
Abstract
Mutations in the rhodopsin gene may cause photoreceptor degeneration in autosomal dominant retinitis pigmentosa (ADRP) by dominant negative or toxic gain-of-function mechanisms. Controversy exists as to the mechanism by which the widely studied P23H mutation induces rod cell dysfunction and death. Inherited disease caused by dominant negative mutations may be amenable to treatment using wild-type gene augmentation. Indeed, prior studies in the RHOP23H, Rho+/− transgenic mouse model of ADRP have suggested that a therapeutic benefit may be achieved when wild-type rhodopsin is overexpressed following subretinal delivery of a recombinant adeno-associated viral (AAV) vector. In this study, we investigated the effect of wild-type rhodopsin supplementation on the rate of retinal degeneration in the more clinically relevant RhoP23H/+ knock-in mouse model of ADRP. Four AAVs carrying the human rhodopsin coding sequence were first designed and compared for efficacy in the rhodopsin knockout mouse. All four vectors were capable of driving expression of the human transgene in the knockout retina with the protein being appropriately trafficked to de novo rod outer segments. The most efficient of these vectors was injected at one of two doses into the subretinal space of RhoP23H/+ mice and the effect on retinal structure and function determined longitudinally by spectral-domain optical coherence tomography and electroretinography, respectively, over a 3-month period. Although significant overexpression of rhodopsin protein was achieved in this model, no beneficial effect on retinal structure or function was observed at either dose. Lack of therapeutic efficacy in this model may be attributable to the relative rapidity of degeneration in the RhoP23H/+ mouse relative to the human disease, over- or under dosing at the level of individual photoreceptors, late timing of the intervention, or a possible predominant toxic gain-of-function mechanism of degeneration.
Introduction
The numerous mutations in the rhodopsin gene (RHO) that are associated with autosomal dominant retinitis pigmentosa (ADRP) may cause rod cell dysfunction and death by dominant negative and/or toxic gain of function mechanisms. 1,2 In the former, mutant protein interferes with the function of the normal protein, whereas in the latter, the mutant protein has a direct toxic effect of the cell. Point mutations that induce cell death by a dominant negative mechanism may be amenable to treatment by way of gene augmentation. 3 Much debate exists as to whether the widely studied P23H mutation in rhodopsin (which falls into Class II according to the classification of Mendes et al. 1 ) causes cell death in this way through dimerization with wild-type protein, 4 –6 or alternatively by direct toxicity. 7 In support of a dominant negative mechanism, the human RHOP23H transgene appears to induce a more rapid retinal degeneration in mice on an endogenous Rho+/− background than on a null Rho−/− background, 8 and additional genomic copies of wild-type rhodopsin attenuate the degenerative phenotype. 9 Other studies have, however, pointed to a toxic gain of function mechanism. In cell culture systems, P23H rhodopsin appears to misfold and accumulate in the endoplasmic reticulum (ER), which may trigger cell death through activation of the unfolded protein response. 10 –12 Further studies have indicated that a proportion of mutant protein reaches the outer segments (OS) in vivo, where it may induce cell death following destabilization of discs. 13 –15
Rhodopsin augmentation is an appealing gene therapy strategy for the treatment of RHO-related ADRP, as it may be effective for any mutation in the gene that causes cell death through a dominant-negative mechanism. 16 In these circumstances, overexpression of wild-type protein might be expected to slow cell death by rendering the ratio of normal to mutant protein more favorable. Alternatively, for a protein expressed at near maximal translational capacity, wild-type gene augmentation has the potential to reduce the overall proportion of mutant protein by increasing the ratio of wild type to mutant mRNA. Through this mechanism, gene supplementation might be expected to slow the progression of a dominant disease regardless of the mechanism of cellular toxicity. Indeed, Mao et al. demonstrated that delivery of additional copies of wild-type rhodopsin by subretinal injection of a recombinant adeno-associated virus (AAV)2/5 could attenuate the degeneration in a RHOP23H transgenic mouse on an endogenous Rho+/+ background. 17 This strategy, however, did not result in a sustained rescue effect in an equivalent transgenic mouse on a Rho+/− background. 18 The background genotype of the animal model used may thus dramatically influence the success or otherwise of the gene augmentation strategy. A knock-in mouse model has recently been developed, which carries the P23H mutation at the endogenous rhodopsin locus on chromosome 6. 19 Since the heterozygous RhoP23H/+ knock-in mouse mimics the genotype of affected patients, this is arguably a more appropriate model in which to test potential therapeutics. The effect of AAV-mediated RHO gene augmentation in this model has not been previously investigated and remains an important question, as demonstration of efficacy may pave the way for human clinical trials for this and other Class II rhodopsin mutations.
In this study, we developed optimized AAVs for rhodopsin gene augmentation and, following subretinal delivery, we assessed their effect on the rate of disease progression in the RhoP23H/+ knock-in mouse model of ADRP.
Materials and Methods
Animal models
All animal experiments were performed in line with the Animals (Scientific Procedures) Act 1986, UK, and with the Association for Research in Vision & Ophthalmology (ARVO) statements on the care of animals in ophthalmic research. Mice were housed in a dedicated facility with a 12-h light/12-h dark cycle and food and water available ad libitum. The Nrl.GFP/+, Rho−/− mouse line was generated by an intercross between the rhodopsin knockout mouse as described by Humphries et al., 20 and a transgenic mouse line expressing enhanced green fluorescent protein (eGFP) under the control of the neural retina-specific leucine zipper (Nrl) promoter [Tg(Nrl-EGFP)], obtained as a kind gift from Professor A. Swaroop, Bethesda, MD, USA. The resulting mice have rods labeled in green that express no rhodopsin, do not elaborate OS, and undergo a rapid degeneration. 21 Mice homozygous for the RhoP23H knock-in mutation [B6.129S6(Cg)-Rhotm1.1Kpal/J] were obtained from the Jackson Laboratories. Nrl.GFP/+, RhoP23H/+ mice were bred by crossing RhoP23H/P23H knock-in with homozygous Nrl.GFP transgenic mice. 21 Animals of both sexes were used throughout. The number of mice used for each experiment was determined by prospective power calculations with alpha of 0.05 and beta of 0.2.
RHO-AAV vectors
Four rhodopsin-expressing AAV2/8 Y733F capsid mutant vectors were designed and cloned for this study (Fig. 1). The AAV genome in all cases included the human rhodopsin coding sequence (CDS) downstream of an 806 bp human rhodopsin promoter and Kozak consensus sequence (GCCACC) at positions −1 to −6 relative to the translational start codon. A bovine growth hormone polyadenylation (polyA) signal sequence was present downstream of the RHO CDS and the transgene cassette was flanked by the 3′ and 5′ inverted terminal repeat (ITR) sequences derived from AAV2 in all vectors. The four AAVs are described below:

Schematics of rhodopsin-expressing recombinant AAV genomes. Size in base pairs is indicated above each element. The locations of restriction sites used for cloning are indicated. The size of the entire genome is indicated to the right of each schematic.
AAV-ssRHO (AAV2/8Y733F.RHOp.RHO): a single-stranded AAV genome containing the human rhodopsin CDS under the control of the human rhodopsin promoter (Fig. 1A).
AAV-WPRE (AAV2/8Y733F.RHOp.RHO.WPRE): as for AAV-ssRHO but with the addition of a 3′ woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) sequence between the RHO stop codon and polyA signal (Fig. 1B).
AAV-Ex/Int (AAV2/8Y733F.RHOp.Ex/Int.RHO.WPRE): as for AAV-WPRE but with the addition of the first exon and intron of the chicken β-actin gene together with the splice acceptor from the rabbit β-globin gene (sequences derived from the CAG promoter) located between the RHOp sequence and Kozak consensus (Fig. 1C).
AAV-scRHO (scAAV2/8Y733F.RHOp.RHO): a self-complementary (sc) version of AAV-ssRHO (Fig. 1D).
Subretinal injection
Before subretinal injection, mice were anesthetized with intraperitoneal ketamine (80 μg/g body weight, Vetalar; Boehringer Ingelheim, Bracknell, United Kingdom) and xylazine (10 μg/g body weight, Rompun; Bayer, Reading, United Kingdom) diluted in sterile 0.9% saline. Tropicamide 1% and phenylephrine 2.5% eye drops (both Bausch & Lomb, Kingston Upon Thames, United Kingdom) were then applied in sequence to achieve mydriasis. A corneal paracentesis was performed using a 33G needle to lower intraocular pressure. Bevelled 35G NanoFil needles mounted on Nanofil 10 μL syringes (both World Precision Instruments, Hitchin, United Kingdom) were used for trans-scleral subretinal injections. A total volume of 1.5 μL was delivered in all cases into the superior subretinal space to create a subretinal bleb under direct visualization using a microsurgical operating microscope. Injections consisted of either phosphate-buffered saline (PBS; “Sham”), or AAV vector at one of two concentrations: 2 × 108 gc/mL (“Low dose”) or 2 × 109 gc/mL (“High dose”). Following completion of the procedure, anesthesia was reversed using intraperitoneal atipamezole (2 mg/kg body weight, Antisedan; Zoetis, Leatherhead, United Kingdom) diluted in sterile 0.9% saline.
Immunohistochemistry
Animals were humanely euthanized by cervical dislocation, and eyes for immunohistochemistry (IHC) extracted and transferred into sterile PBS. The cornea, iris, and lens were carefully removed using microsurgical instruments. The resulting eye cup was fixed in 4% formaldehyde for 30 min and then transferred sequentially into 10%, 20%, and finally 30% sucrose solutions, each for a period of at least 20 min. Eye cups were left in 30% sucrose overnight at 4°C, washed briefly in PBS, dried, and then immersed in optimal cutting temperature compound (VWR International, Lutterworth, United Kingdom) within a mould in the desired orientation. Specimens were then frozen on dry ice, sectioned to a thickness of 18 μm using a cryostat onto polylysine-coated glass slides and left to dry at room temperature overnight. Sections were permeabilized by incubation with 0.2% Triton X-100 in PBS for one 20 min then blocked in 10% normal donkey serum (NDS)-PBS for 1 h. Primary rabbit anti-rhodopsin antibody (ab3424; Abcam, Cambridge, United Kingdom) diluted 1:1,000 in PBS with 1% NDS was applied to the sections, which were left to incubate overnight at 4°C. Secondary fluorescence-tagged donkey anti-rabbit IgG-568 antibody (Alexa-Fluor ab175470; Abcam) diluted 1:500 in PBS was then applied to the sections, which were incubated for 2 h at room temperature. Slides were counterstained with Hoechst 33342 (Thermo Fisher Scientific, Hemel Hempstead, United Kingdom) and mounted with ProLong Diamond Antifade Mounting Medium (Thermo Fisher Scientific). Confocal fluorescence imaging was subsequently performed using the LSM-710 inverted confocal microscope system (Carl Zeiss, Cambridge, United Kingdom).
Protein extraction and western blot
Protein was extracted from neuroretina through lysis in radioimmunoprecipitation (RIPA) buffer (Sigma-Aldrich, Gillingham, United Kingdom) supplemented with protease inhibitor (PI; Roche Diagnostics, Burgess Hill, United Kingdom). Retinal samples were briefly homogenized in 80–100 μL RIPA/PI, then sonicated. Samples were subsequently agitated using a tube rotator set at 20 rpm at 4°C for 30 min before being centrifuged at 13,000 rpm at 4°C for 10 min. The supernatant protein lysate was then transferred to a new microcentrifuge tube and placed on ice. Total protein was quantified from lysates using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) in accordance with the manufacturer's instructions using the iMark™ microplate reader (Bio-Rad, Watford, United Kingdom). A total of 25 μg of protein for each sample was denatured by incubation in Laemmli buffer for 30 min at room temperature and then loaded into wells of precast 10% Criterion™ Tris-Glycine extended (TGX™) gels (Bio-Rad) for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Protein from SDS-PAGE gels was blotted onto polyvinylidene difluoride (PVDF) membranes using the Trans-Blot Turbo transfer system (Bio-Rad) in accordance with the manufacturer's instructions. Blotted PVDF membranes were incubated in Odyssey blocking buffer (TBS; LI-COR, NE) for 45 min at room temperature under agitation. Membranes were then incubated in primary antibodies (mouse anti-rhodopsin [1D4] monoclonal ab5417 and rabbit anti-β-actin polyclonal ab8227; both Abcam) diluted 1:1,000 in PBS with 0.1% Tween-20 (PBST) supplemented with 10% blocking buffer under agitation for a period of 2 h. This was followed by three washes in PBST under agitation for 7 min each. Membranes were then incubated in fluorescence-tagged secondary antibodies (IRDye 800CW donkey anti-mouse and IRDye 680RD donkey anti-rabbit; both LI-COR) diluted 1:10,000 in PBST for 45 min under agitation. The membranes were subsequently washed a further three times as above before being removed and allowed to dry at room temperature. The processed PVDFs were imaged using the Odyssey Fc imaging system (LI-COR), with 700 and 800 nm detection channels and an exposure time of 2 min. Rhodopsin expression relative to β-actin was calculated from acquired images by band densitometry using Image Studio Lite software (version 5.2.5; LI-COR).
Gene expression analysis
For RNA extraction from neural retina, animals were sacrificed and retinal tissue rapidly dissected in sterile PBS. Tissue was then frozen on dry ice and stored at −80°C. The miRNeasy Mini kit (Qiagen, Manchester, United Kingdom) was used for RNA extraction from retinal tissue. In the first step of this process, frozen retinas were thawed on ice and tissue disrupted in 700 μL TRIzol by drawing it up and down through a 25G needle six to eight times. RNA was then isolated in accordance with the manufacturer's instructions. An additional digestion with DNase was included to remove genomic DNA contaminants (RNase-free DNase set; Qiagen). Reverse transcription using the Invitrogen SuperScript III Kit (Thermo Fisher Scientific) was immediately performed using 1 μg RNA.
Real-time quantitative PCR (RT-qPCR) was used to determine levels of gene expression from complementary DNA (cDNA) samples using TaqMan gene expression assays for human rhodopsin, mouse rhodopsin, and EGFP (Thermo Fisher Scientific). RT-qPCR was performed using the CFX Connect™ real-time PCR detection system (Bio-Rad). Thermal cycling conditions consisted of an initial polymerase activation period at 95°C for 10 min, followed by 40 × PCR cycles of denaturation at 95°C for 10 s and annealing/extension at 60°C for 1 min.
Ocular imaging
All retinal imaging was performed using a 55° lens on the Spectralis imaging platform (Heidelberg Engineering, Heidelberg, Germany). Animals were anesthetized and pupils dilated as described above. Hypromellose 0.3% (Blumont Healthcare Ltd., United Kingdom) drops were instilled bilaterally and polymethyl methacrylate contact lenses (Cantor & Nissel Ltd., Brackley, United Kingdom) were applied. The confocal scanning laser ophthalmoscope (cSLO) module was used to acquire near-infrared (815 nm diode laser) and blue autofluorescence (BAF; 486 nm blue diode excitation laser with a 500 nm barrier filter) images of the fundi in the confocal plane of the retinal pigment epithelium (RPE) centered on the optic disc. BAF images were acquired at sensitivity values (which can be set between 31 and 107) of 50, 60, and 70 with normalization switched off. All images were acquired in automatic real-time and high-resolution modes.
Spectral domain ocular coherence tomography (SD-OCT) images were acquired in a radial orientation centered on the optic disc from an average of 25 B-scans per image. For each eye at each time point, eight equally spaced scans were acquired. Photoreceptor layer (PRL) thickness (defined as the distance between the high-contrast outer plexiform layer and RPE bands) was measured from these images using calipers built into the Spectralis software at superior, superonasal, nasal, inferonasal, inferior, inferotemporal, temporal, and superotemporal retinal locations at a distance of 22.5° from the optic nerve margin. Mean PRL thickness for each eye was calculated as the mean of all eight measurements.
Electroretinography
Before electroretinography (ERG), mice were dark adapted overnight (minimum 12 h) within a purpose-built light-tight cabinet. Mice were anesthetized and pupils dilated as described above. The animal was then transferred onto a heated platform set to 38°C within a dark cabinet and ground and reference electrodes placed subcutaneously on the flank and between the eyes, respectively. Following bilateral instillation of hypromellose 1% drops (Alcon, Camberley, United Kingdom), custom contact lenses produced from achromatic ACLAR (Honeywell International, Bracknell, United Kingdom) were used to hold silver thread Dawson, Trick & Litzkow Plus electrodes (Diagnosys, Cambridge, United Kingdom) in place on each cornea. Having completed setup, the platform was maneuvered into the center of a Ganzfeld stimulator (Diagnosys) and the mouse left to acclimatize for a period of 10 min before commencing the ERG protocol using Espion software (Diagnosys). Dark-adapted responses to uniform white flashes of increasing light intensity were recorded, followed by a dark-adapted flicker protocol as per Supplementary Table S1. Mice were subsequently exposed to full-field 30 cd/m2 white background light for a period of 10 min, an intensity greater than that expected to achieve complete rod saturation. Animals were then subjected to further flash and flicker stimuli superimposed upon this background (Supplementary Table S1).
Results
Comparison of RHO-AAVs in Nrl.GFP/+, Rho−/− mice
To quantify and compare the efficacy of each vector in vivo, each was injected initially in the rhodopsin null background, because all rhodopsin can be assumed to be vector derived. Each of the four rhodopsin-expressing AAVs (AAV-ssRHO, AAV-WPRE, AAV-Ex/Int, AAV-scRHO) was injected subretinally at a dose of 2 × 109 gc in postnatal week (PNW)3 Nrl.GFP/+, Rho−/− mice. Four weeks later, animals were subjected to SD-OCT imaging. Mice were then sacrificed and retinas harvested for IHC (n = 6 eyes per vector) and western blot (n = 6 eyes per vector) for human transgene detection. Figure 2A shows retinal cryosections from such mice stained for rhodopsin. All four vectors were capable of driving human rhodopsin protein expression in rods in the murine retina, although at low levels relative to age-matched wild-type mice. Transduced rods elaborated rudimentary OS packed with rhodopsin that were absent in nontransduced eyes. Rhodopsin expression was confined to this layer mimicking the wild-type state and confirming rod promoter specificity and appropriate trafficking. Rhodopsin expression appeared greatest in AAV-Ex/Int and AAV-scRHO-injected eyes.
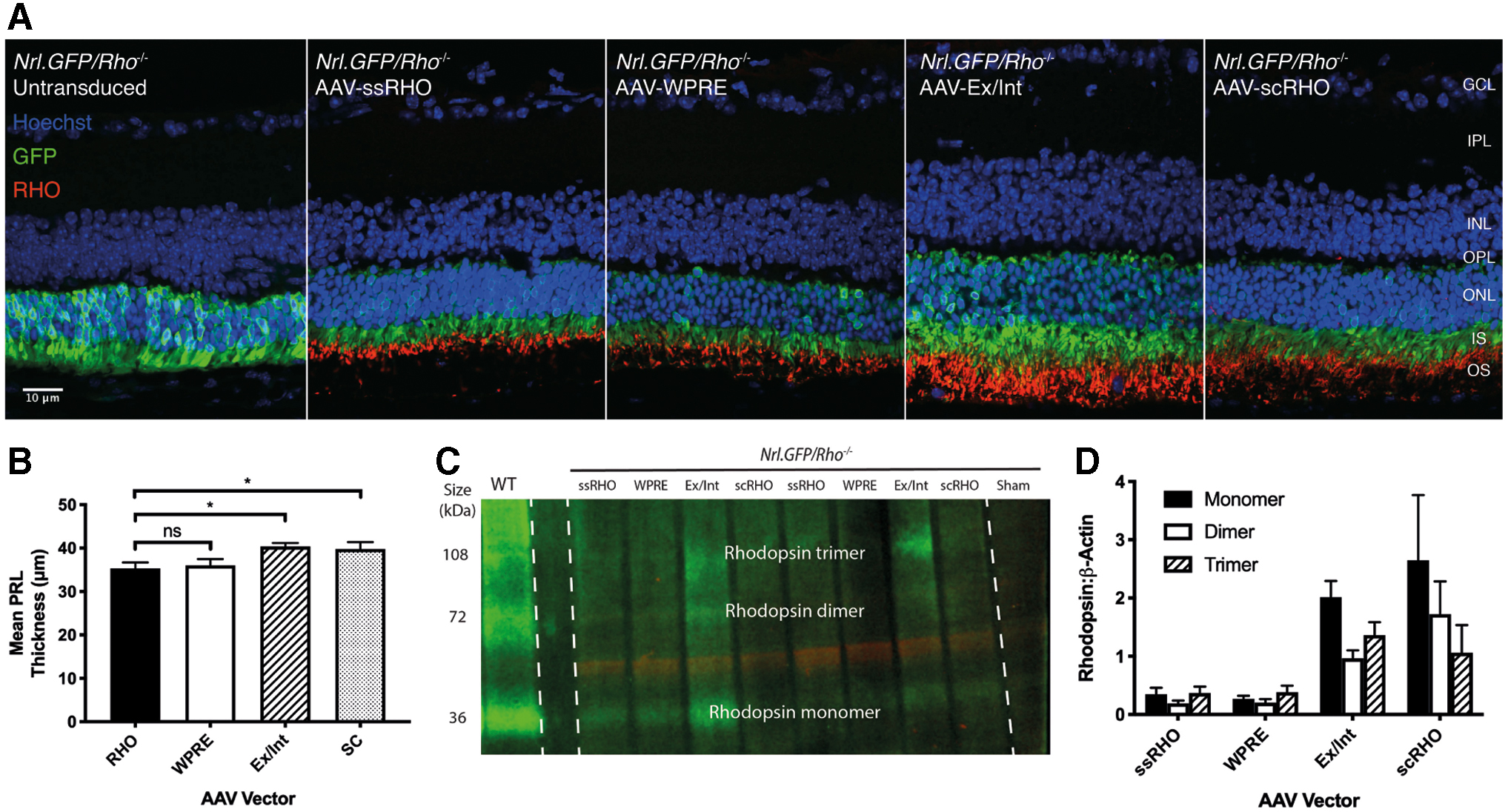
Comparison of RHO-AAVs in Nrl-GFP/+, Rho−/−
mice.
In vivo anatomical assessments of retinal thickness were then performed to confirm physiologic function of the transgenic human rhodopsin. Mean PRL thickness as determined from SD-OCT image analysis was greatest for Nrl.GFP/+, Rho−/− eyes injected with AAV-Ex/Int and AAV-scRHO (Fig. 2B). Similarly, these two vectors induced the highest levels of rhodopsin protein expression in Nrl.GFP/+, Rho−/− eyes as measured by western blot performed using protein lysates derived from transduced neural retinas (Fig. 2C, D). Note that vector-driven expression of rhodopsin in the knockout model was significantly lower than that observed in age-matched wild-type animals (Fig. 2C). However, since whole retina was used, this analysis is likely to underestimate expression levels in treated regions.
Taken together, these data would suggest that the addition of the WPRE and chicken β-actin/rabbit β-globin exon/intron/exon sequences (AAV-Ex/Int; 3.6 kB) can allow similar levels of rod-specific rhodopsin transgene expression to be achieved using a single-stranded construct as that associated with a self-complementary vector (AAV-scRHO; 4.7 kB), but with a significantly smaller AAV genome.
RHO-AAVs induce rhodopsin overexpression in the Nrl.GFP/+, RhoP23H/ + retina
To replicate the clinical scenario of treatment of the dominantly inherited disease, the level of rhodopsin overexpression following subretinal delivery of RHO-AAVs in the Nrl.GFP/+, RhoP23H/+ retina was assessed. We chose the two most efficient vectors- AAV-Ex/Int and AAV-scRHO-, which were injected at a dose of 2 × 109 gc unilaterally into the subretinal space of PNW3 Nrl.GFP/+, RhoP23H/+ mice. Four weeks later, retinas were harvested and RHO protein levels were compared between injected and uninjected eyes by western blot (Fig. 3A–C). A higher level of RHO was detected in injected versus uninjected retinas for both AAV-Ex/Int and AAV-scRHO, although this difference did not reach statistical significance for the latter vector [two-way ANOVA with eye and multimeric structure as factors: F(1, 5) = 10.3, p = 0.023 and F(1, 5) = 2.1, p = 0.21 for AAV-Ex/Int and AAV-scRHO, respectively for effect of eye]. Including both native murine levels and transgenic rhodopsin together, the mean level of overexpression was similar at 151% for AAV-Ex/Int and 153% for AAV-scRHO.
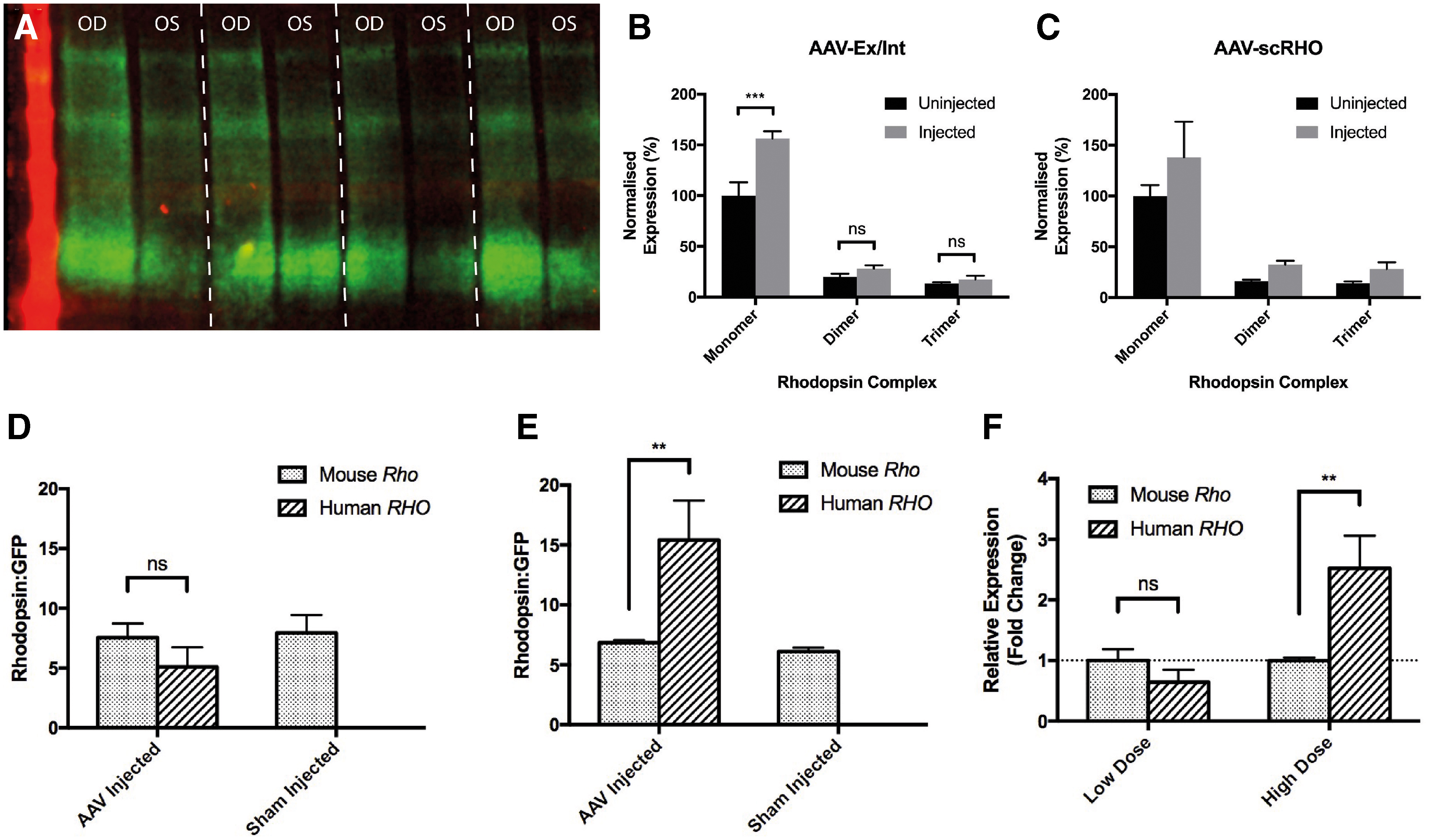
RHO AAVs induce rhodopsin overexpression in the Nrl.GFP/+, RhoP23H/+
mouse.
Gene expression analysis by RT-qPCR was next performed to determine the extent to which levels of AAV-derived human rhodopsin expression compared with those of endogenous mouse rhodopsin. For this, right eyes of Nrl.GFP/+, RhoP23H/+ mice were injected with AAV-Ex/Int at either low dose (2 × 108 gc) or high dose (2 × 109 gc), whereas left eyes received isovolumetric sham injections of PBS to correct for confounding effects of OS damage related to subretinal injection. Retinas were harvested 4 weeks later from which cDNA was derived for RT-qPCR analysis (Fig. 3D–F). At low dose, AAV-Ex/Int achieved a transgenic human RHO expression level that was 0.64 that of endogenous mouse Rho in the sham-injected eye (Fig. 3F; p = 0.66, two-way ANOVA, Sidak's multiple comparison test). At high dose, the expression of the transgene was 2.5 × that of the endogenous mouse Rho in the fellow eye (Fig. 3F; p = 0.0055, two-way ANOVA, Sidak's multiple comparison test).
Effect of rhodopsin supplementation on retinal structure in the Nrl.GFP/+, RhoP23H/ + mouse
To establish whether AAV-mediated RHO supplementation is sufficient to slow the rate of retinal degeneration in the P23H knock-in mouse model, unilateral injections of AAV-Ex/Int at one of two doses, 2 × 108 gc (low dose) or 2 × 109 gc (high dose), were performed in Nrl.GFP/+, RhoP23H/+ mice aged PNW3. A third cohort received isovolumetric unilateral injections of PBS and served as a control group to determine the effect of surgery alone. The impact of these interventions on retinal structure was determined by cSLO and SD-OCT at 1, 2, and 3 months postinjection (Fig. 4).
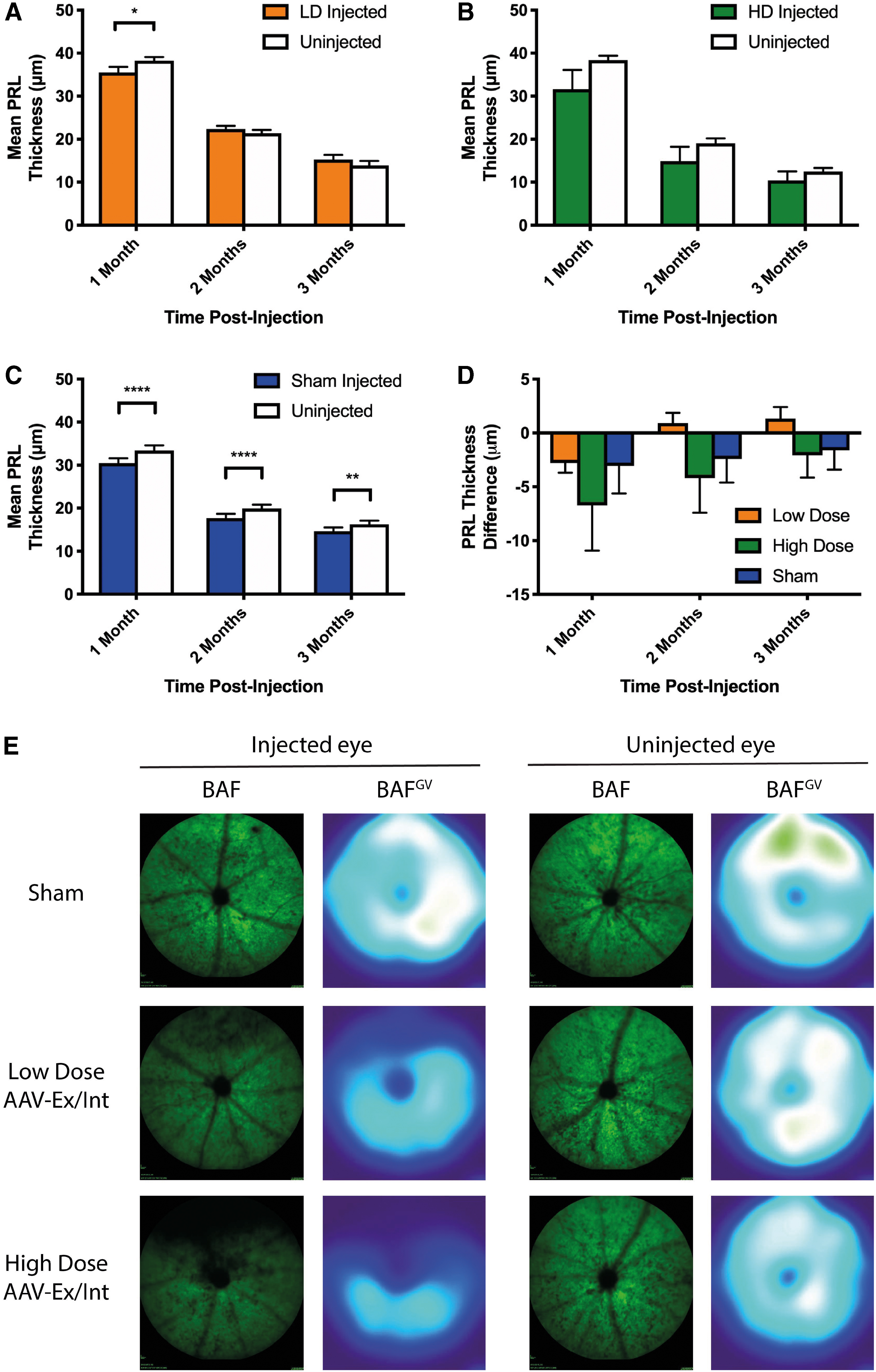
Effect of subretinal injection of AAV-Ex/Int on mean PRL thickness in the Nrl.GFP/+, RhoP23H/+
mouse. In all cases, right eyes of mice aged PNW3 received single superior subretinal injections of either AAV-Ex/Int at LD (2 × 108 gc, n = 7), HD (2 × 109 gc, n = 7), or PBS (sham, n = 13). Left eyes were uninjected as internal controls. Repeated measures two-way ANOVA tests with eye and time as factors were used throughout.
Mean PRL thickness was mildly reduced in injected versus uninjected eyes of animals in the sham-injected cohort (a relative reduction of 8.8%, 11.2%, and 9.9% at 1, 2, and 3 months, respectively). The surgical procedure itself thus induced a small but significant thinning of the PRL. No significant overall difference was however detected between the mean PRL thickness of treated and untreated eyes in either the low- or high-dose cohorts. To enable comparison of the three interventions directly, the numerical difference in mean PRL thickness between injected and uninjected eyes was calculated for each animal for the three cohorts. In this analysis, positive values would suggest a slowing of degeneration in the treated compared with the untreated eye while negative values would indicate an exacerbation of degeneration relative to the fellow eye. The graph in Fig. 4D plots mean PRL thickness difference for the three groups as a function of time. No significant difference between the groups was found and there was no statistical interaction between the factors of experimental group and time.
Given that all injections in this study were performed in the superior retina and that a dorsoventral gradient of degeneration exists in the RhoP23H/+ model, 19 analysis of mean PRL thickness in isolation may mask regional differences. PRL thickness as a function of retinal location was therefore assessed for the three cohorts. In all groups, a thinning of the PRL was apparent in the superior regions of the retina corresponding to the area of injection (Supplementary Fig. S1). This loss of rods was also evident on BAF cSLO imaging (Fig. 4E).
Effect of rhodopsin supplementation on retinal function in the Nrl.GFP/+, RhoP23H/ + mouse
Full-field dark- and light-adapted ERG was performed for low-dose, high-dose, and sham cohorts 1 and 3 months postinjection (Figs. 5 and 6). A significant reduction in dark-adapted responses of injected compared with fellow uninjected eyes was apparent for the high-dose and sham cohorts at 1 month [repeated measures two-way ANOVA with stimulus intensity and eye as factors: F(1, 6) = 14.4, p = 0.009 and F(1, 6) = 7.8, p = 0.0311 for a- and b-waves in the high-dose cohort, and F(1, 11) = 21.1, p = 0.0008 and F(1, 11) = 6.4, p = 0.028 for a- and b-waves in the sham cohort], but these differences failed to reach statistical significance at 3 months. No difference in dark-adapted response was detected between treated and untreated eyes in the low-dose group at either time point (Fig. 5).
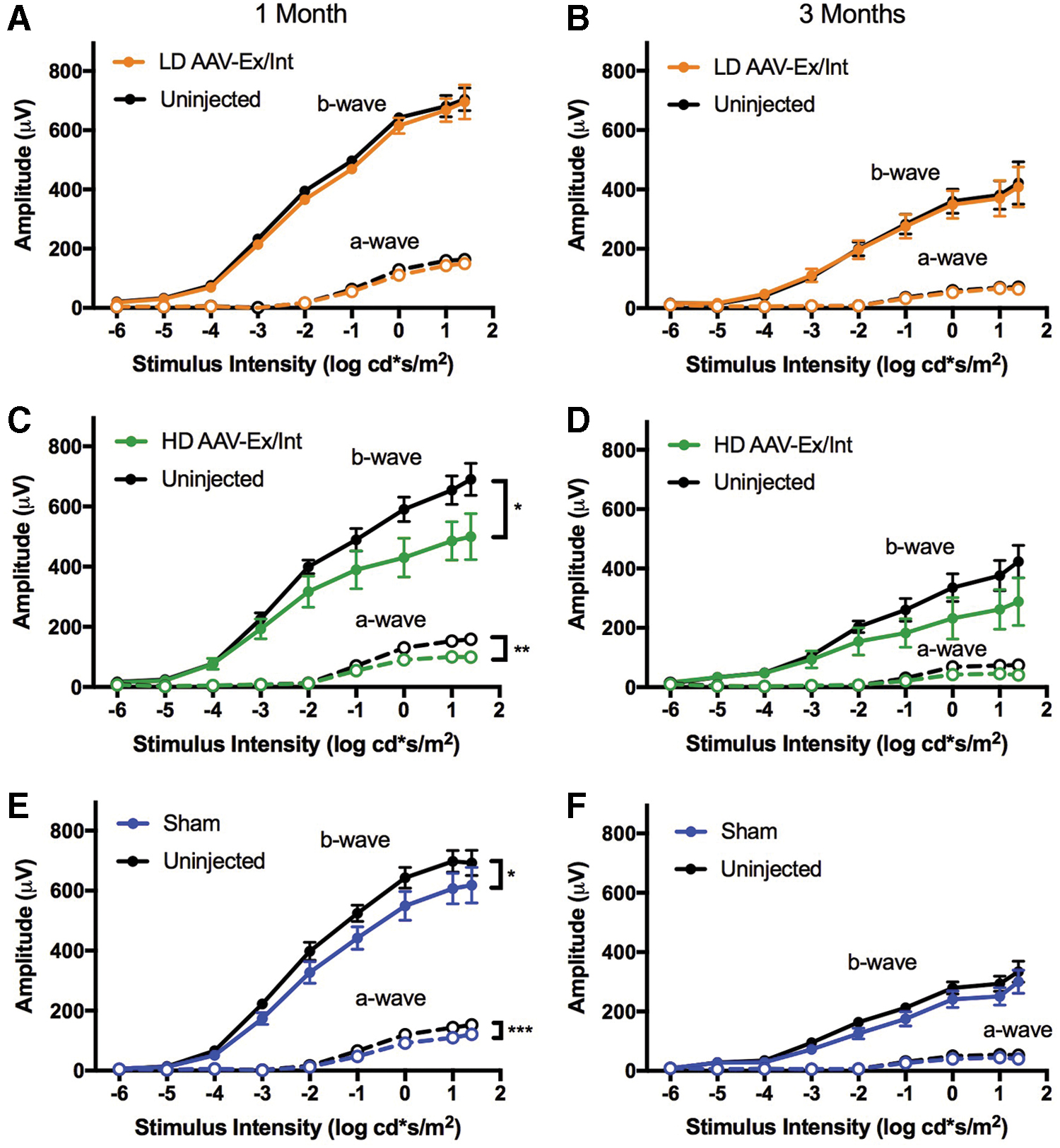
Full-field dark-adapted ERG irradiance-response curves for low- and high-dose AAV-Ex/Int injected and sham-injected cohorts 1 and 3 months postinjection. All mice received a single superior subretinal injection unilaterally at PNW3. All data points represent mean signal ± SEM.
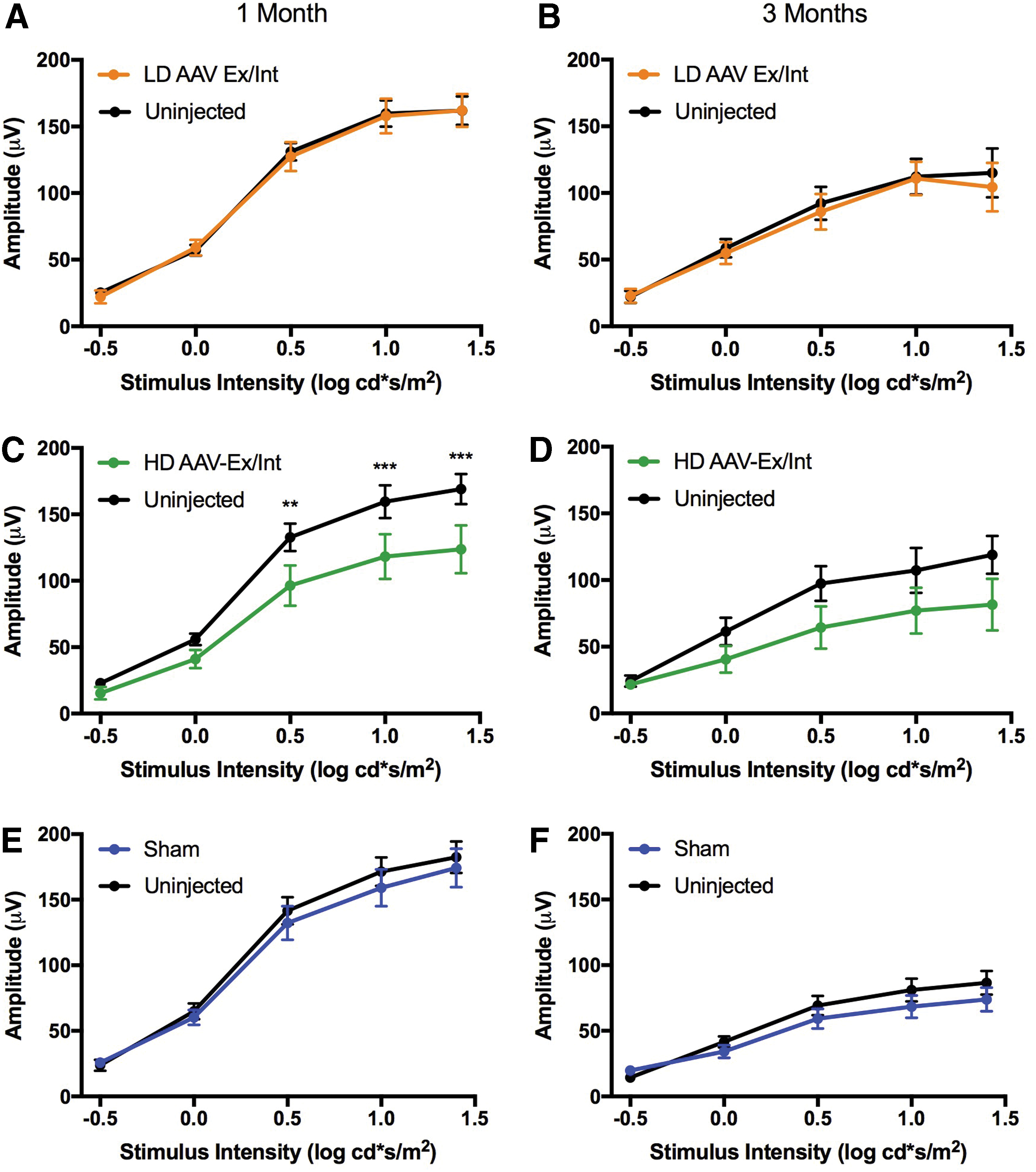
Full-field light-adapted ERG irradiance response curves for low- and high-dose AAV-Ex/Int-injected and sham-injected cohorts. All mice received a single superior subretinal injection unilaterally at PNW3. All data points represent mean signal ± SEM.
For the light-adapted response, no significant difference in overall response between injected and uninjected eyes was present at either 1 or 3 months posttreatment for any of the three experimental cohorts (Fig. 6). A statistical interaction between the factors of stimulus intensity and eye was, however, confirmed by repeated measures two-way ANOVA testing for the high-dose cohort at 1 month [F(4, 24) = 3.6, p = 0.019] and statistically significant differences between right and left eyes were detected at higher stimulus intensities in post hoc analyses (Sidak's multiple comparison test; Fig. 6). This might suggest a detrimental effect of high-dose AAV-Ex/Int injection on overall cone function.
To allow a comparison to be made between groups, further irradiance response curves (IRCs) were plotted using the difference in recorded amplitude between injected and uninjected eyes for the three cohorts (Supplementary Fig. S2). Positive values represent improved signal from injected eyes relative to their uninjected counterparts, whereas the reverse applies for negative values. No statistically significant differences were detected between the groups for dark- or light-adapted responses at 1 or 3 months postinjection (Supplementary Fig. S2). However, a statistical interaction between stimulus intensity and cohort was detected for dark-adapted a- and b-waves 1 month postinjection [F(16, 200) = 2.4, p = 0.0025 and F(16, 184) = 2.5, p = 0.0016, respectively] and post hoc multiple comparison tests (Sidak) showed significant differences between the response of high-dose and low-dose/sham groups for the brightest flash intensities (e.g., a-wave 1.4 cd.s/m2: HD vs. Sham p = 0.0013; HD vs. LD p < 0.0001; and b-wave 1.4 cd.s/m2: HD vs. Sham p = 0.0171; HD vs. LD p = 0.0005).
In summary, no form of injection appeared to improve ERG response compared with the fellow untreated eye (all curves lie below the zero line in Supplementary Fig. S2), and at high dose, the AAV-Ex/Int vector appeared more damaging than an equivalent low dose or sham injection.
Discussion
Overexpression of wild-type rhodopsin has been proposed as a possible treatment for the subset of rhodopsin mutations that act in a dominant negative fashion. 3,17 Others have advocated a mutation-independent “knockdown/replacement” paradigm in which all genomically expressed rhodopsin is targeted for knockdown (using, e.g., RNA interference or CRISPR/Cas9 systems), whilst a resistant version of the gene is codelivered. 18,22 –24 Rhodopsin constitutes around 90% of the total protein found in the rod OS and has a high level of turnover. 25 Correspondingly high levels of RHO expression from an AAV are thus likely to be required if the transgenic wild-type protein is to overcome any dominant negative effect of the genomically expressed mutant. An efficient RHO vector would also be required as part of any “knockdown/replacement” treatment paradigm. 16 In this study, we have compared four optimized AAV2/8Y733F vectors carrying wild-type copies of the human rhodopsin CDS. An in vitro validation of these constructs was not possible since no immortalized cell line is known to have sufficiently strong rhodopsin promoter activity. The murine Rho−/− retina thus represents the most appropriate current model in which to validate and compare rhodopsin-expressing constructs, and was thus used in this study and has been used by others for this purpose. 26,27 These mice do not express rhodopsin, lack OS, and their dark- and light-adapted ERG responses are derived solely from cones. 28 Any rhodopsin expressed following gene therapy in this model must therefore be transgenic in origin. Of the four vectors developed and tested in this study, AAV-Ex/Int and AAV-scRHO appeared to be the most efficient. Self-complementary AAVs are thought to result in a higher level of transgene expression with a more rapid onset as they bypass the rate-limiting step of second-strand synthesis. 29 However, their clinical application may be limited due to the significant loss of packaging capacity associated with their genome structure, as well as lower achievable titers during manufacture and potential increased immunogenicity. 30 AAV-Ex/Int, which includes two cis-acting elements intended to enhance gene expression, 31,32 was able to induce similar levels of rhodopsin protein expression as AAV-scRHO. As a single-stranded vector, AAV-Ex/Int does not suffer from the limitations listed above and has ample remaining packaging space for the subsequent inclusion of additional elements to mediate rhodopsin knockdown in a “knockdown/replacement” vector. It is interesting to note that inclusion of the WPRE in isolation appeared to confer no detectable increase in vector potency (AAV-RHO vs. AAV-WPRE; Fig. 2). Patricio et al. demonstrated that the WPRE could enhance expression of transgenic protein (eGFP or Rab escort protein 1 [REP1]) driven by the CAG promoter in vitro, and within both the mouse and human retina. 32 The WPRE effect may thus be promoter specific, or alternatively may rely on the presence of an intron within the AAV-derived transcript. Context dependency for the WPRE and other regulatory elements has been reported elsewhere and remains an area of controversy. 33,34
In a previous study, Palfi et al. compared six RHO-AAV2/5 vector designs in the Rho−/− model. 26 They found that the highest levels of transgene expression were achieved when the vector included the WPRE sequence and a longer mouse rhodopsin promoter. The authors were additionally able to demonstrate a reconstitution of rod OS following subretinal RHO-AAV injection at postnatal day (PND)0, which was accompanied by a modest rescue of the rod-derived ERG. 26,27 In this study, we have shown that a similar reconstitution of OS with appropriate trafficking of transgenic wild-type human rhodopsin is achievable following injection at PNW3, that is, after the point of terminal rod differentiation. This is an important observation as terminally differentiated rods rather than photoreceptor precursors would be the target for treatment in any human clinical trial.
In this study, we found that subretinal injection of 2 × 109 gc AAV-Ex/Int and AAV-scRHO in Nrl.GFP, RhoP23H/+ mice could achieve total rhodopsin expression levels of 151% and 153% that of untreated fellow eyes, respectively. This level of vector-induced expression appears much greater than that achieved using the same vectors at the same dose in the rhodopsin knockout model (Fig. 2C). The difference in transgenic expression levels may be explained by considering the structural differences between these two models. First, in contrast to the Nrl.GFP, RhoP23H/+ model, the Nrl.GFP, Rho−/− mouse retina develops without OS, suggesting that rhodopsin protein is essential for their formation. When applying gene therapy long after these structures would normally develop, the ability for the outer nuclear layer (ONL) to regrow OS against all the forces of retinal adherence would likely be compromised to some extent, which might limit the volume of the OS generation. In keeping with prior reports, the observed OS lengths in treated knockout mice in our study were significantly shorter than those of wild-type animals, being less than or equal to IS length in all cases. 26 Second, the Nrl.GFP, Rho−/− mouse undergoes a more rapid degeneration than the Nrl.GFP, RhoP23H/+ mouse such that at the point of injection (PNW3) and sacrifice (PNW7), there would be fewer surviving rods to be transduced and express protein, respectively, in the knockout model. 21 Finally, the superior retina in the P23H knock-in model is relatively well preserved until much later in the lifetime of the mouse owing to the dorsoventral gradient of degeneration that exists in this model, 19,21,35 while no such gradient exists in the knockout mouse. 21 Since all injections were performed superiorly, the subretinal injection in the P23H knock-in model would result in transduction of a thicker, more viable ONL when compared with that of the rhodopsin knockout mouse. These anatomical differences prevent any direct comparison between vector-induced protein expression levels in Nrl.GFP/+, Rho−/− and Nrl.GFP/+, RhoP23H/+ mice.
In this study, we investigated whether a rescue effect may be achieved by way of wild-type gene augmentation by subretinal injection of the optimized AAV-Ex/Int vector at two different doses in the Nrl.GFP/+, RhoP23H/+ mouse and found no evidence of either structural or functional benefit. Indeed, at the higher dose tested (2 × 109 gc), a degree of outer retinal toxicity was observed with superior PRL thinning and ERG attenuation relative to the uninjected eye being greater than that observed in sham-injected animals. These results contrast to those found in a similar study by Mao et al., in which wild-type RHO cDNA was delivered by AAV in a transgenic RHOP23H mouse on a Rho+/+ background, where a significant treatment effect was observed. Although a similar level of rhodopsin protein overexpression was achieved, a number of notable differences exist between study of Mao et al. and the work presented here which may, at least in part, explain this discrepancy. First, the mouse model used in this study was a knock-in as opposed to a transgenic species. Although this more faithfully models the human P23H mutation at a molecular level, 19 the rate of degeneration in this mouse is much more rapid than that observed in the transgenic line used by Mao et al., and contrasts with the relatively slow rate of progression which characterizes RHOP23H dominant disease in humans. 36 It is interesting to note that in a subsequent study involving the same transgenic mutation, but on a mouse with Rho+/− genetic background, Mao et al. reported only a small and transient treatment effect from AAV-mediated wild-type rhodopsin supplementation. 18 Any patient who is to undergo gene therapy treatment for rhodopsin-related ADRP is likely to have long established disease at the point of intervention. For this reason, animals were injected here after weaning at PND21 and not at earlier time points when fewer rods would already have degenerated. By contrast, Mao et al. injected mice in their study at PND15. 17 Finally, Mao et al. delivered the mouse rhodopsin gene in their vector while the human coding sequence was used in this study. Although significant homology exists between the two resulting proteins, it is possible that a treatment effect was not observed in the present study due to differences in the affinity for complementation between the two versions of rhodopsin. Indeed, a bias toward overexpression of monomeric forms of rhodopsin was suggested by western blot analysis in this study (Fig. 3B). This may have impacted on the efficiency rhodopsin trafficking from the ER to OS and in turn diminished any possible treatment effect. In summary, the combination of genetic background and rate of natural disease progression in their mouse model, combined with an earlier timing of intervention and use of a species-specific rhodopsin coding sequence by these researchers, may explain why the gene supplementation strategy proved successful in their study, but failed to impart a measurable benefit here.
A degree of toxicity was observed in the treated area of retina when the AAV-Ex/Int vector was injected at high dose. Possible causes of this toxicity include capsid-related immunogenicity or cytotoxicity (although this has not been observed in other studies using AAV2/8Y733F 37 ), or cell death induced by overexpression of the rhodopsin protein. 9,18,38
Nrl.GFP models were used throughout this study to allow noninvasive en face imaging of the surviving ONL by cSLO. One potential limitation of this approach is the possibility of an interaction between the GFP protein and the degenerative process or the gene therapy intervention. Although we feel this is unlikely given that the presence of GFP does not appear to significantly affect the phenotype of wild-type or degenerate models with respect to ONL thickness, 19 –21 this possibility cannot be entirely excluded.
AAV-mediated supplementation has proven ineffective in treating rhodopsin mutations known to mediate cell death by toxic gain of function mechanisms such as the Class I P347S mutation. 22 The fact that no rescue was apparent following overexpression of rhodopsin in the knock-in model in this study might support the hypothesis that the genomic P23H mutation, when present in the heterozygous state, predominantly induces cell death by a toxic gain of function rather than a dominant negative mechanism. However, other factors such as late intervention, or over- or under-dosing at the level of individual photoreceptors as outlined above might offer alternative explanations for these findings. Further in vivo studies are thus likely to be required to definitely elucidate both the mechanism of degeneration, as well as the complex interaction between wild-type and mutant rhodopsin in the P23H retina.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This study was supported by the Medical Research Council (MRC), UK and Fight for Sight, UK, grant reference MR/N00101X/1.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.