
Abstract
Adoptive T cell immunotherapy in combination with gene therapy is a promising treatment concept for chronic infections and cancer. Recently, receptor-targeted lentiviral vectors (LVs) were shown to enable selective gene transfer into particular types of lymphocytes both in vitro and in vivo. This approach might facilitate the genetic engineering of a patient's own T lymphocytes, possibly even shifting this concept from personalized medicine to an off-the shelf therapy in future. Here, we describe novel high-affinity binders for CD8 consisting of designed ankyrin repeat proteins (DARPins), which were selected to bind to the CD8 receptor of human and nonhuman primate (NHP) cells. These binders were identified by ribosome display screening of DARPin libraries using recombinant human CD8 followed by receptor binding analysis on primary lymphocytes. CD8-targeted LVs (CD8-LVs) were then generated that delivered genes exclusively and specifically to human and NHP T lymphocytes by using the same targeting domain. These CD8-LVs were as specific for human T lymphocytes as their single-chain variable fragment-based counterpart, but they could be produced to higher titers. Moreover, they were superior in transducing cytotoxic T cells both in vitro and in vivo when equal particle numbers were applied. Since the here described CD8-LVs transduced primary T lymphocytes from NHP and human donors equally well, they offer the opportunity for preclinical studies in different animal models including large animals such as NHPs without the need for modifications in vector design.
Introduction
As a primary marker of the cytotoxic subset of T lymphocytes, CD8 is in prime focus of ongoing activities in immunotherapy. Particularly, CD8+ cells are frequently used for adoptive cell therapy approaches, often in combination with genetic engineering to express a recombinant T cell receptor or chimeric antigen receptor (CAR).
CD8 is a type-I single-pass transmembrane protein expressed as a disulfide-linked homo- or heterodimeric molecule on the surface of immune cells. The CD8 heterodimer consists of the CD8α and CD8β chain and is only expressed on the surface of immature CD4+CD8+ double-positive thymocytes and mature peripheral cytotoxic αβ T cells. 1 The homodimer consists of two CD8α chains and exhibits expression on a much broader range of immune cells. In addition to classic cytotoxic αβ T cells and thymocytes, it is found on natural killer (NK) T cells, 2 a subset of dendritic cells, 3 and NK cell subpopulations. 4,5 Both CD8αβ and CD8αα can mediate major histocompatibility complex (MHC) class I-restricted binding 1 ; still, the heterodimeric form is more prevalent on the surface of MHC-I-restricted cytotoxic T cells. Of note, CD8ββ-homodimers do not occur naturally. 6
While lentiviral vectors (LVs) pseudotyped with the envelope protein G of the vesicular stomatitis virus (VSV G) have a broad tropism, receptor-targeted LVs using engineered paramyxoviral glycoproteins were proven to specifically transduce only a cell subset of choice dependent on the targeting domain used for pseudotyping. 7 The receptor-targeted LVs described in this study are based on the envelope proteins F and G of the Nipah virus (NiV). Both envelope proteins are truncated to be compatible with LV assembly and the G protein responsible for cell attachment of the wild-type NiV is additionally ablated for natural receptor binding and genetically fused to a targeting domain of choice to mediate cell attachment. 8 Using this system, a broad range of cell surface receptors could already be successfully targeted, including EpCAM and Her2/neu on cancer cells, CD20 on B lymphocytes, as well as CD8 on T lymphocytes. 7
Many CD8-specific antibodies and antibody fragments used for identification, isolation, and functional manipulation of cytotoxic T cells have been described. However, only the monoclonal antibody (mAb) OKT8 has been shown to induce effector functions in CD8+ T cells. 9 Moreover, the single-chain variable fragment (scFv) of OKT8 has been successfully used as targeting domain for a CD8-specific LV to achieve selective CD8+ T cell manipulation both in vitro and in vivo. 8,10 By demonstrating the delivery of a CD19-specific CAR into CD8+ cytotoxic T cells directly in vivo, Pfeiffer et al. outlined the potential and the clinical relevance of this CD8-targeted vector. 11
Despite this success, the overall in vivo transduction efficiency of the CD8-specific scFv-LV was rather low, which suggests exploring strategies to improve gene delivery by vector optimization. In this regard, designed ankyrin repeat proteins (DARPins) may be very well suited as alternative binding domains for vector retargeting. DARPins are artificial binding proteins that have emerged as alternatives to antibodies in biotechnological and, more recently, therapeutic applications. 12,13 Analogous to antibodies, they can be selected to bind virtually any target protein due to their high variability. In addition, DARPins belong to the most stable proteins known so far, 14 a property highly beneficial when it comes to expression in fusion with a type-II transmembrane protein such as paramyxoviral glycoproteins. Indeed, Her2/neu-specific DARPins have been found to be extraordinarily well suited as targeting domains for LVs. 15 Morever, display of a CD4-specific DARPin on LV particles enabled gene transfer exclusively into CD4+ T cells both in vitro and in vivo. 16
To further assess DARPin application in vector retargeting, we have recently developed a novel strategy for the selection of DARPins specific to a target of choice for use as targeting domains on LVs and adeno-associated virus-derived vectors. 17 Using this process, we identified DARPins specific for the interneuron marker GluA4, the endothelial protein CD105, and the NK cell receptor NKp46. All these DARPins mediated efficient and extraordinarily specific gene transfer when displayed on the surface of vector particles.
Here, we show the selection, characterization, and application of DARPins specifically binding to human and nonhuman primate (NHP) CD8 (Fig. 1). Aiming at selecting DARPins preferentially recognizing CD8 on cytotoxic T cells, we chose to generate heterodimeric CD8αβ-Fc as bait protein for ribosomal selection. After ribosome display, we characterized the selected DARPins for specific CD8 binding in a binding assay using primary human and NHP peripheral blood mononuclear cells (PBMCs). Further, binders were assessed for their targeting capacity on receptor-targeted LVs in PBMC transduction assays. Thorough characterization of the three most promising candidates revealed a higher yield in functional transducing particles compared with the established CD8-specific scFv-LV at similar selectivity. In vivo, the candidate 53F6-LV displaying a CD8-specific DARPin was found to be three times more potent than the CD8-specific scFv-LV when applied at equal particle numbers. This highly potent, NHP cross-reactive novel CD8-DARPin-LV can now be used to assess cytotoxic T cell targeting in large animal models, which will be especially important with regard to the further development of in vivo CAR T cell generation.
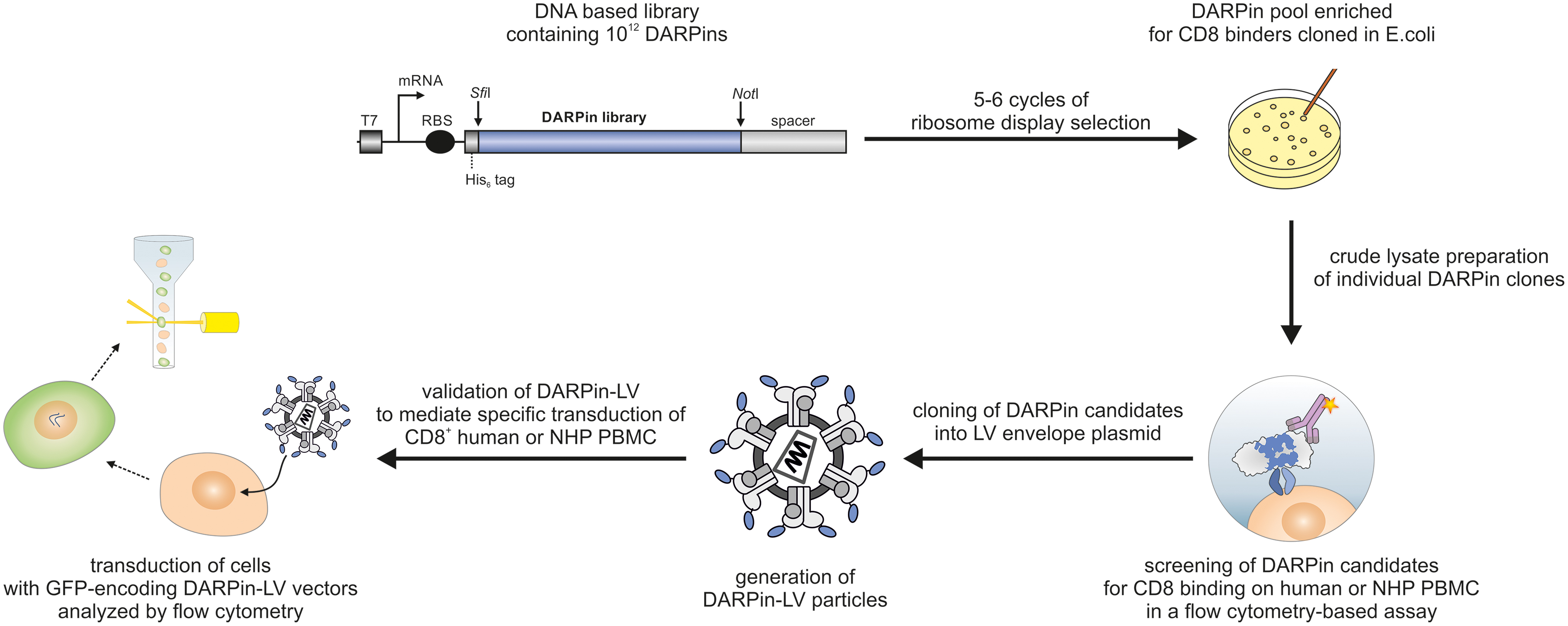
Overview of the selection process to identify CD8-specific DARPins for receptor-targeted LVs. CD8-specific DARPins were selected by ribosome display using DARPin libraries. A detailed description of the ribosome display process is described by Hartmann et al. 17 After five to six cycles of ribosome display, a DARPin pool enriched for CD8 binders was obtained from which individual DARPin clones were expressed in Escherichia coli. In the next step, crude protein lysates containing DAPRins were screened for their ability to bind to human and NHP PBMC in a flow cytometry-based assay. Promising DARPin candidates were cloned in fusion to the G envelope protein of Nipah virus and used to produce DARPin displaying LV particles encoding GFP as a transgene. LV stocks were evaluated for their ability to mediate specific transduction of CD8-positive human and NHP PBMC as analyzed by flow cytometry. DARPins of vector stocks transferring GFP specifically into the CD8-positive fraction of cells were considered as suitable targeting domains. DARPins, designed ankyrin repeat proteins; GFP, green fluorescent protein; LVs, lentiviral vectors; mRNA, messenger RNA; NHP, nonhuman primate; PBMC, peripheral blood mononuclear cell; RBS, ribosome binding site. Color images are available online.
Materials and Methods
Ethics statement
All animal experiments were carried out in accordance with the regulations of the German animal protection law and the respective European guidelines.
Generation of CD8-Fc expression plasmids
To generate soluble, recombinant CD8 proteins, human CD8α residues 22
To generate CD8αα-Fc, the extracellular CD8α coding region (excluding disulfide residues responsible for dimerization) was PCR amplified, purified, and cloned into phuFc-Avi via digestion with SfiI/XbaI to obtain phuCD8α-huFc-Avi. The extracellular CD8β coding sequence (excluding disulfide residues responsible for dimerization) was cloned into phuFc-Avi harboring an additional XbaI restriction site downstream of the Avi tag (phuFc-AviX) via digestion with BssHII/NheI to obtain phuCD8β-huFc-Avi.
For generation of CD8αβ-Fc heterodimers, a strategy described by Merchant et al. 18 relying on knobs-into-holes mutations in the constant region of human IgG1 harboring a C-terminal Avi-tag (Fc) was used. Introduction of the mutations into the Fc domain was carried out by overlap extension PCR. To introduce the knob mutations, primer combination 2148 and 2147 in conjunction with primer combination 2121 and 2149 were used while applying phuFc-Avi as template. To introduce the hole mutations, primer combination 2121 and 2151 in conjunction with primer combination 2150 and 2153 and primer combination 2152 and 2147 were used while applying phuFc-Avi as template. DNA fragments were purified and ligated into the plasmid phuFc, which is a derivate of pCMV-hIgGI-Fc-XP 19 harboring an SfiI restriction site downstream of the signal peptide, via SfiI/XbaI to yield phuFc-Avi(Knob) and phuFc-Avi(Hole). Finally, the CD8α domain of phuCD8α-huFc-Avi was cloned into phuFc-Avi(Knob) via NcoI/NotI restriction sites to generate CD8α-Fcknob and the CD8β domain of phuCD8β-huFc-Avi was cloned into phuFc-Avi(Hole) via BssHII/NheI restriction sites to generate CD8β-Fchole.
All PCR reactions were carried out with the Phusion High-Fidelity Polymerase (New England Biolabs, Frankfurt, Germany). Primer sequences are provided in Supplementary Table S1.
Cell culture
HEK 293T cells (ATCC CRL-11268) were cultivated in Dulbecco's modified Eagle medium (DMEM; Biowest, Nuaillé, France) supplemented with 10% fetal bovine serum (FBS; Biochrom, Berlin, Germany) and 2 mM L-glutamine (Sigma-Aldrich, Munich, Germany). Molt4.8 and JE6.1 cells were grown in complete RPMI medium (RPMI 1640 [Biowest] supplemented with 10% FBS and 2 mM L-glutamine), whereas J76S8ab cells (kindly provided by I. Edes) were cultured in complete RPMI containing 1 × nonessential amino acids and 1 mM sodium pyruvate (both Sigma-Aldrich). Routine testing for mycoplasma contamination was carried out twice a year.
Human PBMC were isolated from fresh blood of healthy anonymous donors who had given informed consent. PBMC were cultivated in RPMI complete supplemented with 0.5% penicillin/streptomycin, 25 mM HEPES (Sigma-Aldrich), and 100 U/mL human recombinant interleukin (IL)-2 (Miltenyi Biotec, Bergisch Gladbach, Germany). For cell activation, PBMC were seeded into plates coated with 1 μg/mL anti-human CD3 mAb (clone OKT3; Miltenyi Biotec) and the medium was supplemented with 3 μg/mL anti-human CD28 (clone 15E8; Miltenyi Biotec). After 72 h of culture, PBMC were used in DARPin screening and transduction assays.
Macaca mulatta (Rhesus monkey) and Macaca nemestrina (pig-tailed monkey) PBMC were isolated from fresh blood of healthy individuals as previously described. 16 NHP T cell activation was carried out by using the T Cell Activation/Expansion Kit, NHP (Miltenyi Biotec) according to the manufacturer's instructions. After 72 h, NHP T cells were resuspended in fresh RPMI complete medium supplemented with 0.5% penicillin/streptomycin, 25 mM HEPES, and 4 μg/mL Proleukin S (kindly provided by J. Seifried [Paul-Ehrlich-Institut]) or polyethylenimine) or 25 U/mL recombinant human IL-7 (Miltenyi Biotec) before use in DARPin screening and transduction assays.
Recombinant protein expression and purification
Target proteins for ribosome display were produced by transient transfection of the CD8 constructs in the presence or absence of the biotinylation constructs pDisplay-sBirA and pDisplay-BirAER into HEK 293T cells followed by purification of the proteins from the cell culture supernatant as previously described. 17
Briefly, 1 × 107 cells were seeded into T175 flasks one day before transfection. On the day of transfection, the culture medium was replaced by DMEM supplemented with 15% fetal calf serum (FCS) and 2 mM L-glutamine. To prepare the transfection mix, 35 μg of DNA was mixed with DMEM without additives and added to DMEM containing 140 μL of 18 mM polyethyleneimine (PEI).
The mix was incubated for 20 min at room temperature before addition to the seeded cells. After 24 h, the transfection medium was replaced by DMEM supplemented with 5% Panexin NTA (Pan Biotech, Aidenbach, Germany) and 2 mM L-glutamine and 10 μM biotin for biotinylated constructs. Two days post-transfection, the culture supernatant was harvested by passing through a 0.45-μm filter followed by purification via Protein A-Sepharose (Thermo Fisher Scientific, Darmstadt, Germany) columns according to the manufacturer's instructions. If required, size exclusion chromatography was performed by using a Superdex 200 HighLoad 16/600 column (GE Healthcare, Germany) in a high-pressure liquid chromatography (ÄKTAxpress; GE Healthcare) system according to the manufacturer's instructions.
The purified recombinant protein was concentrated by using 50 kDa cut-off spin concentrators, supplemented with 5% glycerol and protease inhibitor (cOmplete ULTRA; Roche, Mannheim, Germany) and stored at −80°C until use. Protein concentration was determined by Bradford assay. Plasmid combinations used to produce the various recombinant CD8 proteins are provided in Supplementary Table S2. Analysis of recombinant proteins by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and subsequent Coomassie staining or immunoblot analysis were carried out as previously described. 17
Ribosome display
Ribosome display selection was carried out as described by Hartmann et al. 17 Briefly, for the first three selection rounds, the translated VV-N3C 17 and S-N3C 17,20 DARPin library were subjected to pre-panning steps with immobilized neutravidin or streptavidin (both 20 μM). For on-target selection, the libraries were incubated with immobilized CD8αβ-Fc (50 nM). The resulting DARPin libraries were amplified and used as a template for the next selection round. After three rounds with immobilized target protein, the fourth round of selection was carried out with target in solution. Before on-target selection, a preselection step using 0.9 pmol unbiotinylated CD8αα-Fc was performed. Then, the libraries were exposed to biotinylated CD8αβFc target protein (0.65 pmol) and unbiotinylated CD8ααFc in 100-fold molar excess (65 pmol). Selection round five was again performed by using immobilized proteins and included a preselection step with immobilized CD8αα-Fc (20 nM) before incubation with CD8αβ-Fc. Finally, a sixth round of selection was carried out in solution as follows: Preincubation with 0.9 pmol soluble CD8αα-Fc was followed by a combined off-rate and counter-selection step, where the libraries were co-incubated with biotinylated target protein (0.65 pmol), an excess of unbiotinylated CD8αα-Fc and unbiotinylated CD8αβ-Fc (both 65 pmol). After the fifth and sixth selection round, DARPin-encoding DNA fragments were analyzed for CD8 binding by single clone analysis.
DARPin expression and crude lysate preparation
To test selected DARPins for specific binding to CD8 after the fifth and sixth selection round, DNA fragments were cloned into the bacterial expression vector pQE-HisHA and transformed into Escherichia coli XL1-blue as described earlier. 17
Single clones were picked and cultured overnight in 600 μL 2YT medium (2YT, 1% glucose, 100 μg/mL ampicillin) at 37°C before cultures were diluted to an OD600 of 0.1, and expression of the DARPins was induced by addition of 100 mL of 5.5 mM isopropyl-β-D-thiogalactopyranosid in 2YT medium. After culture for 5 h at 37°C, bacteria were harvested by centrifugation and pellets were stored overnight at −80°C. The next day, pellets were thawed on ice and lysed by addition of B-PER solution and subsequent incubation at room temperature for 2 h. The lysate was pelleted to remove cell debris; the supernatant containing crude DARPin was aliquoted and stored at −80°C until use in cellular binding assays.
Protein content was determined by Bradford assay. Crude lysate preparations were always handled on ice and subjected to a maximum of three freeze
Cellular binding assays
To analyze specific binding of DARPins to human and NHP CD8, cellular binding assays using Molt4.8 and J76S8ab cells as well as primary human and NHP PBMC (isolated and activated as described) were carried out. In brief, 1 × 105 cells were washed once with wash buffer (phosphate-buffered saline [PBS], 2% FCS, 0.1% NaN3) and incubated with 10 μL of crude DARPin extracts in a total volume of 200 μL for 60 min at 4°C. After incubation, cells were washed twice with wash buffer, stained with fluorescently labeled antibodies, and analyzed by flow cytometry. Screening for CD8 binding by cellular binding assays using cell lines and validation by binding to human and NHP PBMC was carried out at n = 1. Gating strategy for primary cells is depicted in Supplementary Fig. S1.
LV production
LVs were produced in HEK 293T cells via PEI transfection. One day before transfection, 2 × 107 HEK 293T cells were seeded into T175 cell culture flasks in complete culture medium. On the day of transfection, the culture medium was replaced by DMEM supplemented with 15% FCS and 2 mM L-glutamine.
To produce HIV-derived LVs, 0.9 μg of the envelope plasmid pCG-NiV-Gd34mut-L3-targeting domain (encoding one of the eleven targeting domains in conjunction with the modified NiV Gmut) and 4.5 μg of pCAGGS-NiV-Fd22 (encoding for NiV F) were co-transfected along with the packaging construct pCMV-dR8.9 (15.2 μg) and transfer plasmid pSEW-GFP (14.4 μg). For simian immunodeficiency virus (SIV)-derived LVs, pCG-NiV-Gd34mut-L3.-targeting domain (0.9 μg) and pCAGGS-NiV-Fd22 (4.6 μg) were co-transfected with the SIV packaging plasmid pSIV10+ (14.4 μg) and the SIV transfer plasmid pGAE-SFFV-GFP-WPRE (15.1 μg). VSV-LVs were produced by co-transfection of 6.1 μg pMD2.G along with the packaging and transfer plasmids (17.5 and 11.4 μg, respectively).
The DNA was mixed with 2.3 mL DMEM without additives and added to 2.2 mL DMEM containing 140 μL of an 18 mM PEI solution. After an incubation of 20 min, the DNA mix was added to HEK 293T cells. Medium was changed 6–16 h post-transfection to complete DMEM. Two days post-transfection, the vector-containing supernatant was harvested, filtered through a 45-μm filter, and either centrifuged for 5 min at 1,000g to remove cell debris and stored at 4°C, or subjected to concentration by low-speed centrifugation (4,500g) through a 20% (w/v) sucrose cushion for 24 h at 4°C. After concentration, supernatants were discarded; vector pellets were resuspended in PBS and stored at −80°C until further use. Unconcentrated vector supernatants were stored for a maximum of 24 h and used in transduction experiments beforehand.
All DNA amounts and volumes apply to transfection in a T175 cell culture flask. For screening, one T175 flask was used for vector stock preparation whereas large-scale stocks were produced in at least two T175 flasks.
Titration of LV particles and determination of particle numbers
For titration of HIV-derived LV particles, Molt4.8 cells were seeded at 3 × 104 cells per 96 well, subsequently transduced with serial dilutions of vector particles in culture medium, and analyzed for transgene expression by flow cytometry four days later. Titers were calculated based on the dilutions showing linear correlation with the dilution factor. To obtain particle numbers of concentrated HIV-derived or SIV-derived LV stocks, either a p24-specific enzyme-linked immunosorbent assay (ELISA) (HIV Type 1 p24 Antigen ELISA; ZeptoMetrix Corporation, Franklin, MA) or a p27-specific ELISA (SIV p27 Antigen ELISA; ZeptoMetrix Corporation) was performed according to the manufacturer's instructions.
Transduction of cell lines and primary PBMC in vitro and in vivo
Molt4.8 and JE6.1 cells were seeded at 2 × 104 cells per well into 96-well plates before transduction. For transduction of primary human PBMC, 4 × 104 cells per well were seeded into 96-well plates in culture medium with 100 U/mL IL-2, but without activation antibodies. To transduce NHP PBMC, cells were seeded into u-bottom 96-well plates at 1 × 105 cells per well in culture medium supplemented with cytokines and 4 μg/mL protamine sulfate (Sigma-Aldrich) or IL-7. Vectors were prediluted in culture medium with additives or directly added to the cells followed by centrifugation at 850g for 90 min at 32°C for transduction of primary cells. Analysis of transduction efficiency and specificity by flow cytometry were performed at days 4 and 7 post-transduction.
For assessment of in vivo gene transfer efficiency, 6-week-old NSG mice (NOD.Cg.PrkdcscidIL2rgtmWjl/Szj; Charles River Deutschland GmBH, Sulzfeld, Germany) were intravenously (i.v.) injected with 5 × 106 activated human PBMC, followed by an i.v. injection of a total volume of 200 μL of vector or PBS as vehicle control. Seven days post-vector application, blood was taken before sacrifice of the animals, the spleen was removed, and single-cell suspensions were prepared by meshing the spleens through a 45-μm cell strainer. Single-cell suspensions from blood and spleens were stained, subjected to erythrocyte lysis by using PharmLyse buffer (BD Biosciences, Heidelberg, Germany), and analyzed by flow cytometry.
Flow cytometry
For flow cytometry analysis, up to 1 × 105 cells were used for staining. Cells were washed twice with wash buffer (PBS, 2% FCS, 0.1% NaN3) and stained with fluorescently labeled antibodies.
For human PBMC, CD8-APC (clone: BW135/80; Miltenyi Biotec or RPA-T8; BD Biosciences), CD3-PerCP (clone: BW264/56; Miltenyi Biotec), CD3-PacificBlue (clone: UCHT1; BD Biosciences), CD4-PE-Vio770 (clone:VIT4; Miltenyi Biotec), CD4-APC-Cy7 (clone:RPA-T4; BD Biosciences), and/or CD45-VioBlue (clone: 5B1; Miltenyi Biotec) were used. NHP cells were stained with CD8-BV421 (clone: RPA-T8), CD4-PE (clone: L200; both BD Biosciences), and CD3-PE-Vio770 (clone: 10D12; Miltenyi Biotec).
Dead cells were excluded by staining with Fixable Viability dyes (Invitrogen) or Zombie dyes (BioLegend, Koblenz, Germany). After staining, cells were washed twice by using wash buffer and fixed by using PBS supplemented with 1% formaldehyde. Data acquisition was performed on a MACSQuant Analyzer 10 (Miltenyi Biotec) or LSRII (BD Biosciences) instrument, and data analysis was carried out by using the FCS Express, version 6 software (DeNovo Software, Glendale, CA).
Results
Generation of recombinant CD8αα-Fc and heterodimeric CD8αβ-Fc
Before selection of CD8-specific DARPins by ribosome display, soluble, recombinant CD8 proteins had to be produced in a native form as bait for the selection process. Two different bait proteins were generated: a soluble, homodimeric and a soluble, heterodimeric version. The soluble, homodimeric version (CD8αα-Fc) consisted of the extracellular immunoglobulin variable-like domain as well as parts of the hinge region of CD8α fused to the constant region of human IgG1 harboring a C-terminal Avi-tag (Fc). For the generation of a soluble, heterodimeric CD8 receptor (CD8αβ-Fc), so called “knob-into-hole” mutations 18 were introduced into the Fc part (Fcknob and Fchole) avoiding homodimer formation of two Fcknob or Fchole protein chains and cloned in conjunction with the extracellular part of either CD8α (CD8α-Fcknob) or CD8β (CD8β-Fchole) (Fig. 2A, B). After expression of CD8αβ-Fc and CD8αα-Fc in HEK 293T cells in a biotinylated and unbiotinylated form, they were purified to homogeneity by protein A affinity and subsequent preparative size exclusion chromatography. The resulting recombinant Fc proteins showed high purity and homogeneity (Fig. 2C, D). Metabolic biotinylation of CD8αβ- and CD8αα-Fc was confirmed by immunoblot analysis (Fig. 2E). A biotin-specific ELISA revealed that >75% of the proteins were biotinylated. All proteins were produced at yields sufficient for ribosome display and subsequent analyses.
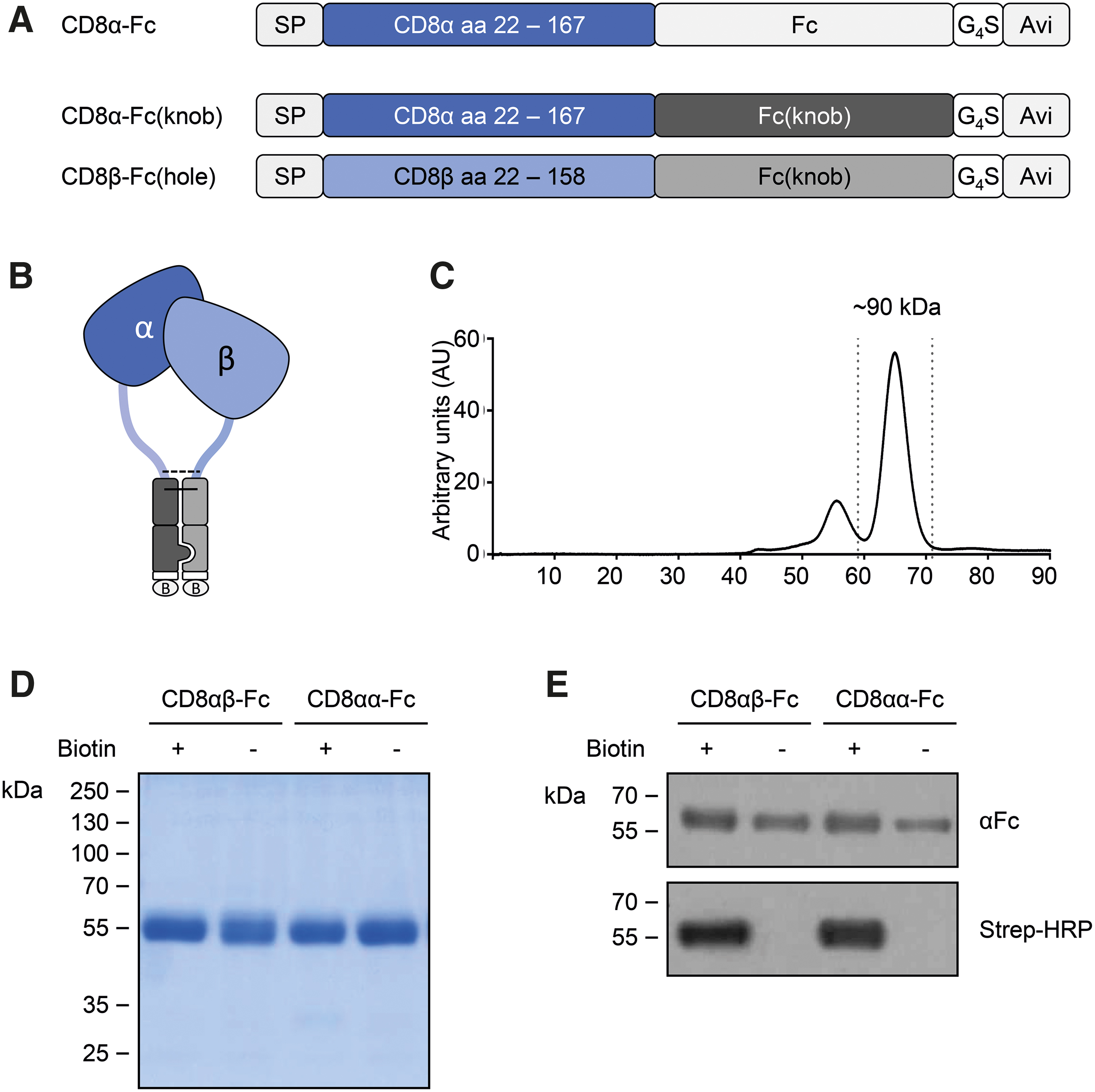
Expression and purification of recombinant human CD8αα-Fc and CD8αβ-Fc fusion proteins.
Selection of CD8-specific DARPins
The previously described combinatorial DARPin libraries VV-N3C 17 and S-N3C 17,20 were screened for CD8-specific DARPins by ribosome display using the generated recombinant CD8 proteins as bait. In total, up to six selection rounds were carried out. The first three as well as the fifth round were performed with immobilized CD8αβ-Fc as bait protein, including preselection steps against neutravidin, streptavidin, and immobilized Fc protein to exclude selection of DARPins with unwanted specificity. The fourth and sixth round were carried out in solution and included a counter-selection with unbiotinlyated CD8αβ-Fc and CD8αα-Fc to select binders with high affinity for CD8αβ.
To identify the best CD8-specific DARPins for targeted gene transfer, the output repertoire was screened in a two-step procedure. First, CD8 binding was evaluated in cell-based assays. In the second step, the gene transfer activity of 10 clones was assessed. In total, 94 DARPin clones obtained from the fifth and sixth selection round were expressed in E. coli and tested for binding to Molt4.8 cells (express CD8αα) and to J67S8ab cells (express CD8αβ). Of these, 30 individual DARPin clones that bound both cell lines equally well (Supplementary Fig. S2) were analyzed for binding to primary human and simian lymphocytes.
All candidates specifically bound to human CD8+ cells, whereas CD8− cells were not decorated above background (Fig. 3A, B). Notably, variations in the cell staining intensity across the DARPin candidates were observed (Fig. 3A). Accordingly, DARPins were grouped into low-, medium-, and high-intensity binders. In the next step, cross-reactivity of these DAPRins to macaque PBMC was assessed. All the identified human CD8-specific DARPins also bound to CD8 on NHP PBMC (Fig. 3C). Of the identified CD8 binders, five DARPins binding human CD8 with an intermediate mean fluorescence intensity (MFI) and five candidates binding with a high MFI were selected for further characterization. Sequencing revealed that each of these 10 candidates had a unique amino acid sequence (Supplementary Fig. S3).
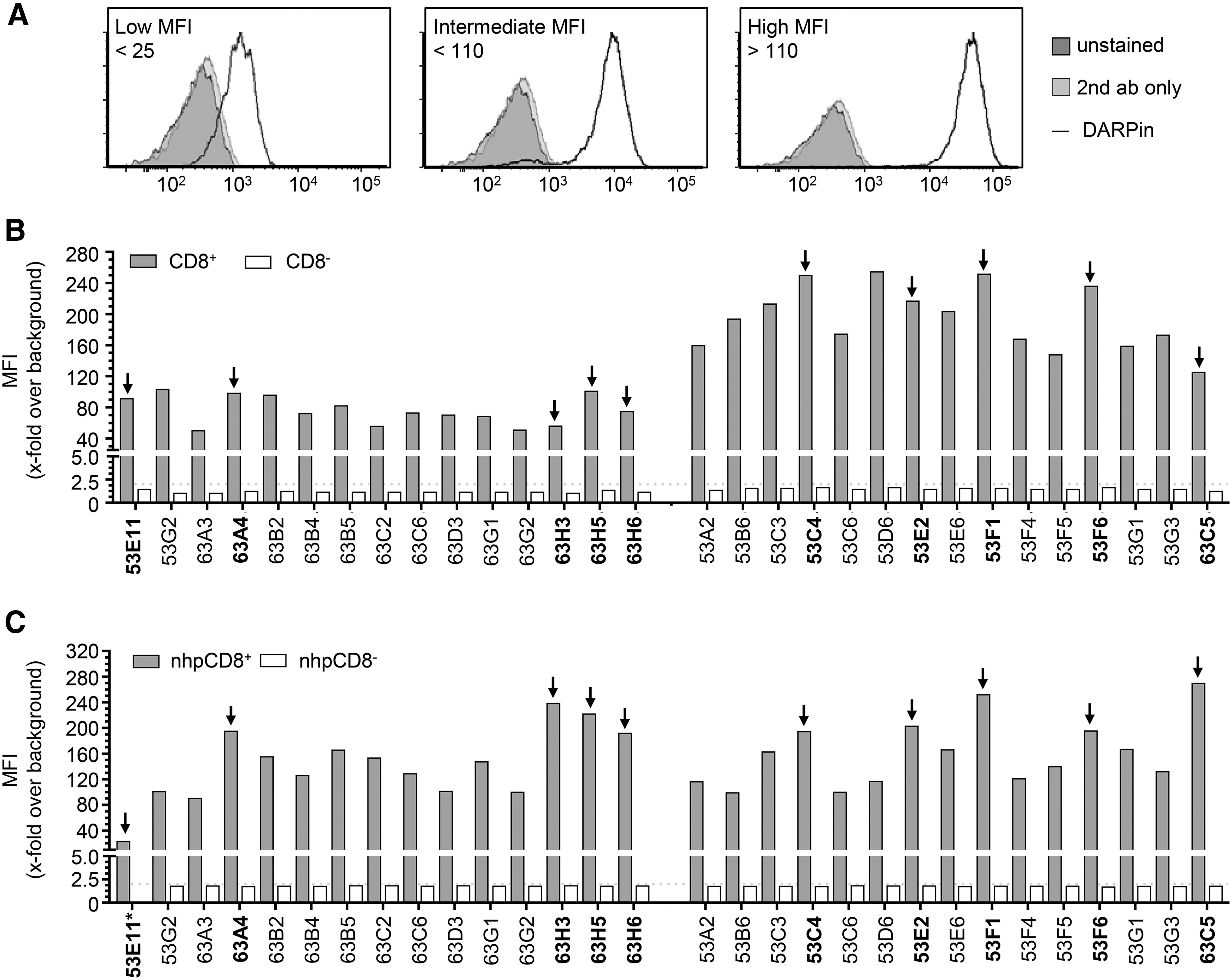
Identification of human and NHP CD8-specific DARPins by cellular binding assays. To identify DARPins specifically binding to CD8, crude Escherichia coli lysates of randomly picked DARPin clones were analyzed for binding to primary human and NHP PBMC via flow cytometry.
In the next step, the capacity of the ten selected DAPRins to mediate gene delivery into CD8+ cells when displayed on the surface of LVs was assessed. For this purpose, the DARPins were C-terminally fused to the modified NiV glycoprotein G, which was then incorporated into HIV- and SIV-derived LV particles along with the NiV fusion protein (F) (Fig. 4A). The HIV-derived vectors were used to transduce activated primary human PBMC, and gene transfer efficiency was evaluated 7 days post-transduction. All selected DARPins were able to mediate gene transfer of the corresponding LV particles, with transduction rates ranging from 6.5% to 25.3% (Fig. 4B). Notably, four out of the ten DARPin-LV particles (53F6-LV, 63A4-LV, 63C5-LV, and 53E11-LV) exhibited gene transfer activities coming close to that of the previously published CD8-targeted LV displaying the OKT8-derived scFv (scFv-LV, 25.7%) under these screening conditions.
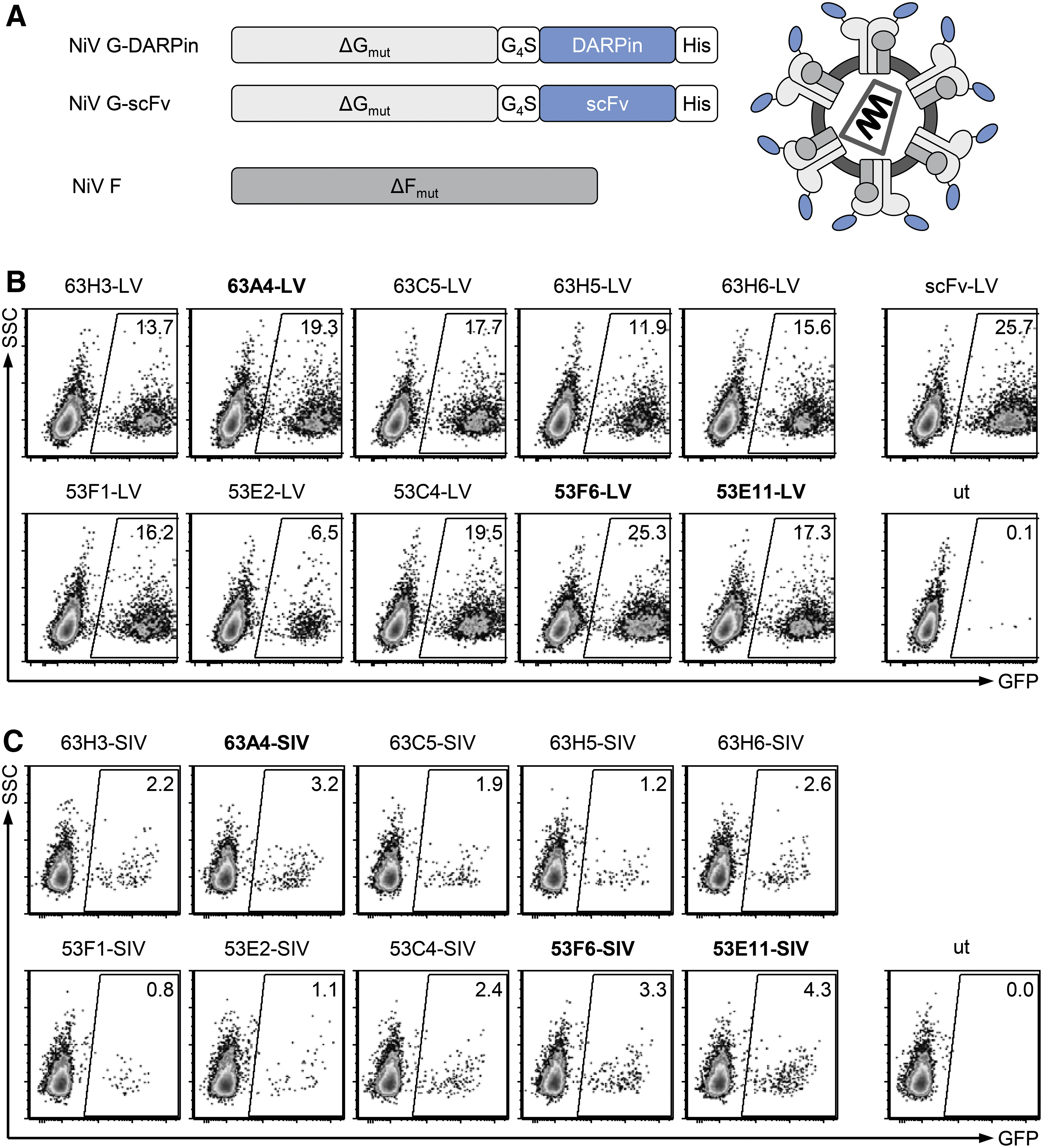
Screening CD8-specific DARPins for transduction of primary human and NHP PBMCs. Identified CD8-binding DARPins were C-terminally fused to the truncated and mutated NiV glycoprotein G (ΔGmut) and subsequently used to produce HIV- or SIV-derived CD8-targeted vector particles. The produced particles were screened for efficient transduction of primary human and NHP PBMC and compared against a CD8-targeted LV displaying the CD8-specific scFv derived from OKT8 (scFv-LV).
Next, the SIV-derived counter parts were evaluated on activated NHP PBMCs. All ten DARPin-SIVs transduced NHP PBMC (Fig. 4C). The highest transduction rates were reached by 63A4-SIV, 53F6-SIV, and 53E11-SIV (3.2%, 3.3%, and 4.3% transduced cells, respectively).
Display of CD8-specific DARPins on LVs increases titers and transduction efficiency in vitro
Based on the screening results, we decided to further evaluate DARPins 63A4, 53F6, and 53E11 as targeting ligands for HIV- and SIV-derived LV particles. Large-scale HIV-derived vector stocks were produced first. Transducing units (TU) present in unconcentrated supernatant and in concentrated stocks were determined on the CD8+ cell line Molt4.8. For comparison, the scFv-LV was produced and titrated alongside the DARPin-LVs.
The unconcentrated titers of the three DARPin-LVs were all clearly above 105 TU/mL, with 53F6-LV and 53E11-LV exhibiting statistically significantly higher titers than the scFv-LV (Fig. 5A, left panel). This increase in titers compared with the scFv-LV was even stronger after concentration, especially for 53F6-LV and 63A4-LV (Fig. 5A, right panel). Some DARPin-LV stocks yielded above 108 TU/mL, which represents an almost one-log increase in titer over the scFv-LV vector. Importantly, the particle number was comparable for all concentrated vectors (Fig. 5B), thus excluding a higher concentration of LV particles as reason for the increase in titer. In addition, a similar degree of targeting domain incorporation into the vector particles was demonstrated by immunoblot blot analysis (Supplementary Fig. S4A).
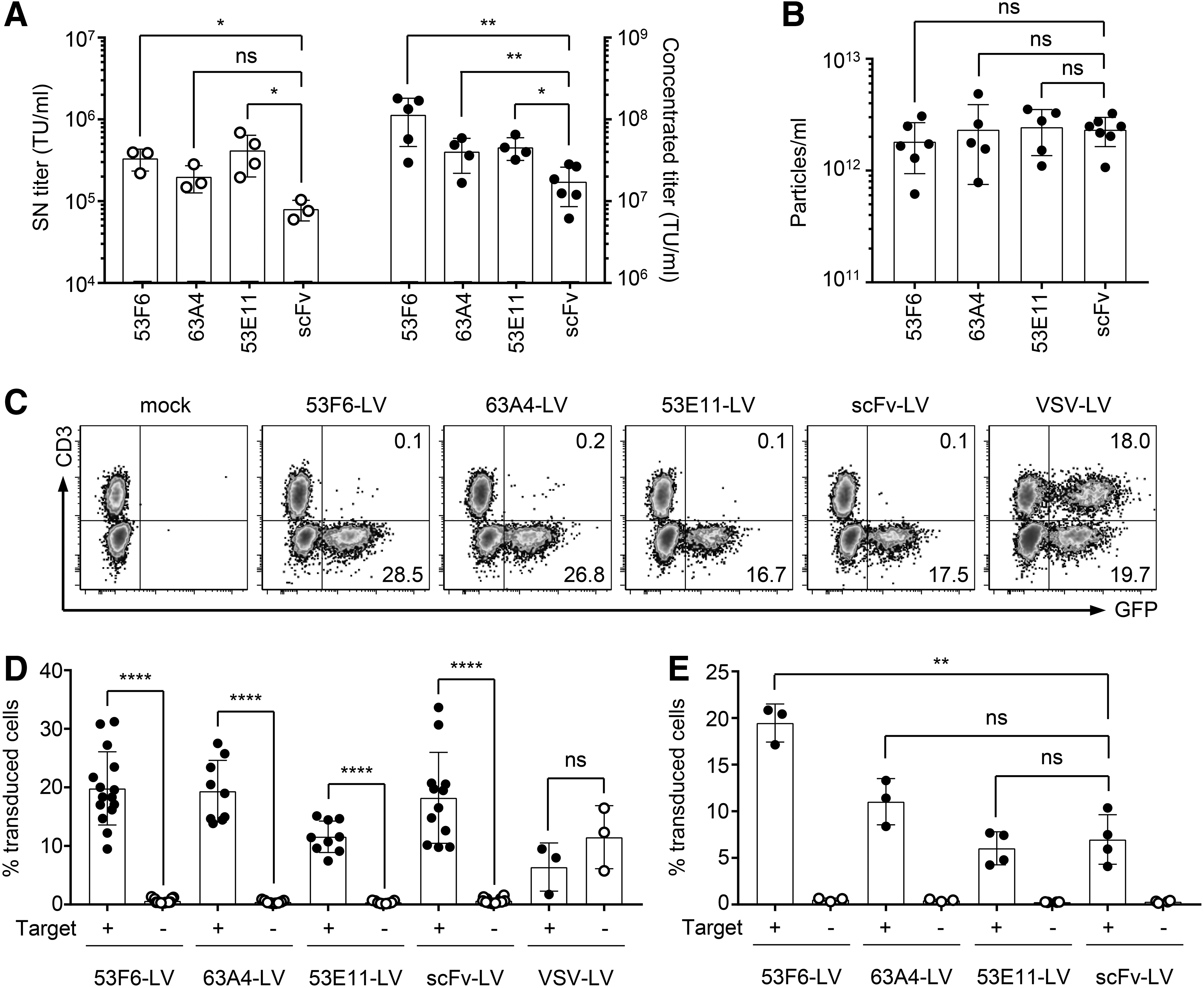
Characterization of candidate DARPin-LVs.
Further on, the selectivity of the vectors was evaluated and compared with scFv-LV on cell lines and primary cells. In the first step, selectivity was assessed by transduction of a mixture of CD8+ Molt4.8 and CD8− JE6.1 cells. CD3 was used as a marker to discriminate between Molt4.8 (CD3−) and JE6.1 (CD3+) cells. All three candidate DARPin-LVs as well as the scFv-LV proved to be selective for CD8+ cells, as they exclusively transduced the CD8+, CD3− Molt4.8 cells (Fig. 5C). In contrast, the non-targeted control vector VSV-LV, pseudotyped with the VSV G protein, transduced both cell populations equally well (Fig. 5C). The selectivity of the DARPin-LVs was further confirmed on activated primary human PBMC by using CD4 as a marker for counterstaining, whereas VSV-LV resulted in equal transduction of target positive and target negative cells (Fig. 5D and Supplementary Fig. S4B). Although differences in transduction rates between the CD8-LVs were minimal when applied at equal multiplicity of infection, they were more pronounced at equal particle numbers. Then, transduction by 53F6-LV resulted in significantly more green fluorescent protein (GFP) positive target cells compared with 53A4-LV and 53E11-LV as well as the scFv-LV benchmark vector (Fig. 5E; Supplementary Fig. S4C). Notably, the small population of CD4+ cells transduced by all targeted vectors are likely CD4/CD8 double-positive cells present in the PBMC culture.
DARPin-LVs can efficiently transduce NHP cells in vitro
Having shown successful transduction of human CD8+ PBMC mediated by the selected DARPins, we next compared their activities with those of the scFv in the context of SIV-derived vectors and the transduction of primary CD8+ NHP cells. As observed for their HIV-derived counterparts, particle numbers in the SIV-derived vector stocks did not significantly differ (Fig. 6A), which indicates that production of all vectors was similarly efficient. Transduction of primary NHP cells with equal particle numbers revealed that all DARPin-SIVs mediated stable GFP expression exclusively in the CD8+ cell compartment 7 days post-transduction (Fig. 6B, C). Again, the vector displaying DARPin 53F6 showed the highest transduction efficiency, with an average of 6.4% transduced CD8+ T cells. The other two candidate vectors transduced 2% to 3% of the CD8+ population. Surprisingly, the SIV vector displaying the CD8-specific scFv did not transduce the NHP cells at all. Thus, all three DARPins can be used as a targeting domain to selectively transduce the CD8+ cell compartment of human and NHP PBMCs. Vectors displaying DARPin 53F6 outperformed the other evaluated LVs in terms of potency. Consequently, this DARPin was chosen as a targeting domain for further in vivo assessment.

Characterization of candidate DARPin-SIV-LVs. The three candidate DARPins 53F6, 63A4, and 53E11 were used for SIV retargeting to CD8+ NHP cells.
53F6-LV shows a threefold-increased in vivo gene delivery efficiency compared with scFv-LV
Next, the capacity of 53F6-LV to improve in vivo gene delivery on i.v. application was assessed and compared with the benchmark scFv-LV by using GFP as a reporter. For this purpose, NSG mice were transplanted with activated human PBMC followed by systemic vector application one day later (Fig. 7A). Two different doses of 53F6-LV were injected. The first group of animals received 8.2 × 106 TU of either scFv-LV or 53F6-LV in a total volume of 200 μL PBS. This corresponded to the maximal available dose of the scFv-LV. The second group was injected with 200 μL of 53F6-LV stock per animal, which corresponded to 2.5 × 107 TU. Notably, this dose corresponded in its total physical particle number to that of the scFv-LV group (4.7 × 1011 physical particles). PBS served as a vehicle control. Seven days post-vector application, animals were sacrificed and blood and spleen were analyzed for transduction by flow cytometry. As expected, GFP expression could be detected exclusively in the CD8+ compartment of scFv-LV-treated animals in both organs. Likewise, 53F6-LV treatment led to exclusive transduction of CD8+ cells in blood (Fig. 7B, C) and spleen (Fig. 7B, D), showing that the vector is as specific as its scFv-based counterpart also in vivo. When equal TU had been administered, transduction efficiencies in scFv-LV- and 53F6-LV-treated groups were similar, with an average of 5% of all CD8+ cells in blood and 7% in spleen being GFP+. When the maximal dose of 53F6-LV was injected, however, the transduction rate increased significantly to almost 20% of all CD8+ cells, with a very low variability between the individual animals. This corresponded to a more than threefold increase in transduction rate compared with the scFv-LV. Importantly, specificity was retained, with virtually no transduction detected in the CD8− compartment in both tested organs. Thus, 53F6-LV is superior to scFv-LV in transducing CD8+ cells not only in vitro but also in vivo. Taking into account also its ability to transduce NHP cytotoxic T cells, 53F6 clearly outperformed the already established scFv as a targeting domain for HIV- and SIV-derived vectors.
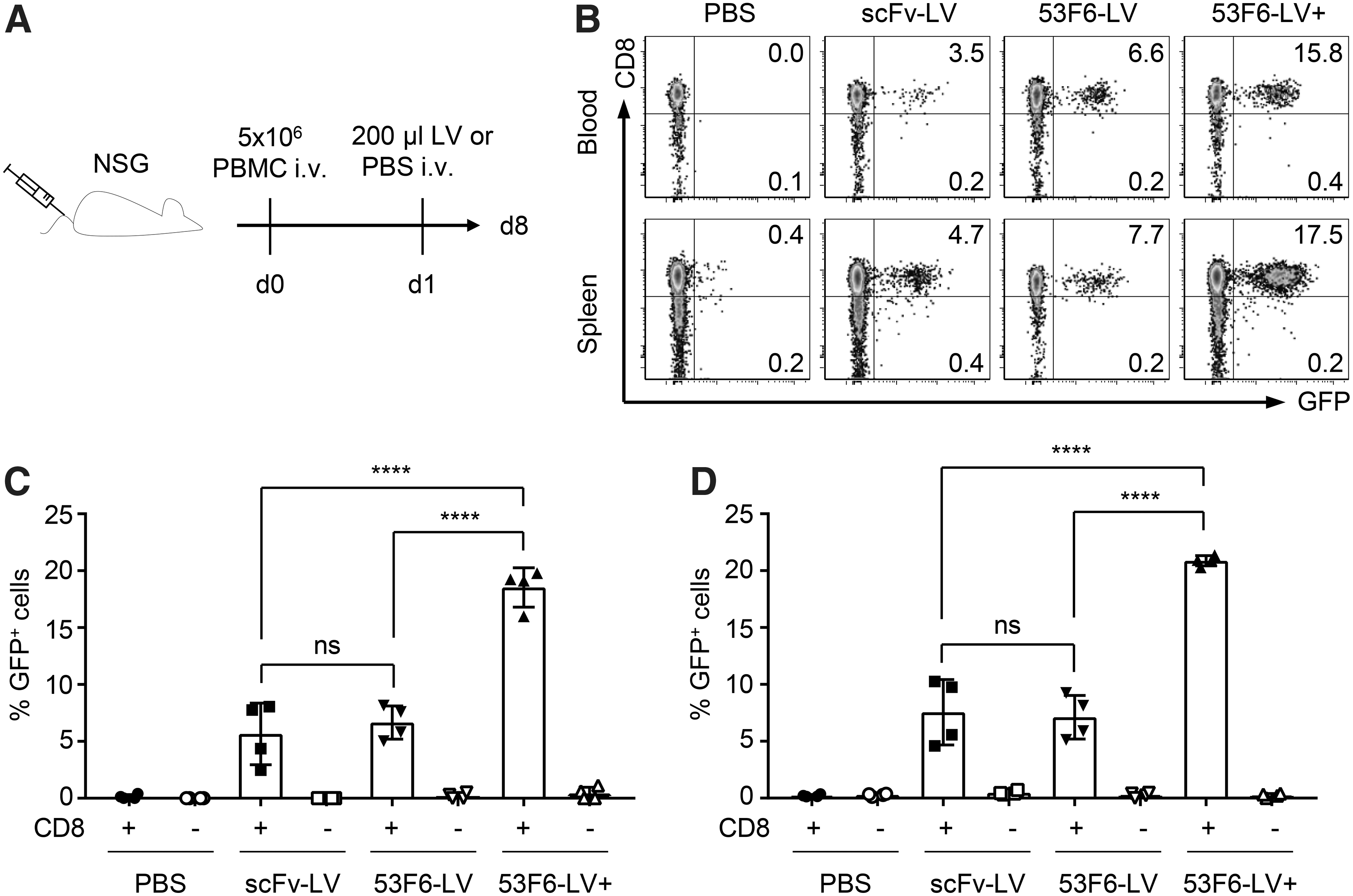
Highly efficient and selective in vivo gene delivery by 53F6-LV.
Discussion
This is, to our knowledge, the first description of CD8-recognizing DARPins. Offering natively folded CD8 as bait in the selection and involving primary human and NHP lymphocytes for the screening of the output pool of potential binders were crucial for the successful outcome. Although the selected DARPins may be of value for many applications in immunotherapy, we focused here on their use for targeted gene delivery to CD8+ T lymphocytes.
Based on the overall high sequence homology of CD8 between human and NHP (over 90%), the likelihood to identify cross-reactive binders was expected to be high. Indeed, our screening procedure was very effective in enriching CD8-specific DARPins from the library that were cross-reactive for NHP CD8. Although we used CD8αβ as bait in the selection, the vast majority of selected candidates recognized the CD8α chain. However, potential CD8β chain binders were present in our selection as well and will be evaluated for their suitability as a targeting domain on LVs in future (Supplementary Fig. S2).
N-terminal fusion of DARPins to the glycoprotein used for vector pseudotyping and expression in a mammalian cell line can impair protein surface expression or the binding behavior of the DARPin to its respective receptor, as demonstrated during the selection process of GluA4-specific DARPins. 17 Interestingly, all ten selected CD8-DARPins allowed generation of functionally active LV particles that were able to transduce human and NHP T cells. This was not the case for the LV displaying the CD8-specific scFv, which mediated transduction of human but not NHP T lymphocytes. This was quite surprising since the parental OKT8 antibody is described to be cross-reactive with rhesus CD8. 21 Whether the OKT8-derived scFv lost its ability to recognize rhesus CD8 or whether the functional titer of the generated scFv-SIV vector stocks compared with their DARPin counterparts was too low to result in NHP T cell transduction remains to be elucidated.
Besides the cross-reactivity, substantially increased functional titers were achieved with the selected DARPins. A possible explanation for this phenomenon could be the greater stability of DARPins in comparison to scFvs. In particular, scFvs derived from unstable antibody subgroups tend to aggregate and require protein engineering to increase their stability. 19 Indeed, the OKT8-derived scFv framework residues had to be optimized to act as an LV targeting domain. 10 In contrast, DARPins are known to be very stable even under elevated temperatures and are less prone to aggregation or unfolding. 14 Consequently, the concentration procedure could be better tolerated by DARPin-LVs compared with scFv-LVs, resulting in more functionally active LV particles. This is supported by the fact that the particle numbers of the concentrated stocks of all targeted vectors were in the same range, thus excluding a higher concentration of LV particles as reason for the increase in titer.
Vector stocks with a high functional titer are especially important with regard to in vivo applications. In this setting, the volume that can be applied per treatment is limited and rather small. With a higher functional titer, more functional active particles can be applied per injection, resulting in increased in vivo gene transfer efficiency. In this regard, the true potential of the novel selected CD8-specific DARPin-LV 53F6-LV was revealed while comparing equal particle numbers in in vitro and in vivo experiments. Notably, for in vitro experiments, the transduction efficiency of 53F6-LV can be even further increased by using the transduction enhancer Vectofusin-1. This enhancer was recently shown to substantially improve the gene transfer of retargeted LV vectors while retaining their target receptor specificity. 22
Taken together, the selected 53F6-LV has an improved transgene delivery rate to human cytotoxic T cells while retaining its selectivity and is the first described vector that is able to specifically transduce NHP cytotoxic T cells. This achievement is especially important in light of the recent success in delivering CARs directly in vivo into CD8+ cytotoxic T cells. 11 Thus, 53F6-LV is not only suited to improve gene transfer rates in such experiments, but it will also enable testing the feasibility and safety of in vivo CAR delivery in an NHP model in the future. Importantly, in combination with our CD4-targeted LV, 16 genetic engineering of CD8 as well as CD4 T cells could be achieved in vivo. This is especially relevant with regard to the superior antitumor reactivity of CAR T cells derived from CD8 and CD4 cells compared with products consisting of CD8+ CAR T cells alone. 23
Footnotes
Acknowledgments
The authors like to thank Markus A. Seeger for providing the S-N3C library and helpful discussions.
Author Disclosure
A.M.F., J.H., and C.J.B. are listed as inventors on a patent application related to this manuscript. All other authors declare that no competing financial interests exist.
Funding Information
This work was supported by grants from the European Union (Horizon 2020 Framework Programme [H2020], CARAT [667980]) and the LOEWE Center Frankfurt Cancer Institute funded by the Hessen State Ministry for Higher Education, Research, and the Arts (III L 5—519/03/03.001—[0015]).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.