
Abstract
Friedreich's ataxia (FA), an autosomal recessive disorder caused by a deficiency in the expression of frataxin (FXN), is characterized by progressive ataxia and hypertrophic cardiomyopathy. Although cardiac dysfunction is the most common cause of mortality in FA, the cardiac disease remains subclinical for most of the clinical course because the neurologic disease limits muscle oxygen demands. Previous FXN knockout mouse models exhibit fatal cardiomyopathy similar to human FA, but in contrast to the human condition, untreated mice become moribund by 2 months of age, unlike humans where the cardiac disease often does not manifest until the third decade. The study was designed to create a mouse model for early FA disease relevant to the time for which a gene therapy would likely be most effective. To generate a cardiac-specific mouse model of FA cardiomyopathy similar to the human disease, we used a cardiac promoter (αMyhc) driving CRE recombinase cardiac-specific excision of FXN exon 4 to generate a mild, cardiac-specific FA model that is normal at rest, but exhibits the cardiac phenotype with stress. The hearts of αMyhc mice had decreased levels of FXN and activity of the mitochondrial complex II/complex IV respiratory chain. At rest, αMyhc mice exhibited normal cardiac function as assessed by echocardiographic assessment of ejection fraction and fractional shortening, but when the heart was stressed chemically with dobutamine, αMyhc mice compared with littermate control mice had a 62% reduction in the stress ejection fraction (p < 2 × 10−4) and 71% reduction in stress-related fractional shortening (p < 10−5). When assessing functional cardiac performance using running on an inclined treadmill, αMyhc mice stayed above the midline threefold less than littermate controls (p < 0.02). A one-time intravenous administration of 1011 genome copies of AAVrh.10hFXN, an adeno-associated virus (AAV) serotype rh10 gene transfer vector expressing human FXN, corrected the stress-induced ejection fraction and fractional shortening phenotypes. Treated αMyhc mice exhibited exercise performance on a treadmill indistinguishable from littermate controls (p > 0.07). These αMyhc mice provide an ideal model to study long-term cardiac complications due to FA and AAV-mediated gene therapy correction of stress-induced cardiac phenotypes typical of human FA.
Introduction
Friedreich'
With an incidence of 1 in 50,000, FA is the most common inherited ataxia. 6 –9 The onset of neurologic symptoms is typically between 5 and 15 years. 7,10 –12 The clinical manifestations of FA involve the central nervous system and the heart. 1,6,9,11 While the mortality associated with FA is most commonly due to cardiac failure, the cardiomyopathy usually remains subclinical for most of the clinical course, likely because affected individuals have limited mobility and low oxygen demands. 9,11,13 –16 The evaluation of treatment strategies for cardiac manifestations of FA is currently limited to mouse models with severe cardiomyopathy, which are not cardiac specific and do not accurately represent the subclinical manifestations typical of human cardiac disease. 17,18
To study therapies for cardiac manifestations of FA in a mouse model that more accurately reconstitutes the functional characteristics of the human disease, we used the suboptimal Cre-Lox recombination to partially excise FXN exon 4 specifically in the hearts of C57bl/6JxCBA mice. As in the human condition, no clinical signs in this model are apparent at rest. However, with chemical- and exercise-induced stress, these mice exhibit significant cardiac dysfunction that can be effectively treated with an adenoassociated virus (AAV)-gene therapy using AAVrh.10, a serotype known to effectively deliver the FXN coding sequence to the myocardium following intravenous administration. 18 –21
Methods
Generation and genotyping of the murine model
Clustered, regularly interspaced, short palindromic repeats (CRISPR) technology combined with homologous recombination was used to add loxP sites surrounding exon 4 of the FXN gene in C57Bl/6xCBA mice (Supplementary Fig. S1). The guide RNA TGCTATGCAACTCAGGCATC gRNAC55, Cas9 nuclease, and DNA template were coinjected into the pronucleus of mouse zygotes using conventional techniques (Mouse Genetics Core, Memorial Sloan Kettering Cancer Center). Polymerase chain reaction (PCR) products, denatured and reannealed, were treated with T7 endonuclease I to cleave heteroduplexes between wild-type and loxP-modified FXN genes, and individual mice were screened for fragmented PCR products that indicate successful recombination. Sequencing confirmed the loxP insertions.
A cross between homozygote FXN exon 4 flox mice and B6.FVB-Tg(Myh6-cre)2182Mds/J mice (Jackson Laboratory, Bar Harbor, ME) was used based on the logic that the αMyhc promoter is a cardiac-specific promoter. 22 The result was in partial expression, generating mice containing both the FXN exon 4 flox and the Myh6-cre transgene. A second cross of heterozygote FXN/loxp/Myh6-cre and homozygote FXN/loxp then produced the cardiac-specific mouse model of FA, the αMyhc mouse, in which the partial expression of CRE leads to a subclinical FA mouse model (Fig. 1A). Genotype analysis on tail biopsies was performed at a commercial laboratory (TransnetYX, Germantown, TN). All mice were housed in microisolator cages and maintained according to standard guidelines and protocols reviewed and approved by the Weill Cornell Institutional Animal Care and Use Committee.

αMyhc mouse model.
Biochemical and expression analysis
Hearts from αMyhc and control mice sacrificed at 24 weeks of age were excised, following anesthesia and phosphate-buffered saline (PBS) cardiac perfusion, and homogenized in 750 μL of lysis buffer (10 mM HEPES-KOH, pH 7.4, 5 mM mannitol, and 1% Triton X-100 in water) with 5-mm steel beads using a TissueLyser LT (Qiagen, Germantown, MD) for 10 min at an oscillation of 50 per second. The homogenates were aliquoted and immediately stored at −80°C. Respiratory chain enzyme complex succinate dehydrogenase (SDH, complex II) and cytochrome c oxidase (complex IV) activities were assayed using commercial kits (Abcam, Cambridge, MA).
For Western analysis, hearts and skeletal muscle from the αMyhc and control mice sacrificed at 24 weeks of age were collected, following anesthesia and PBS cardiac perfusion, and homogenized in 750 μL of lysis buffer (10 mM HEPES, pH 7.4, 5 mM mannitol, 1% deoxycholate, and 1% Triton X-100 in water) with 5-mm steel beads using a TissueLyser LT for 60 min, 50/sec. Tissue homogenates were clarified by centrifugation (6 min, 13,000 rpm) and the supernatant protein concentration was measured using the bicinchoninic acid protein assay (Thermo Fisher, Somerset, NJ). An equal amount of protein (40 μg) was denatured at 90°C for 2 min and electrophoresed on 4–12% NuPAGE Bis-Tris gels (Thermo Fisher).
The proteins were then transferred to a polyvinylidene difluoride membrane (Thermo Fisher) by electroblotting, followed by incubation in blocking solution (10% nonfat dry milk) for 20 min. The membrane was immunoblotted with the following antibodies: primary antibodies: rabbit anti-FXN (Abcam) and antiglyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, Danvers, MA); and secondary antibody: goat anti-rabbit IgG-horseradish peroxidase (Santa Cruz, Dallas, TX). Proteins were detected using the ECL (electrochemiluminescence) SuperSignal™ kit (Thermo Fisher) following the manufacturer's guidelines.
Characterization of cardiac function
Echocardiography was carried out on 4- to 8-week-old mice that were shaved on the chest, anesthetized with isoflurane (2.5%, with 2% oxygen), and placed on an imaging platform at 42°C in the supine position. Echocardiogram measurements were taken through ultrasound transmission gel with a 40-MHz linear probe (Fujifilm VisualSonics, Toronto, Canada). Two-dimensional images were recorded in long- and short-axis projections in motion mode (M-mode) at the midventricular level in both views.
The ejection fraction was calculated by (end-diastolic volume − end-systolic volume)/end-diastolic volume, and left ventricular (LV) fractional shortening was calculated by (LV internal dimension diastole − LV internal dimension systole)/LV internal dimension diastole. Each of these parameters, as well as LV mass, was output from Fujifilm VisualSonics vevo software. LV mass and echocardiogram measurements were done at rest and 15 min after intraperitoneal administration of the β-receptor agonist, dobutamine (4.5 μg/g body weight).
Exercise-induced phenotypes in mice were assessed at 10 weeks of age on a four-lane treadmill (DogTread, Ogden, UT) equipped with shock bars at the rear of the belt to stimulate the mice to run. Mice were run at 12 m/min for 20 min at an inclination of 15°, followed by quantification of the time over a 3-min period above the treadmill midpoint line.
AAVrh.10 vectors and treatment of the cardiac disease
The adeno-associated viral vector, AAVrh.10hFXN, comprised the Rhesus serotype 10 capsid proteins and the AAV2 inverted terminal repeats (ITRs) surrounding the AAV2 packaging signal (Ψ), cytomegalovirus (CMV) enhancer, chicken β-actin promoter and splice donor and rabbit β-globin intron with splice acceptor (CAG), and cDNA for the normal human FXN coding sequence, followed by the rabbit β-globin polyA. 23,24 AAVrh.10hFXN vectors were produced by cotransfection into 293T cells of a plasmid (pAAV2hFXN), consisting of ITRs surrounding the CAG promoter and FXN expression cassette, and the AAVrh.10 packaging plasmid, pPAKMArh.10, providing Ad helper functions and AAV rep and cap genes. Transfection was achieved with the PEI transfection reagent (Polysciences, Warrington, PA). Transfected cells were harvested at 72 h, and crude viral lysate was prepared using five cycles of freeze/thaw.
The lysate was clarified by centrifugation to remove cell debris, followed by iodixanol gradient centrifugation and QHP anion exchange chromatography (GE Healthcare, Piscataway, NJ). The purified vector was concentrated with a BioMax 100K membrane concentrator (Millipore, Billerica, MA) and stored in PBS, pH 7.4, at −80°C. Vector genome titers were determined by TaqMan real-time PCR using a CMV–chicken β-actin promoter (CAG)-specific primer–probe set, forward primer: GTCAATGGGTGGAGTATTTACGG and reverse primer: AGGTCATGTACTGGGCATAATGC (Applied Biosystems, Foster City, CA). The capsid proteins of AAVrh.10hFXN were digested with proteinase K in the presence of 0.5% sodium dodecyl sulfate (SDS) and 25 mM ethylenediaminetetraacetate at 70°C for 1 h, followed by protease inactivation at 95°C for 15 min.
A standard curve for TaqMan analysis used an AAV-CAG-hCLN2 plasmid DNA of known copy number. 25 Before use, the AAVrh.10hFXN vector was tested for purity by SDS-PAGE (polyacrylamide gel electrophoresis), endotoxin, and sterility. The function of AAVrh.10hFXN was demonstrated in an in vivo potency assay that evaluated vector DNA, transgene mRNA, and FXN protein in the liver. 26 Individual lots were titered by quantitative PCR and shown to be sterile and contained no detectable levels of endotoxin.
αMyhc mice (6-week-old, male, n = 4) were administered a single dose of 1011 genome copies of AAVrh.10hFXN intravenously through the tail vein. Untreated αMyhc and C57Bl/6 male mice were used as controls. Mice were evaluated by echocardiography 5 times at 2-week intervals and for exercise-induced stress response on a treadmill at 10 weeks postvector administration. Each mouse was evaluated only once on the treadmill to avoid bias due to habituation.
Results
Biochemical characterization of αMyhc mice
Heart tissues of 24-week-old αMyhc mice had 51% lower levels of FXN protein compared with 24-week-old C57Bl/6 wild-type controls (Fig. 1B; p < 0.05; Supplementary Fig. S1). In contrast, FXN protein levels were indistinguishable in skeletal muscle of αMyhc and wild-type mice (p > 0.7; Fig. 1C).
Reduced FXN levels negatively impacted the complex II activity of the heart mitochondrial electron transport chain of αMyhc mice (Fig. 2B). Complex II activity was normalized to the complex IV electron transport chain function, which is not affected in the human disease. 6 Wild-type mice had a complex II/IV ratio of 2.6 ± 0.3 (mean ± standard error), whereas αMyhc mice had a complex II/IV ratio of 1.5 ± 0.2 (mean ± standard error), a 56% decrease in the ratio of II/IV activity (p < 0.04).

Heart mitochondrial complex II activity in αMyhc mice.
Cardiac function in the αMyhc mouse
Ejection fraction and fractional shortening of αMyhc mice were assayed by echocardiography at rest and after dobutamine-induced stress (Fig. 3). At rest, ejection fraction and fractional shortening of αMyhc mice were similar to wild-type mice (p > 0.1). Similarly, there was no significant difference of LV mass at rest between wild-type and αMyhc mice (Supplementary Fig. S2A). In contrast, αMyhc mice exhibited a marked inability to respond to dobutamine stress. At rest, wild-type mice had an ejection fraction of 63.9% ± 0.7%. With dobutamine-induced stress, the ejection fraction was 85.5 ± 1.0. In contrast, αMyhc mice had an ejection fraction of 64.7 ± 0.8 at rest, and with dobutamine-induced stress, the ejection fraction was 74.5 ± 1.9 (Fig. 3A).
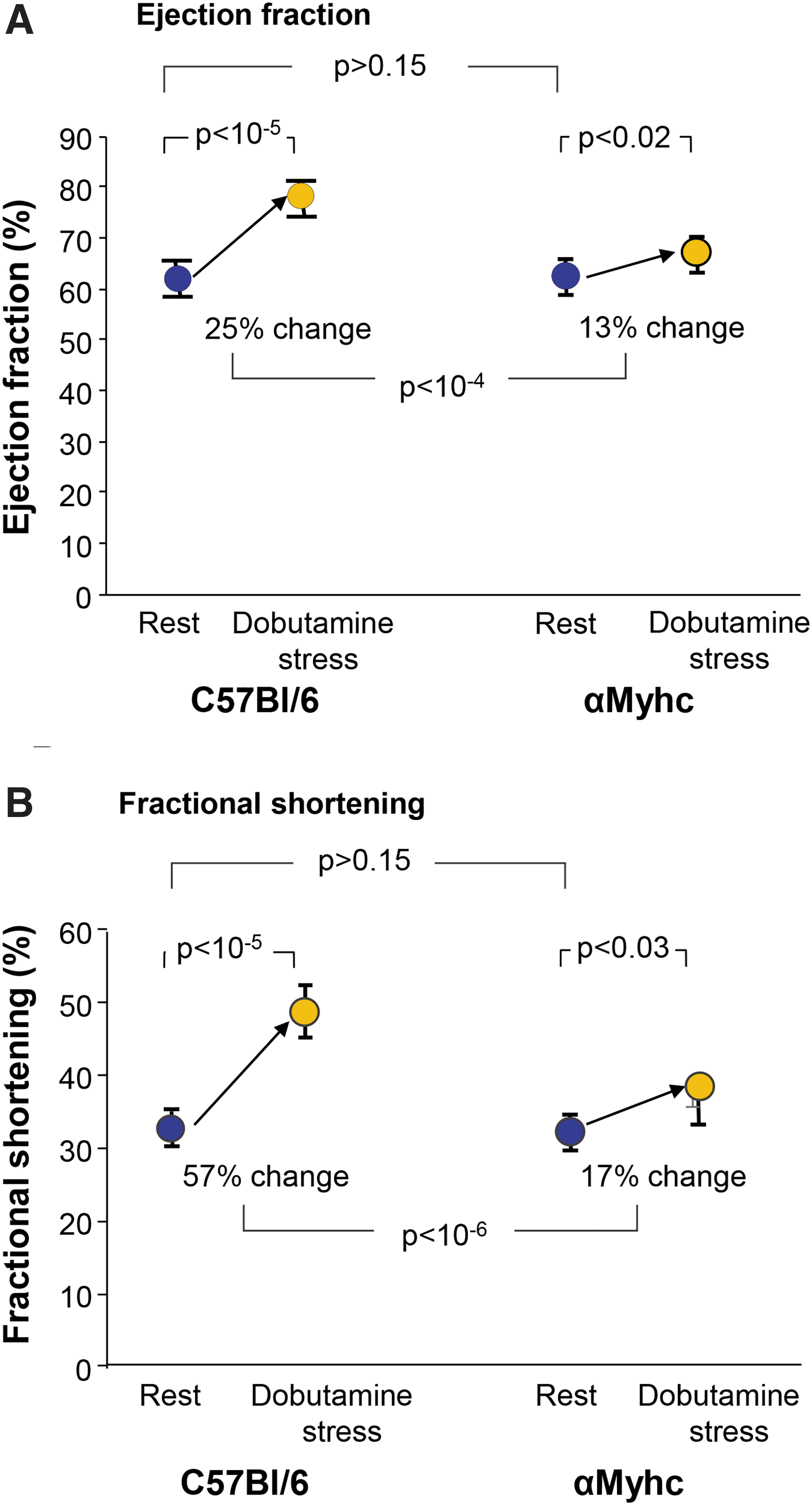
Echocardiograph evaluation of the cardiac ejection fraction and fractional shortening of αMyhc versus C57Bl/6 mice at rest and under dobutamine stress.
At rest, wild-type mice had a fractional shortening of 34.7% ± 0.5% and with dobutamine-induced stress, it was 54.6% ± 1.4%. In contrast, at rest, αMyhc mice had a fractional shortening of 35.5% ± 0.5%, and with dobutamine-induced stress, the fractional shortening was 43.1% ± 1.7% (Fig. 3B). The differences between the changes in response to stress of the αMyhc and wild-type mice were significant for both ejection fraction (p < 10−4) and fractional shortening (p < 10−6), each demonstrating reduced response to stress without any significant difference in LV mass (Supplementary Fig. S2B), as expected by the design of this mouse model.
AAVrh.10hFXN therapy
The capacity to mediate expression to the heart by intravenous administration of AAVrh.10 was evaluated with an expression cassette encoding an HA-tagged FXN. Western analysis for the HA tag confirmed expression in the heart and liver (Supplementary Fig. S3). Correction of the limited stress response of the cardiac ejection fraction and fractional shortening in the αMyhc mouse was as evaluated by echocardiography. αMyhc mice, intravenously administered 1011 genome copies of AAVrh.10hFXN, along with untreated αMyhc mice and normal C57Bl/6 male mice at 6 weeks age, were evaluated with and without dobutamine-induced stress over the following 10 weeks. There was a 26.4% ± 2.5% dobutamine-induced increase in ejection fraction for the C57Bl/6 mice, while αMyhc mice displayed only an increase of 9.9% ± 3.1%.
In contrast, AAVrh.10hFXN-treated αMyhc mice responded to stress with an increase in the ejection fraction of 34.5% ± 3.6% that was significantly greater than untreated αMyhc mice (p < 10−5) and indistinguishable from normal C57Bl/6 mice (p > 0.07, Fig. 4A). There was a 45.8% ± 3.6% increase in fractional shortening with stress for C57Bl/6 mice, while αMyhc mice displayed only a 13.2% ± 4.1% increase. In contrast, AAVrh.10hFXN-treated αMyhc mice responded to stress with an increase in fractional shortening of 53.1% ± 5.2% that was significantly greater than untreated αMyhc mice (p < 10−5) and indistinguishable from normal C57Bl/6 mice (p > 0.2, Fig. 4B). Results for all echocardiography parameters are provided in Supplementary Table S1.
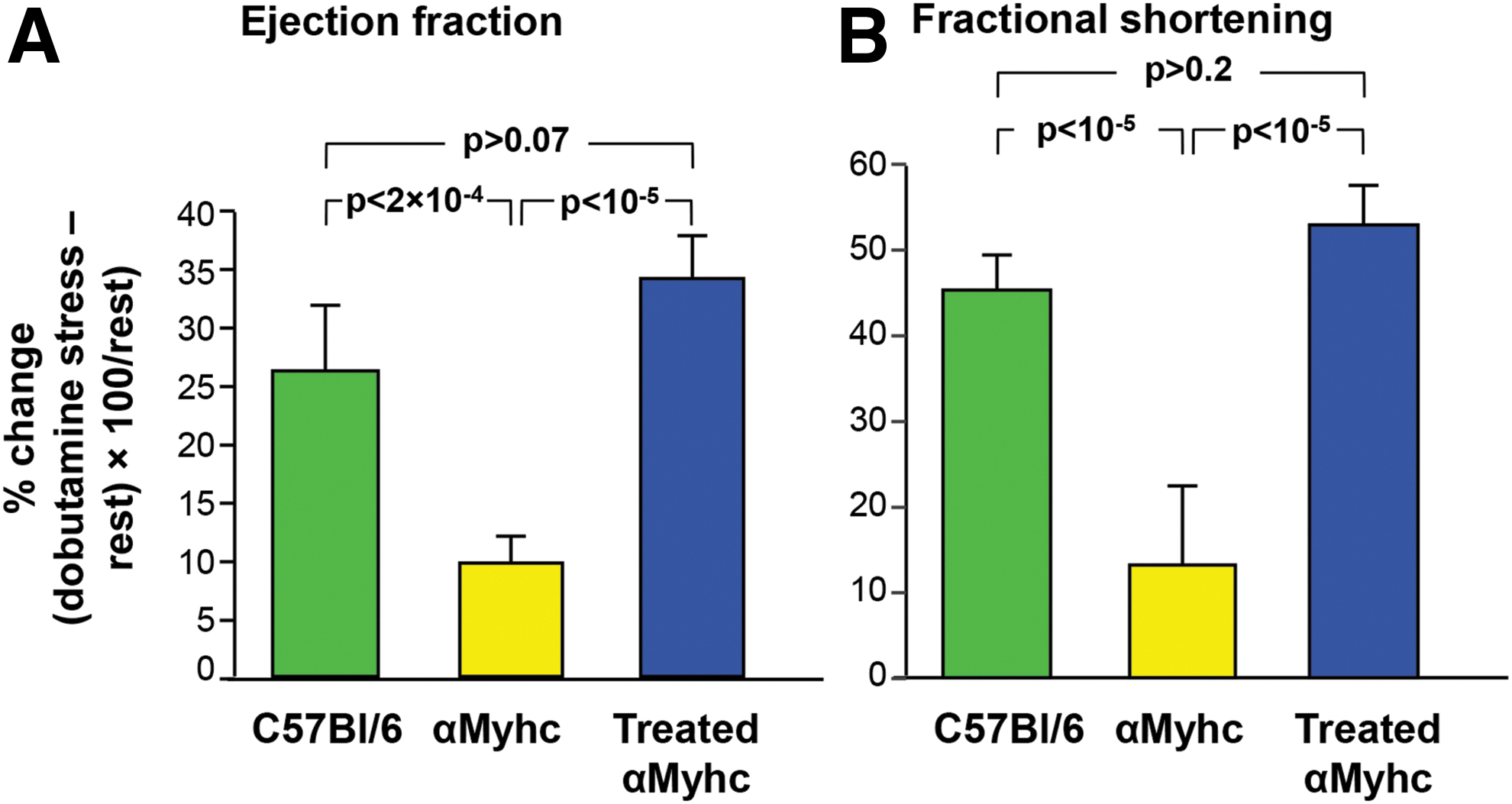
Echocardiograph assessment of cardiac function of αMyhc mice following treatment with AAVrh.10hFXN.
Exercise-induced stress restricted the untreated αMyhc mice from maintaining the same pace on a treadmill as wild-type C57Bl/6 mice. During the 180-s evaluation period, wild-type mice remained above the treadmill midpoint for 150.3 ± 6.8 s, whereas the αMyhc mice spent significantly less time above the treadmill midpoint (44.8 ± 21.4 s; p < 0.02). In contrast, 10-week-old αMyhc mice treated intravenously with 1011 genome copies of AAVrh.10hFXN at 6 weeks of age averaged 148.7 ± 17.0 s above the midpoint line of the treadmill and were indistinguishable from wild-type mice (p > 0.9) and significantly greater than untreated αMyhc mice (p < 0.03; Fig. 5, see Supplementary Video S1 of the treadmill evaluation for wild-type, αMyhc, and treated αMyhc mice).
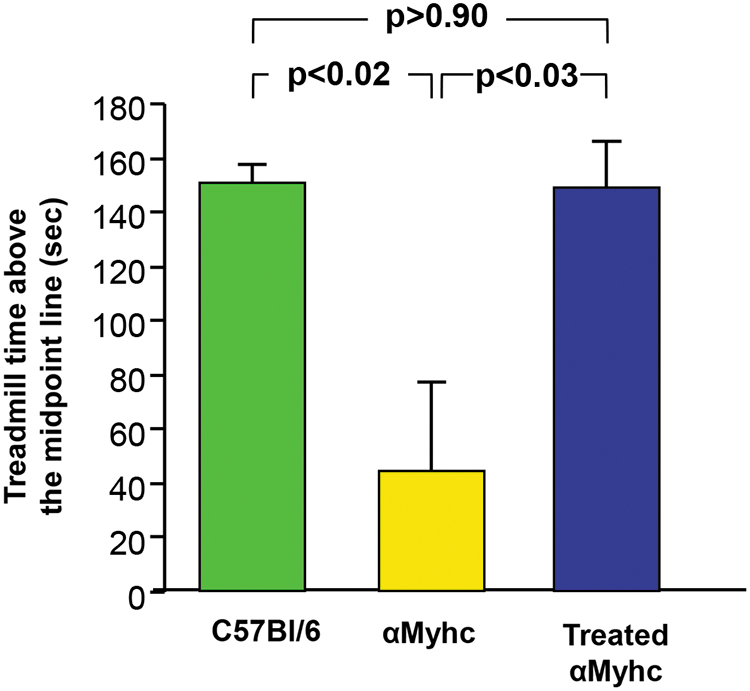
Impact of treatment with AAVrh.10hFXN on the exercise ability of αMyhc mice. Wild-type C57Bl/6, αMyhc, and AAVrh.10FXN-treated αMyhc mice were subjected to stress over a 3-min evaluation period on a treadmill for the ability to stay above the midline. The time each group remained above the midline during the 3-min interval is shown in seconds ± standard errors. Groups (n = 3–4/group) are compared by two-tailed paired t-tests. See Supplementary Video S1 for a video of mice on the treadmill. Color images are available online.
Discussion
FA is a rare hereditary disorder manifested by neurodegenerative pathology and cardiomyopathy due to a decrease in the function of mitochondrial oxidative phosphorylation and formation of iron cluster proteins of the respiratory chain. 1 –3,18,27 Individuals with FA have progressive cerebellar ataxia, atrophy of the optic nerve, sensory-neural deafness, and foot abnormalities and require wheelchairs about 11 to 25 years after symptom onset. 1,7,9,15,28,29 While the neurologic manifestations have a major impact, cardiac failure is the most frequent cause of mortality. 9,11,13 –16,30,31 Despite this, for most of the early clinical course, the cardiac manifestations remain subclinical as exogenous stress is limited due to the inactivity associated with the neurologic disease, limiting oxygen demands. 1,7,8,11,14,32,33 These limited energy demands limit the stress on the heart and the cardiac-associated clinical sequelae are typically delayed. 9,15 In this context, we hypothesized that assessment of new therapies directed at FA-associated cardiac disease would benefit from an animal model that reconstitutes the mild cardiac pathology that manifests only with stress and without the neurological disease component. To achieve this goal, we created αMyhc mice, a cardiac-specific, partial FXN knockout model that exhibits normal cardiac function at rest (ejection fraction and fractional shortening), but significantly diminished cardiac function with stress (dobutamine and treadmill exercise). In contrast, when treated with intravenous administration of 1011 genome copies (a dose that can be safely scaled to humans), the αMyhc mouse cardiac function was normalized, with similar cardiac function as the C57Bl/6 controls when stressed with dobutamine or exercise. While it is possible that some reduction of FXN levels undetected by Western analysis contributes to the stress-induced reduction of treadmill performance, the stress-induced impact on echocardiography parameters and clear reductions in myocardial FXN levels establish significant cardiac involvement.
Cardiac disease associated with FA
A substantial number of patients with FA develop severe cardiomyopathy and 60% of patients die from cardiac causes, typically in the third to fourth decade. 11,33,34 There is a correlation between the GAA repeat number and the onset and severity of clinical symptoms with higher repeat numbers signaling an earlier onset and more severe cardiomyopathy. 13,14,30,32,35 The cardiomyopathy associated with FA is characterized by arrhythmias, heart failure, and intolerance of cardiovascular stress, such as surgery. Histologically, the LV myocardium is commonly moderately to severely hypertrophied and has a high degree of fibrosis and cardiomyocyte loss at the time of death. 1,33,34,36,37 To date, despite trials of antioxidant and iron chelation therapies, there is no effective therapy for the cardiac manifestations of FA. 10,38,39
Models of FA cardiac disease
The αMyhc mouse described in the present study has, at rest, normal cardiac function and mass, but similar to humans with FA, diminished cardiac function with stress. The stress-induced cardiac functional abnormalities can be corrected with AAV-mediated, systemic gene therapy. Six other FA murine models have been described (MCK, NSE, PRP, KIKO/KIKI, YG8R/YG22E, and YAC); of these, three (MCK, NSE, and YAC) exhibit a cardiac phenotype. 17,40
The Mck model has a complete deletion of FXN in the heart and skeletal muscle. 41 These mice develop LV hypertrophy, starting at 4 to 5 weeks, with a rapidly associated and progressive geometric remodeling. The loss of systolic function begins by 6 to 7 weeks and leads to a severe decrease in resting cardiac output, which is fatal by the age of 12 weeks. Iron–sulfur enzyme deficiencies begin before the onset of cardiac dysfunction, with intramitochondrial iron accumulation occurring at the end stage of the disease. The Mck mouse is a stress-independent model of disease with heart dysfunction at rest. 41 Like the αMyhc model, the Mck model can be corrected with intravenous administration of AAVrh.10hFXN. 18
The FXN YAC mouse has the human FXN gene with GAA repeat expansions in the context of the mouse Fxn−/− background. 42 This model exhibits reduced levels of FXN mRNA and protein expression, decreased heart aconitase activity, and sensitivity to oxidative stress and has both progressive neurodegenerative and very mild cardiac pathological phenotypes. 43 Although these are not knockout models, there are reduced levels of enzyme activity with less severe disease, but they do not require stress to demonstrate phenotypes, as in the earlier human disease or the αMyhc mouse model.
The NSE model is developed by expressing a CRE recombinase behind a neuron-specific promoter, NSE, in a similar process as that for the Mck mouse. 41 In part, because the NSE promoter is leaky, these mice have severe neurological phenotypes, as expected, and severe mitochondrial defects in the heart, leading to cardiomyopathy with mortality at around 25 days, unlike the late development of the human cardiac disease.
Gene therapy for cardiac manifestations of FA
The first demonstration of effective gene therapy for FA was with AAVrh.10hFXN treatment of the Mck mouse model. Intravenous administration by the retro-orbital route with 5.4 × 1013 vector genomes/kg demonstrated expression and correction of the mitochondrial defect in cardiomyocytes, prevention, and reversal of the cardiac phenotype. 18 The correction reversed the mitochondrial abnormalities and the biochemical Fe-S protein deficit at 1 week after treatment, with cardiac recovery and function reaching normality within 4 to 5 weeks. This successful correction used a high vector dose to correct a significantly greater disease phenotype than the human condition. This observation suggests that gene therapy would be effective for successful treatment of mild, early human disease with a lower vector dose that would be safe for human therapies.
While the additional analytical techniques could provide an AAVrh.10hFXN dose response for treatment of the mouse model, the relevance to FA disease remains to be evaluated in human clinical studies.
FXN functions in the iron–sulfur cluster assembly, which is an important component of the respiratory electron transport chain for complexes I (NADH ubiquinone oxidoreductase), II (SDH), and III (ubiquinone cytochrome c oxidoreductase). While the exact role of FXN in the Fe-S assembly is not clear, it is known that FXN deficiency in human cells reduced the Fe-S cluster-dependent complex II activity by 60%, but not that of complex IV. As a primary measure of the FXN deficit in the αMyhc mouse, AAVrh.10hFXN therapy normalized the ratio of complex II activity to complex IV.
Echocardiogram assessment of the αMyhc mouse demonstrated that the cardiac ejection fraction and fractional shortening in the αMyhc mouse were similar at rest to wild-type mice. Data also show no changes to the LV mass, as seen in the early human disease. In contrast, the chemically stressed αMyhc mice could not respond with appropriate increased ejection fraction or fractional shortening, as observed in wild-type mice. A one-time intravenous administration of AAVrh.10hFXN to the αMyhc mice fully restored the change in fractional shortening and ejection fraction to stress. To evaluate the stress more typically affecting FA individuals, we examined the impact of exercise on the αMyhc mouse.
While untreated αMyhc mice had significantly less ability to stay above the treadmill midpoint line than wild-type mice after exercise-induced stress, administration of AAVrh.10hFXN enabled the αMyhc mice to exercise in a manner indistinguishable from wild-type littermate controls. It is possible that some improvement to the stress-based phenotype is due to off-target noncardiac expression of FXN; given that this mouse model is based on a heart-specific deficit, and previous data demonstrate that AAVrh.10hFXN treatment corrects the cardiac myocyte function, 18 enhancement of FXN levels in other tissues would be expected to be minor. In the context that FA disease affects all tissues, such off-target enhancement is likely to be advantageous to a therapy.
In conclusion, partial cardiac-specific excision of FXN exon 4 in the heart using the Cre-Lox recombination generated an FA cardiac-specific mouse model with a mild phenotype that mirrors the early human clinical cardiomyopathy of FA where the clinical cardiac phenotype requires stress. A single intravenous administration of AAVrh.10hFXN relieves the phenotypic outcomes of FA cardiomyopathy. This observation, together with the AAVrh.10hFXN correction of the abnormal cardiac phenotype of the MCK mouse, 18 provides proof of concept for AAVrh.10hFXN therapy for cardiac manifestations of FA.
Footnotes
Authors' Contributions
C.O.S., K.J., C.J., L.A., G.C., and B.P.D. performed experimental work. C.O.S., J.R., K.S., S.M.K., and R.G.C. provided the experimental design. S.M.K., C.O.S., K.J., and G.C. performed data analysis. M.C., S.M.K., and R.G.C. designed the animal model. C.O.S., G.C., D.S., R.G.C., and S.M.K. reviewed the manuscript for content. C.O.S., S.M.K., and R.G.C. conceived the study and performed data analysis/interpretation.
Acknowledgments
The authors thank Jonathan Rosenberg, Jasmine Reid, and Clotell Hanks for assistance with animal studies and vector production; Scott Rodeo lab, Hospital for Special Surgery, for the use of the treadmill; and N. Mohamed for editorial assistance.
Author Disclosure
No competing financial interests exist.
Funding Information
These studies were supported by the Department of Genetic Medicine, Weill Cornell Medical College.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Video S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.