long-term immunosuppression since receiving AAV vectors, and the potentially beneficial effect of immunosuppression on the observed clinical benefit cannot be ruled out because neuroinflammation is a main player in disease pathology. 92 The ongoing long-term follow-up of these MPSIIIA patients (NCT02053064) should provide further information regarding efficacy. Recently, a new, open-label, single-arm, multicenter, phase II/III clinical trial has started in MPSIIIA patients (NCT03612869) administered a simplified version of the therapeutic vector, which does not include the SUMF1 gene present in the original vector. 85
As a technical improvement to the approach used for the original MPSIIIA study, a more recent trial for MPSIIIB was conducted with 2 additional deposits in the cerebellum, resulting in vector deposition simultaneously in 16 sites (8 burr holes). 93 Four young MPSIIIB patients have been treated thus far using this method, with concomitant immunosuppression. 93 The procedure was well tolerated and initial evidence of improved cognitive outcomes is suggestive of efficacy, with the same caveat than for the MPSIIIA trial regarding the use of chronic immunosuppression. 93
Interestingly, a similar intraparenchymal approach using a new AAV vector variant with a putative better pharmacokinetic profile, AAV-TT, is being developed for MPSIIIC, although in this case, the deficient enzyme is not secretable and cross-correction cannot occur. 96 Treatment with AAV-TT-HGSNAT was able to correct the neurological phenotype of a mouse model of MPSIIIC. 96 A clinical trial has been planned to test the approach in MPSIIIC patients. †
Intravascular delivery of AAV vectors to treat MPSIII
Of the several ways of delivering genes to the CNS, the simplest approach exploits the ability of certain AAV serotypes, such as AAV9, to cross BBB and reach the CNS when delivered intravenously (IV). 97,98
Systemic delivery of AAV9 has proven successful in the treatment of spinal muscular atrophy type 1 (SMA1). SMA1 is caused by a mutation in the survival motor neuron 1 (SMN1) gene that leads to severe degeneration and loss of lower motor neurons, resulting in muscle atrophy. 99 Death occurs during infancy (before 2 years of age). 100 Intravascular AAV9-SMN1 gene therapy delivered the SMN1 gene to motor neurons of SMA1 patients, was safe, and resulted in longer survival, superior achievement of motor milestones, and better motor function than in historical cohorts. 99 The highly positive results obtained in these clinical studies (NCT02122952 and NCT03955679) 99 have led to the recent marketing approval by the Food and Drug Administration (FDA) of the new gene therapy product Zolgensma.
Treatment of MPSIII disease requires targeting of the whole CNS, and this may be more difficult to achieve after systemic delivery of AAV9 vectors than targeting of motor neurons, as in the case of SMA. Proof-of-concept studies in mouse models of MPSIIIA and MPSIIIB following IV delivery of AAV9 vectors encoding for deficient enzymes have shown reduction in brain GAG accumulation and neuroinflammation, as well as correction of behavioral deficits. 82,84,101,102 Based on the demonstrated efficacy in rodent models, clinical studies are presently underway for MPSIIIA and MPSIIIB.
In an open-label, dose-escalation, phase I/II gene transfer trial, MPSIIIA patients were IV administered self-complementary AAV9 vectors carrying the hSGSH gene under the control of the ubiquitous U1a promoter (scAAV9.U1A.hSGSH) (NCT02716246). All subjects received immunosuppression, consisting in a tapering course of prophylactic enteral prednisone or prednisolone.
It has been shown in several clinical trials that systemic exposure to high doses of AAV vectors may trigger activation of CD8+ T cell responses directed against the viral capsid in a dose-dependent manner. 103 –106 Short-term (few weeks) steroid treatment is used to prevent the immune-mediated elimination of vector-transduced cells, resulting in long-term expression of therapeutic genes. 52,103
No product-related, serious adverse events have been reported to date in this MPSIIIA trial. Efficacy data have not been published yet, but some interim data have been made available by the sponsor Abeona Therapeutics. A dose-dependent and sustained reduction in levels of heparan sulfate in the CSF has been reported for all three cohorts of patients treated with low (5 × 1012 vg/kg), medium (1 × 1013 vg/kg), or high (3 × 1013 vg/kg) vector doses. Moreover, the three youngest patients enrolled in the high-dose cohort (cohort 3)—14–26 months of age at dosing—continued to track within normal age-equivalent development 12–18 months post-treatment ‡ .
Based on these encouraging results, the same sponsor has initiated a new trial using the IV AAV9 approach for treatment of MPSIIIB (NCT03315182). This is an open-label, dose-escalation phase I/II trial in MPSIIIB patients aged 6 months to 2 years, or older than 2 years with a minimum cognitive development quotient of 60 or above. In this case, the AAV9 vector carried the hNAGLU gene under control of the strong viral cytomegalovirus promoter (AAV9.CMV.hNAGLU). All dosed subjects have received immunosuppression, consisting in oral prednisolone from the day before gene transfer until at least 60 days after. 107 Two doses of AAV vectors are being tested, 2 × 1013 vg/kg (cohort 1) and 5 × 1013 vg/kg (cohort 2), which are higher than those administered in cohorts 1 and 2 of the MPSIIIA trial. Dosing of cohort 1 has been completed and the first patient in cohort 2 has received the vector. §
Although clinical evidence supports that systemic administration of AAV vectors results in therapeutic benefit for MPSIII patients, this route of administration may not be of choice to reach the CNS in the subset of patients who are seropositive for anti-AAV antibodies, which can greatly limit the efficacy of in vivo gene transfer upon systemic administration. 104,108,109
Serum neutralizing antibodies (NAbs) to AAVs are highly prevalent in humans. 110 The prevalence and magnitude of seropositivity, however, vary with the AAV serotype. While about 60% of the adult population has anti-AAV2 NAbs at high titers, only about 30% of healthy individuals have detectable anti-AAV9 antibodies. 110 Moreover, there is a pattern of seroconversion with age since children under 1 year are seronegative, or seropositive at very low titers, but then titers progressively increase up to around 5–6 years of age, reflecting an increase in socialization, which favors natural infection by wild-type AAVs. 111,112 The same tendency was observed in healthy and MPSIIIA- and MPSIIIB-affected children. Serum NAb titers against AAV2 were generally higher than those against AAV9 and they tended to increase with age. 51,101
Aside from the limitations imposed by anti-AAV pre-existing immunity and the potential risk of cytotoxic CD8+ T cell responses, the consistently high doses required for brain transduction and efficacy following IV administration of AAV vectors 38,82,84,113,114 may suppose a manufacturing challenge from the technical and economic points of view, although great efforts are being devoted to scaling up and improving the yield of AAV manufacturing by different platforms.
Intra-CSF administration of AAV vectors to transduce the CNS
As an alternative to local delivery of vectors to the CNS, several groups have begun to administer AAV vectors—particularly AAV9—directly to the CSF and have achieved efficient CNS transduction in different animal species (mice, rats, cats, dogs, pigs, and nonhuman primates [NHPs]) at relatively lower vector doses when compared with those used when the vector is injected systemically. 49,51,53,54,56,115 –119 Upon administration to the CSF, the vector is diluted in a fluid that circulates through CNS compartments, bathing the whole encephalon and spinal cord, allowing for widespread vector distribution, including deep structures. 51,119 As a result, the pattern of vector transduction differs considerably from that obtained following intraparenchymal injection at multiple sites, which results in uneven distribution of vector genomes, with high copy numbers at the point of injection quickly decreasing with distance. 83,91,120
When vectors carry genes encoding for secretable proteins, such as in MPSIIIA and IIIB, transduced cells secrete these proteins to the CSF at relatively high levels, 49,51 which could further increase efficacy by distribution of the therapeutic protein throughout the whole CNS. Furthermore, at least in animal models, upon intra-CSF AAV9 delivery, a portion of the vector escapes to the circulation and transduces the liver, which can secrete the therapeutic protein to the bloodstream. 49,51,53,54,56,116
Whole-body correction of LSD was observed following delivery of AAV9 vectors to the CSF of several MPS mouse models. 49,51,53,54,56 In all cases, administration of the therapeutic vector mediated clearance of accumulated GAGs, resolution of lysosomal pathology and neuroinflammation in the brain, correction of behavioral deficits, and significant extension of life span.
Proof-of-concept studies in dogs and pigs have shown that the delivery of vectors to the CSF is scalable to larger brains. 49,51,119 In most of these studies, vectors were administered to the cisterna magna, a large subarachnoid space between the caudal part of the cerebellum and the medulla oblongata, filled with CSF produced in the fourth ventricle that will distribute the vectors to the whole CNS. 121 Intracisternal injection, however, is not a common route of administration to the CSF in clinical pediatric practice because of the relatively smaller size of the cisterna magna in humans compared with animals and its proximity to vital centers, which adds risk to the procedure in case of accidental puncture. 121
An alternative route for CSF delivery is lumbar puncture. Although this is a common clinical procedure, limited distribution of products in widespread CNS areas is achieved through this route. 122,123 Preclinical data in pigs show that AAV9-derived gene expression following local lumbar intrathecal administration remains restricted to areas of injection and that widespread spinal cord transduction requires several administrations of the vector at the cervical, thoracic, and lumbar regions, suggesting that the vector penetrated mostly at the vicinity of the catheter tip used for delivery. 122 Global CNS gene delivery following intrathecal AAV administration was reported in cynomolgus macaques, an NHP of small size (∼3–7 kg), using AAV vectors diluted in a hyperosmotic buffer. 116 Although this study provides evidence that the approach is potentially feasible, detailed confirmatory studies in larger animal models are needed.
Ventriculostomy is a standard surgical procedure to deliver drugs (oncology drugs, antibiotics, and antifungal agents, etc.) to the CSF for treatment of different diseases or for management of hydrocephalus. 123,124 Although intracerebroventricular (ICV) access does require unilateral trepanation of the skull, the trajectory to reach the ventricle is well defined and goes through mute areas of the brain. 123 Using a surgical procedure that has been in place for many years has the advantage of providing a solid safety record as well as a well-characterized list of potential complications and protocols for their management. It also simplifies clinical translation as the technique is known to pediatric neurosurgeons worldwide and would not require specific training.
Compared with intraparenchymal administration, another advantage of ICV administration is that by delivering the vector to the CSF, a relatively large volume of vector can be supplied within a brief period of time, thus shortening the duration of surgery and providing flexibility in terms of vector concentration and formulation. In dogs, ICV administration demonstrated to be highly efficacious in delivering AAV vectors to the whole encephalon and spinal cord. 51
An open-label, dose-escalation, phase I/II clinical trial was initiated recently in MPSIIIA patients older than 2 years, which uses the ICV procedure to deliver AAV9 vectors carrying the human SGSH gene (AAV9-CAG-cohSGSH) into the CSF (EU Clinical Trials Register 2015-000359-26). This is a safety, tolerability, and initial efficacy clinical trial.
Although it has been suggested that the BBB is disrupted in patients affected by LSDs, 41 asymmetrical distribution of anti-AAV NAbs across the BBB of patients suffering from Sanfilippo syndrome is preserved even in severely affected children. 51
The evidence showing that very low titers of NAbs are sufficient to completely block transduction upon systemic administration 104,108,109 prompted several laboratories, including ours, to evaluate CNS transduction after intra-CSF delivery in large animals with anti-AAV pre-existing immunity. In NHPs with serum AAV9 NAb titers of 1:128, successful CNS gene transfer was observed after intra-CSF administration of vectors. 116 Similarly, in healthy dogs preimmunized by IV administration of noncoding AAV9 vectors leading 1 month later to high anti-AAV9 NAb serum titers (1:100–1:1000), intra-CSF delivery of AAV9 vectors encoding for green fluorescent protein resulted in efficient transduction of the brain and spinal cord at levels comparable with those achieved in animals naïve to AAV9. 51
These findings were further confirmed following intra-CSF administration of a therapeutic transgene (canineNaglu) to seropositive dogs with high AAV9 NAb titers in plasma (1:100–1:1000). 49 Again, similarly high levels of NAGLU activity were detected in the CSF of both naïve and seropositive animals upon intra-CSF AAV-mediated gene transfer. 49 These studies suggest that CNS efficacy would not be compromised in seropositive patients when vectors are delivered to the CSF. As expected, transduction of the liver is completely abolished following vector delivery to the CSF of animals with circulating NAbs. 51
Given that the main target organ for MPSIII is the CNS, the lack of peripheral efficacy would still allow for a significant degree of disease correction. Further clinical studies should establish the safety of gene transfer to the CSF of seropositive individuals.
Concluding Remarks
Several AAV vector-mediated, in vivo, gene therapy approaches for treatment of MPSIII have been developed in recent years, some of which are already under clinical investigation. Different therapeutic strategies use different routes of vector administration to reach the CNS, the most affected organ.
Studies in small and large animal models of MPSIII diseases have provided evidence supporting the efficacy and safety of these strategies and are expected to be predictive of possible efficacy in humans. Results from the ongoing clinical trials should confirm, or not, these expectations. Although direct comparisons of clinical outcomes of these studies cannot be made, due in part to differences in inclusion criteria, protocol design, or vector manufacturing, the ongoing studies should shed light on which gene transfer strategy maximizes efficacy in the CNS while minimizing delivery-associated risks, leading to higher clinical benefits.
Of importance, clinical trials in which the vector is delivered directly to the CNS (intraparenchymal or intra-CSF) should inform on the potential to achieve amelioration of somatic disease in humans, a fact that would become more relevant as patients live longer due to improved neurological outcomes.
Finally, these studies should provide information on the tolerability, safety, and efficacy of AAV-mediated CNS gene transfer not only for treatment of MPSIII but also of other neurodegenerative diseases.
Footnotes
Author Disclosure
F.B., V.H., and S.M. are coinventors on patent applications for the use of AAV vectors for treatment of MPSIII.
Funding Information
Work in the authors' laboratory relevant to this review has been supported by grants from Plan Nacional I+D+I from the Ministerio de Ciencia, Innovación y Universidades, and the European Union through Regional Development Funds (ERDF) (INNPACTO IPT-2012-0772-300000, SAF 2014-54866-R, and SAF2017-86166-R), Generalitat de Catalunya (ICREA Academia Award to F.B.), MPS España Foundation, and it is part of a public–private partnership on gene therapy between UAB and ESTEVE Pharmaceuticals, Spain.