
Abstract
Tight control of transgene expression is key to ensure the efficacy of a wide range of gene therapy interventions, in which the magnitude and duration of gene expression have to be adjusted to therapeutic needs, thereby limiting secondary effects. The development of upgraded strategies to link transgene expression to pathological stress episodes is an unmet need in gene therapy. Here, we propose an expression strategy that associates transgene expression to an intracellular stress coping mechanism, the unfolded protein response. Specifically, we harnessed the cis elements required to sustain the noncanonical splicing of X-box binding protein 1 (XBP1) messenger RNA (mRNA) in response to the dysfunction of the endoplasmic reticulum (ER), a situation commonly known as ER stress, to drive the expression of heterologous genes. Since ER stress features a wide variety of pathological conditions, including viral infections, cancer, or metabolic disorders, this new expression module stimulates the synthesis of therapeutic genes as a response to cellular damage, and ensures their expression only when necessary. Validation of this inducible expression system was performed in vitro and in vivo, and its potential to limit/inhibit viral infections has been shown in proof-of principle experiments.
Introduction
The regulation of recombinant gene expression is a major challenge in cell biology and gene therapy. 1 Most inducible systems are triggered by exogenous stimuli, demanding an active exogenous intervention. Aimed to restore cellular and tissue homeostasis, gene therapy can (1) complement the lack of specific gene products in diseases caused by loss-of-function mutations, 2,3 (2) prevent the pathogenic effects of gene mutations, 4 (3) boost or modulate the immune response, 5,6 or (4) it can serve to enhance cellular/neuronal survival. 7
Regardless of the therapeutic benefit achieved, gene therapy interventions frequently require the tight tuning of transgene production, such that its expression meets the therapeutic needs in time and amplitude. In fact, excessive or untimely expression of transgenes has been shown to compromise the efficacy of gene therapy displaying unwanted, toxic side effects. 8,9 In addition, in the context of certain pathologies such as viral infections, treatments based on general antiviral cytokines such as interferons (IFNs) can have a detrimental effect on the patients. 10 –13 Therefore, the development of a new generation of gene therapy vectors able to constrain the gene expression to cellular stress is essential to optimize such adjustment.
Deficiencies in protein folding within the endoplasmic reticulum (ER) lead to a dysfunctional state, known as ER stress, a process shared by a wide range of pathologies, including cancer, metabolic and neurodegenerative diseases, or viral infections. Protein folding deficiencies promote a set of ER-to-nucleus signaling mechanisms known as the unfolded protein response (UPR). 14 At the core of the UPR, the noncanonical processing of UPR transcription factor X-box binding protein 1 (XBP1) messenger RNA (mRNA) enables the establishment of a transcriptional response to upgrade protein folding and sustain cell survival and proliferation. 15
XBP1 splicing is initiated by the ER stress sensor/transducer IRE1α. IRE1α is an ER-resident multifunctional transmembrane protein that contains kinase and endonuclease domains at its cytosolic side. ER stress triggers the clustering of IRE1α kinase in ER foci (which results in the autophosphorylation of the protein) and leads to the activation of its RNAase domain, such that IRE1α is then able to excise a nonconventional 26-nucleotide intron within the open reading frame of the XBP1 mRNA. 15 The resulting exons are then ligated by the transfer RNA (tRNA) ligase RtcB, thereby generating an mRNA with a new open reading frame that allows efficient expression of XBP1s. 16 This molecular splicing mechanism was used in the past to develop fluorescent/luminescent ER stress reporters. 17 Up to date, all inducible systems used in gene therapy have been based on transcriptional regulation. Here, we propose to harness a posttranscriptional mechanism, the splicing of XBP1 mRNA, to enforce expression of the transgene under conditions in which the ER is dysfunctional.
Over 257 million people worldwide are chronically infected with hepatitis B virus (HBV) and 1 million of them die every year due to progression of liver disease. 18 Current therapeutic options are far from optimal, require lifelong treatment and are associated with important side effects. 19 Thus, more efficient and safer therapeutic strategies are needed. Upon infection of hepatocytes, HBV hijacks the protein folding machinery of the ER for the efficient production of viral glycoproteins, thereby triggering UPR activation. 20 At the same time, HBV components inhibit innate immune signaling and prevent the development of effective antiviral responses. In HBV patients, pegylated interferon alpha (IFNα) provides an effective treatment with durable benefits; however, systemic IFNα administration can induce severe adverse side effects, which establish the need to regulate therapeutic IFNα levels. 19
Adeno-associated viruses (AAVs) constitute one of the best current options for gene therapy as the combination of AAV serotypes and tissue-specific promoters ensures specific, safe, and durable transgene expression in target organs. 21 In this study, we have generated a set of recombinant AAVs that allowed the expression of reporter or therapeutic genes in the liver of mice in an XBP1-ER stress-dependent manner. We chose to tune the expression of an antiviral factor, murine IFN-α1, and assess its capacity to combat HBV infection in a transgenic HBV (HBV-Tg) mouse model. We demonstrate that reporter or therapeutic transgenes can be induced effectively in hepatocytes in a way that links transgene expression to liver damage.
Materials and Methods
Cell culture experiments
HEK293T and HeLa cells lines were obtained from ATCC. Cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Fisher Scientific, Madrid, Spain) supplemented with 10% fetal bovine serum, 1% Na-pyruvate, 1%
Luciferase reporter assay
To determine luciferase (LUC) reporter expression, HEK293T cells were seeded in 12-well plates (2–5 × 105 cells/well) and, using Lipofectamine 2000, were transfected with the plasmid expressing luciferase, as well as the control plasmid pRL-TK that drives the expression Renilla luciferase. Transfections were performed in triplicates. Cells were lysed 24 h posttransfection and the luminescence of firefly luciferase and Renilla was determined using the dual-luciferase assay kit (Promega) according to the manufacturer's instructions. The relative pathway activation was calculated as the ratio between the firefly and Renilla luminescences.
RNA isolation and real-time polymerase chain reaction
RNA was extracted from cells by the guanidine isothiocyanate and phenol/chloroform method (TRIzol; Invitrogen). Liver RNA was obtained by magnetic separation (Maxwell 16V RNA isolation kit; Promega) from 20 mg of frozen tissue. One to two micrograms of total RNA was treated with DNAse I (Qiagen) and used for subsequent retrotranscription. Fifty to one hundred nanograms of total complementary DNA (cDNA) was used in real-time polymerase chain reaction (PCR) using SYBR Green (BIORAD, Madrid, Spain). The sequence of primers used in real-time PCR is detailed in Supplementary Table S1.
Production of AAVs
Serotype 8 AAV vectors were produced in HEK293T (ATCC CRL-3216) packaging cells as described. 4 Briefly, for each production, the shuttle vector for AAV and the packaging plasmid pDP8.ape (Plasmid factory) were cotransfected into HEK293T cells. The cells and supernatants were harvested 72 h after transfection and the virus was released from the cells by three rounds of freezing–thawing. Crude lysate from all batches was then treated with DNAse and RNAse (0.1 mg per p150 culture dish) for 1 h at 37°C and then kept at −80°C until purification. Purification of crude lysate was performed by ultracentrifugation in OptiPrep Density Gradient Medium-iodixanol (Sigma-Aldrich). Thereafter, iodixanol was removed and the batches concentrated by passage through Amicon Ultra-15 tubes (Ultracel-100K; Merck Millipore). For virus titration, viral DNA was isolated using the High Pure Viral Nucleic Acid kit (Roche Applied Science). Viral titers in terms of viral genome per milliliter (vg/mL) were determined by quantitative PCR (Applied Biosystems) using specific primers.
The integrity of single-stranded AAV genomic DNA packaged into viral particles was determined in alkaline DNA electrophoresis. Briefly, 1.2 μg of genomic DNA was loaded into 0.8% agarose alkaline gels and run overnight at 4C and 20 V. Then, gels were equilibrated for 1 h in 50 mM Tris pH 8.0 at room temperature, and for 2 more h in 100 mM NaCl. DNA was stained with SYBR Gold stain for 30 min and visualized in a ChemiDoc apparatus (BIORAD).
Animals and manipulations
HBV-Tg mice were kindly provided by Francis V. Chisari. 22 Mice were bred and maintained under pathogen-free conditions at the animal facility of the University of Navarra. For experiments, they were matched for age (6–10 weeks), sex (male), and levels of HBV DNA and HBV surface antigen (HBsAg) in serum. Age-matched C57BL/6 wild-type males were purchased from Harlan Laboratories (Barcelona, Spain). The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra.
For all procedures, animals were anesthetized by intraperitoneal injection of a mixture of xylazine (Rompun 2%; Bayer) and ketamine (Imalgene 500; Merial) (1:9, vol/vol). HBVTg mice were injected intravenously with AAV at a dose of 1 × 1012 vg/kg.
Blood was collected by bleeding from the retro-orbital plexus, and serum samples were obtained by centrifugation of total blood. IFNα levels were determined by using the Mouse IFN Alpha ELISA Kit (PBL Assay Science).
Immunohistochemical analyses
Liver sections were fixed in 4% paraformaldehyde (Panreac), embedded in paraffin, sectioned (thickness, 5 μm), and stained with hematoxylin and eosin. Immunohistochemistry for HBV core antigen (HBcAg; B0586; Dako, Glostrup, Denmark) was performed using the EnVision system (Dako) according to the manufacturer's recommendations.
Statistical analysis
Statistical analysis was performed using PRISM version 5.0 (GraphPad). Data are presented as mean ± standard deviation. Comparisons between two groups were made using a two-tailed unpaired t-test. Multiple groups were compared using analysis of variance followed by the Bonferroni posttest. Statistical significance was assigned to p-values <0.01 (***), <0.1 (**), or <0.5 (*).
Results
Development of a novel gene expression cassette to link transgene expression to ER stress
We optimized a gene expression vector based on XBP1 splicing to condition transgene expression to ER stress. The chimeric mRNA contains the following coding elements (from 5′ to 3′): (1) the FLAG epitope, in frame with (2) a cDNA fragment encompassing nucleotides 372–597 of human XBP1 mRNA, which contains the intron processed by IRE1α, as well as the flanking sequences needed for its efficient splicing under ER stress conditions, (3) the autoproteolytic peptide 2A of the foot and mouth disease virus (FMDV), and (4) the transgene coding sequence (Fig. 1A). The open reading frame (ORF) of the transgene was placed in such a way that it can only be translated after the mRNA is spliced. 15 The XBP1 intron is recognized by IRE1α as a highly conserved, double hairpin structure. Of note, transcription of this chimeric mRNA is driven by the constitutive, ER stress-insensitive elongation factor 1 alpha (EF-1α) promoter, and therefore, the regulation of transgene expression is strictly posttranscriptional.
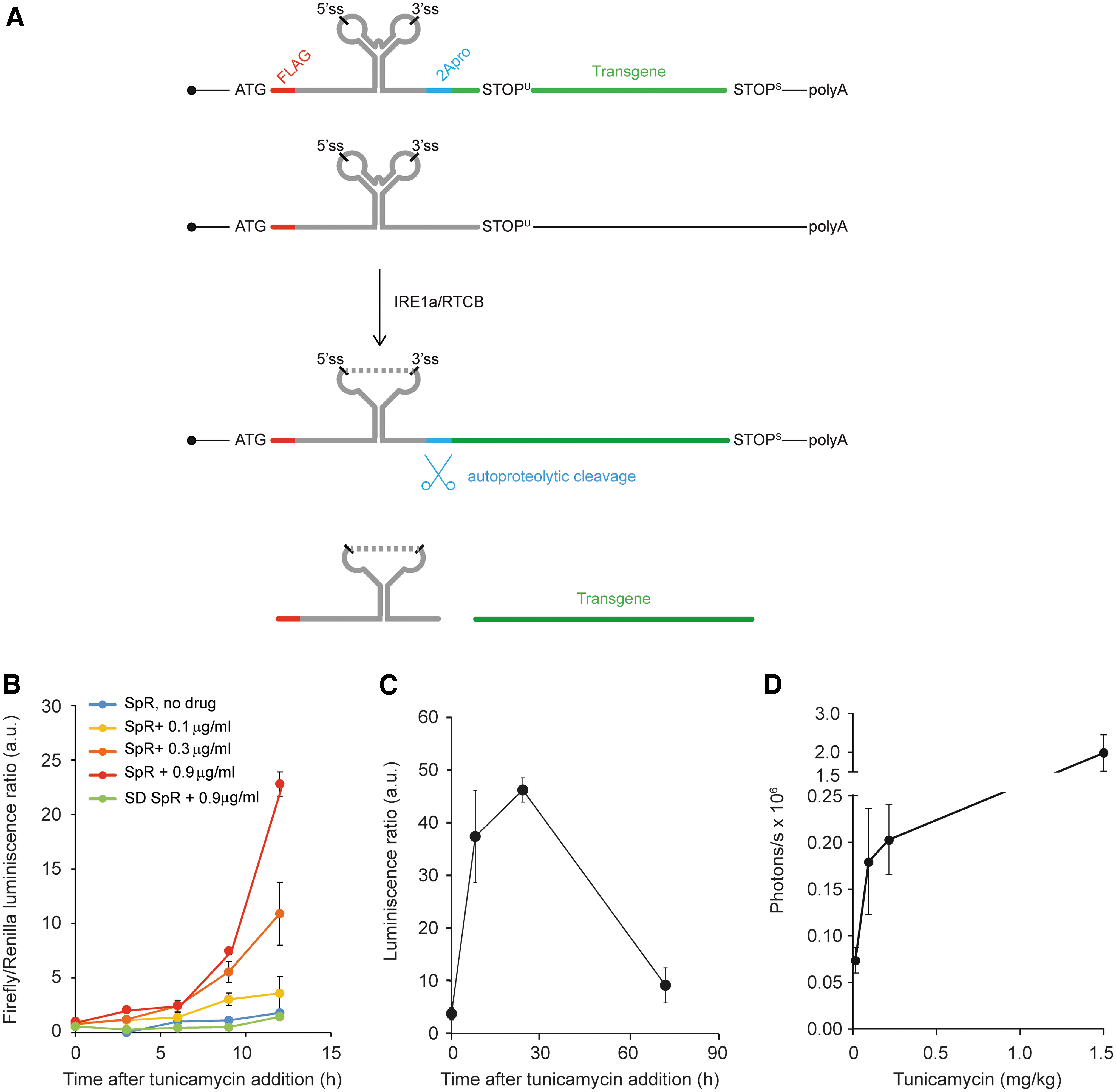
A UPR splicing-based reporter vector to longitudinally monitor hepatic ER stress.
For proof-of-principle validation of its regulatory properties, we used the firefly luciferase reporter as a transgene. The polypeptide translated from the spliced form of this mRNA contains a short N-terminal peptide derived from the translation of the XBP1 splicing element. To release the reporter/therapeutic protein of interest as an untagged polypeptide, a 2A autoproteolytic site originating in the FMDV was inserted right after the XBP1 minimal sequence. As a final modification of the XBP1-2A-LUC construct, we mutated two internal AUG sequences located within the XBP1 sequence and at the beginning of AUG codon in the luciferase ORF to prevent the residual translation of luciferase protein from spurious AUG codons. We named this vector splicing reporter (SpR). A splicing-deficient version of this reporter bearing point mutations at the 5′ and 3′ splice sites was synthesized as control and named SD. When transfected in HEK293 cells, SpR yielded a robust increase in LUC-derived luminescence in response to a well-established inducer of ER stress, the N-linked glycosylation inhibitor tunicamycin (Fig. 1B). This upregulation of LUC levels was a bona fide consequence of SpR UPR splicing, since the SD version only produced background levels of luminescence. Importantly, the kinetics of luminescence production was dependent on the dose of tunicamycin administered.
We then generated the recombinant hepatotropic AAV8 vector carrying the SpR expression cassette that was injected intravenously into C57BL/6 wild-type mice at a dose of 1 × 109 vg/g. The sequence specifications of this construct are described in the Supplementary Data. Two weeks later, luciferase expression was analyzed before and 10, 28, and 72 h after tunicamycin administration. A single administration of tunicamycin elicited a sharp increase in hepatic luminescence (Fig. 1C). In fact, SpR-derived luminescence was proportional to the dose of tunicamycin administered (data not shown). Together, these experimental results validated the capacity of this expression cassette to express hepatic transgenes only under ER stress conditions.
Expression of a therapeutic gene under ER stress conditions
Once the capacity of SpR AAV vector to drive LUC expression in vivo was confirmed, we replaced the LUC ORF by the coding sequence for murine IFN-α1 (NM_010502.2). To control the specific UPR-driven expression, the first IFN-α AUG was removed to force the translation of a single Flag-XBP-1-2A-IFN-α1 ORF.
To test whether XBP1-IFNα could specifically trigger the production of IFNα upon ER stress induction, we monitored the serum levels of IFNα in mice transduced with AAVs expressing either the splicing competent version of the XBP1-IFNα, named SC XBP1-IFNα, or a splicing-deficient version, named SD XBP1-IFNα. Both vectors were administered intravenously to C57BL/6 mice at a dose of 1 × 1012 vg/kg. Two weeks later, half of the mice (n = 5) received an intraperitoneal injection of tunicamycin and half of the mice received vehicle (dimethyl sulfoxide [DMSO]). Mice were sacrificed 8 h later and the levels of unspliced (XBP1-IFNu) and spliced XBP1-IFN (XBP1-IFNs) mRNA were determined. In control mice (DMSO), the levels of unspliced XBP1-IFNα mRNAs were slightly higher in SD XBP1-IFNα than in SC XBP1-IFNα, indicating a similar transduction efficacy for both vectors (Fig. 2A, left panel). As expected, tunicamycin treatment triggered the splicing of SC XBP1-IFNα, which resulted in the reduction in the levels of unspliced XBP1-IFNα levels, and the concomitant increase of spliced RNA levels (Fig. 2A, right panel). On the contrary, RNA from the SD mutant RNA was not spliced and higher levels of the XBP1-IFNu precursor RNA accumulated. In line with this finding, an increase in the levels of serum IFNα levels was observed only in mice receiving AAV SC XBP1-IFNα and levels were strongly boosted by an acute treatment with tunicamycin. AAV SD XBP1-IFNα did not yield detectable levels of IFNα (Fig. 2B).
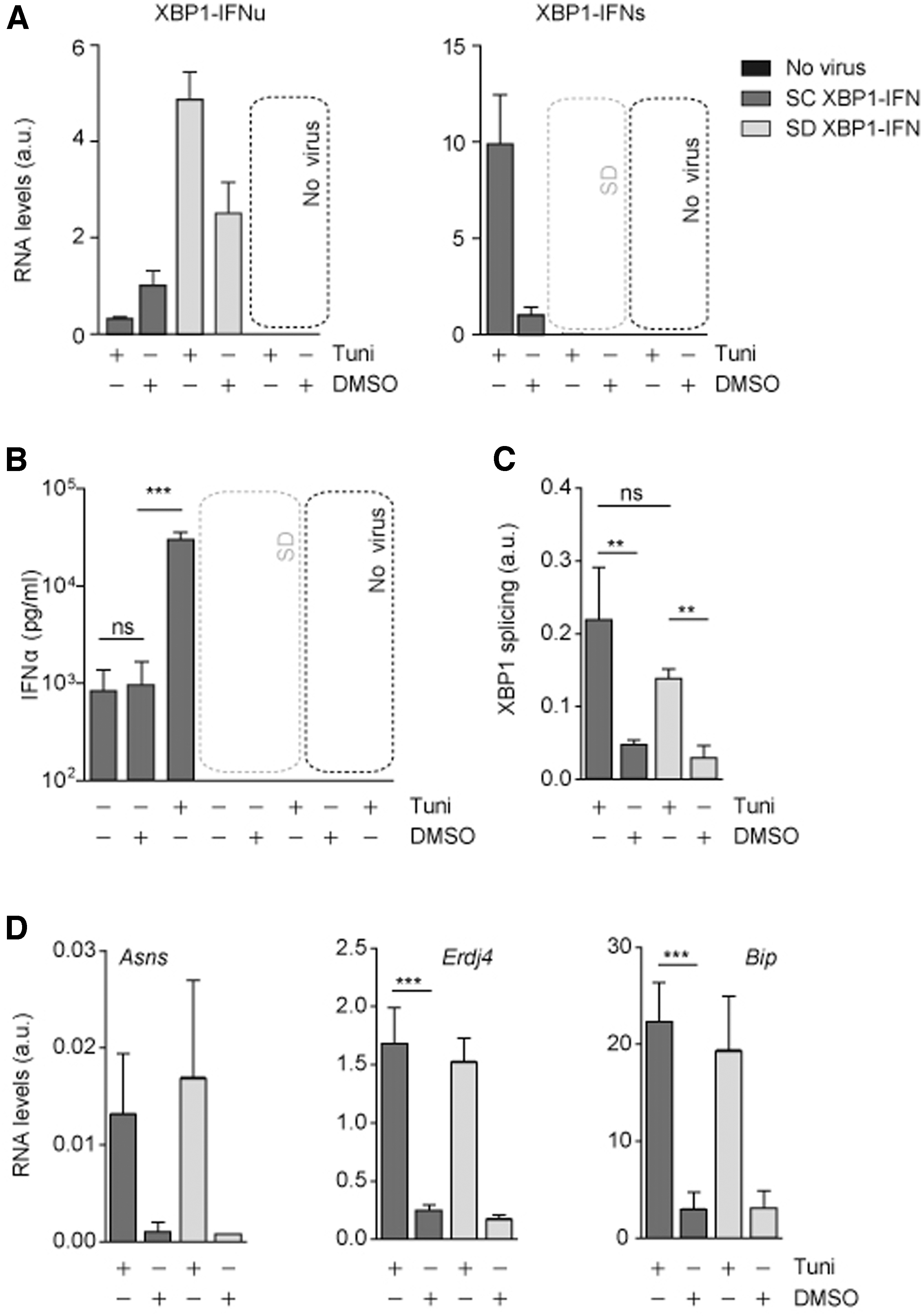
XBP1 splicing-dependent expression of IFNα does not interfere with the hepatic response to ER stress.
The ectopic overexpression of XBP1-like mRNA molecules may interfere with the endogenous response to ER stress (for instance, by competing with the endogenous XBP1 mRNA for the splicing reaction). If so, these vectors could compromise physiological responses to ER stress. To rule out this possibility, we examined the processing of endogenous XBP1 by quantitative reverse-transcriptase followed by polymerase chain reaction (RT-PCR) (Fig. 2C), and confirmed that endogenous XBP1 occurred with similar efficiencies in the livers transduced with either SC XBP1-IFNα or SD AAV XBP1-IFNα variants. In line with this observation, the mRNA levels of well-established UPR target genes such as Asns, Erdj4, or Bip displayed a similar increase after tunicamycin transduction both in SC XBP1-IFNα- and SD AAV XBP1-IFNα-transduced animals (Fig. 2D).
UPR-regulated IFNα expression has no effect over the inducibility of the system but induces an effective antiviral response
As the capacity of AAV XBP1-IFNα to produce IFN under ER stress conditions was established, we decided to test if XBP1-IFN expression could affect the transduction or stability of therapeutic AAV vectors. To that aim, we either transduced AAV SpR alone or we cotransduced it with AAV SC XBP1-IFN and tested whether luciferase expression from the SpR AAV could be affected by XBP1-IFN. As expected, stimulation of ER stress with tunicamycin promoted a sharp increase in the serum levels of IFNα only in AAV SC XBP1-IFNα-transduced mice (Fig. 3A). We observed a similar upshift in the levels of SpR-derived luminescence in all SpR- and SpR+SC XBP1-IFNα-treated mice (Fig. 3B), indicating that IFNα does not interfere with the inducibility of its own promoter. This is in line with the notion that IFNα expression does not interfere with endogenous ER stress responses. Furthermore, the robust expression of IFNα promoted a significant increase in the mRNA levels of IFN-stimulated genes, such as Ip10, Isg15, Oas2, or Pkr (Fig. 3C).
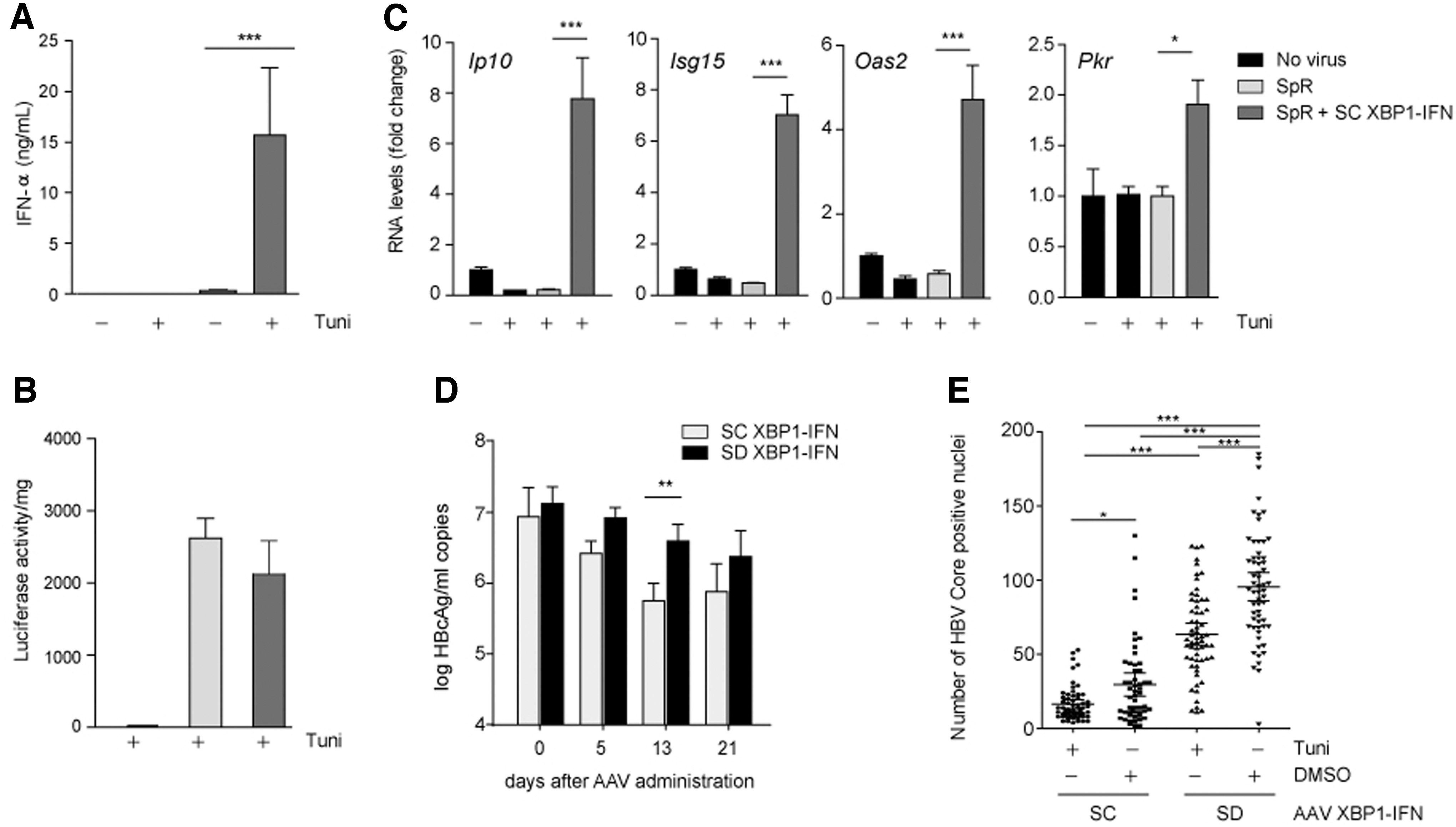
UPR splicing-dependent expression of IFNα elicits robust antiviral responses.
We last explored the capacity of our inducible vector to inhibit viral replication in a transgenic mouse model of HBV. In this context, virus replication determines a condition where UPR activation has been well documented. 20 Groups of HBV-transgenic were injected with AAV SC XBP1-IFNα or AAV SD XBP1-IFNα at a dose of 1 × 1012 vg/kg. HBV viremia in circulation was analyzed before and 5, 13, and 21 days of vector administration. In both groups, we observed a decrease in viremia with time, significantly lower in the AAV SC XBP1-IFNα group (Fig. 3D). On day 21 after vector injection, the animals were separated into two groups, and one received a tunicamycin injection and the other one only the vehicle DMSO.
Eight hours later, the animals were sacrificed and the number of hepatocytes expressing HBcAg was quantified. We observed significantly lower levels of HBcAg-positive cells in the mice receiving AAV SC XBP1-IFNα than in the mice receiving AAV SD XBP1-IFNα. This reduction was improved upon tunicamycin treatment (Fig. 3E).
Discussion
We here describe the development, characterization, and a possible application of an inducible system that does not rely on gene induction as described for other inducible systems 1 : induction is posttranscriptional and not affected by possible dysfunctional scenarios at the transcriptional level. Furthermore, our system associates transgene expression with cellular events that cause the disruption of protein folding in the ER, provoking ER stress and the activation of the UPR. More specifically, we have harnessed the cis elements required to sustain the noncanonical splicing of XBP1 mRNA in response to ER stress to drive the expression of heterologous genes.
Although other groups have described the use of the minimal XBP1 region to regulate the expression of reporter genes to monitor ER stress, to our knowledge this is the first time that this system is used to activate the synthesis of recombinant proteins with a therapeutic function. ER stress can be artificially induced or endogenously as a consequence of ER alterations associated with pathologies such as conformational diseases that are caused by mutations altering the protein or a microbial/viral infection. These ER stress episodes/states are intrinsic to the severity of the pathology and therefore could serve to tune not only the therapeutic dose of the transgene but also its availability over time. Still, ER stress can also be pharmacologically induced to boost the therapeutic response, if necessary, as shown here (Fig. 3).
Inducible systems require a tight control of protein expression since they are commonly used to express therapeutic genes that might be toxic if overexpressed or if constitutively expressed. 9 Previous description of the XBP1 minimal splicing element fused to a reporter protein did not take into account internal AUGs that can escape the splicing requirement. Here, the induction system was optimized to avoid spurious use of AUG in the XBP1 coding sequence, which may cause the translation of the therapeutic transgene from the unspliced mRNA. We mutated these codons without affecting the splicing or the expression of the inducible protein and proved that there is no leakage of protein expression from the unspliced messenger, as no expression was observed in the constructs containing mutations at the XBP1 splicing sites.
Most viruses manipulate the transcriptional and/or translational machinery during the infection process. Some viruses rely on the ER protein maturation machinery for proper folding, posttranslational modifications, or protein Sorting. In many instances, viral protein folding at the ER causes ER stress. In parallel, viruses have evolved to block the induction of type I IFN by preventing the recognition and activation of the IFN-β induction cascade. The viral antagonistic proteins involved in this inhibition could be conceptualized as a short circuit of the antiviral signal. As a consequence, the cellular response slows down and viruses gain time to replicate and spread faster. The inducible system presented here could bypass these antagonistic viral mechanisms by restoring antiviral signaling in response to the ER stress that results from viral replication. We showed that this therapeutic principle was applicable to HBV infection, in which the ER protein folding machinery is hijacked for the efficient production of viral glycoproteins that in turn trigger activation of the UPR.
Current treatment for chronic HBV infection includes the administration of pegylated IFNα with a finite duration. The main disadvantages of PegIFNα treatment are the high variability of the response and its unfavorable safety profile, both making a significant number of patients non-eligible for or unwilling to receive this type of treatment. 19 Using our inducible system, we linked IFNα expression to HBV replication. With this strategy, IFNα expression is constrained to the presence of the virus, and therefore, the toxic effect of the drug is reduced. Importantly, the incorporation of the inducible system into AAV vectors allowed the efficient delivery of the therapeutic system to the liver of mice. The administration of the vector, which produced IFNα upon the induction of XBP1 splicing, resulted in a significant antiviral effect. Of note, we also observed a significant antiviral effect associated with tunicamycin administration, most likely due to the impact of N-linked glycosylation in viral protein folding.
In summary, the inducible tuning of transgene expression opens new avenues for the future treatment of liver conditions resulting in episodes of ER stress such as HBV infection, where transgene expression will occur just when/where needed.
Footnotes
Acknowledgments
We thank Professor Frank Chisari for providing us with essential reagents for our study. We are grateful to David Salas and Daniel Moreno for technical assistance, CIMA's animal facility staff for animal care and vivarium management.
Author Disclosure
No conflict of interest with respect to this article.
Funding Information
Grant support: SAF2015-70028-R and RTI2018-101936-B-I00 to G.G-A. from Secretaria de Estado de Investigación, Desarrollo e Innovación, Ministerio de Economia y Competitividad and Ministerio de Ciencia y Tecnología. Spanish MINECO Fellowship JCI-2011-09179, the ISCIII, cofounded by FEDER, grant No. PI11/01534 and the European Marie-Curie IRG-2010-277172 to EN-V. BFU2017-90043-P and BFU2013-48703-P to T.A., from Secretaría de Estado de Investigación, Ministerio de Economía y Competitividad and Ministerio de Ciencia y Tecnología.
Supplementary Material
Supplementary Data
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.