
Abstract
After more than two decades since clinical trials tested the first use of recombinant adeno-associated virus (rAAV) to treat cystic fibrosis (CF) lung disease, gene therapy for this disorder has undergone a tremendous resurgence. Fueling this enthusiasm has been an enhanced understanding of rAAV transduction biology and cellular processes that limit transduction of airway epithelia, the development of new rAAV serotypes and other vector systems with high-level tropism for airway epithelial cells, an improved understanding of CF lung pathogenesis and the cellular targets for gene therapy, and the development of new animal models that reproduce the human CF disease phenotype. These advances have created a preclinical path for both assessing the efficacy of gene therapies in the CF lung and interrogating the target cell types in the lung required for complementation of the CF disease state. Lessons learned from early gene therapy attempts with rAAV in the CF lung have guided thinking for the testing of next-generation vector systems. Although unknown questions still remain regarding the cellular targets in the lung that are required or sufficient to complement CF lung disease, the field is now well positioned to tackle these challenges. This review will highlight the role that next-generation CF animal models are playing in the preclinical development of gene therapies for CF lung disease and the knowledge gaps in disease pathophysiology that these models are attempting to fill.
Introduction
Cystic fibrosis is a multiorgan disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. 1 CFTR is an anion channel that conducts Cl− and HCO3 − across several types of epithelium in the lung, intestine, pancreas, liver, and gallbladder. Each of these organs shares commonalties in pathology, including impaired hydration of the epithelial surface, excessive mucus production and obstruction, and reduced pH of secretions. 2 In the cystic fibrosis (CF) lung, these abnormalities impair clearance and innate immunity, and they lead to chronic bacterial infections. 3 –7 Since lung disease is the most life-threatening component of CF, it is considered the primary initial target organ for gene therapies.
The first CF clinical trials with recombinant adeno-associated virus (rAAV) utilized a rAAV-2 serotype to express CFTR in the maxillary sinuses. 8 Results from this initial dose-escalation toxicity study demonstrated it was safe and that the vector genome accumulated in the sinus epithelium. A subsequent does-escalation study demonstrated in 2 of 10 patients that the CFTR transgene was expressed and the transepithelial potential difference, an index of CFTR-mediated Cl− transport, was partially corrected. 9,10 These studies paved the way for testing the rAAV2-CFTR vector through aerosol administration to the lungs of CF patients 11 –13 and culminated in a repeat-dosing aerosol administration study, which examined forced expiratory volume (FEV1) as the primary endpoint. 13 Although the rAAV2-CFTR vector was proven safe in each of these studies, there was no improvement in the primary endpoint of the repeat-dosing study and it was subsequently terminated. Results from this repeat administration study demonstrated persistence of vector genomes, but no expression of CFTR transgene-derived mRNA.
Reasons for the failure of these initial rAAV2-CFTR clinical trials targeting the lung were difficult to predict at the time, but they emerged with more basic research over the subsequent decade. First, the original rAAV2-CFTR vector system (called tgAAVCF) utilized the inverted terminal repeat (ITR) within the vector genome as the promoter. Although this promoter was weak, it was necessary to fit the full-length CFTR cDNA into the vector without exceeding the packaging capacity. 14 Later improvements in vector design utilized short synthetic promoters with the full-length CFTR cDNA 15 or smaller mini-gene versions of CFTR with a deleted region that did not alter channel function. 16 –18 Second, despite efficient entry of rAAV2 from the apical membrane of airway epithelia, this serotype—and many others for that matter—encounters a proteasome-dependent block in nuclear uptake. 19 –24 This barrier was later circumvented when it was discovered that proteasome inhibitors can significantly enhance apical transduction of rAAV in airway epithelia and thus also enhance complementation of Cl− transport in CF epithelia by using rAAV-CFTR vectors. 15,21 –24 Lastly, the preclinical rhesus monkey studies that supported the initial rAAV2-CFTR clinical trial demonstrated efficient transduction of the airway, 25 but it was later found that the tropism of rAAV2 for the apical surface of polarized human primary airway epithelia was 100-fold lower than that observed in polarized rhesus monkey primary airway epithelia. 26 These studies emphasized the need for a concrete understanding of vector/target cell biology and the importance of choosing a preclinical animal species that has a similar vector tropism to humans.
It is clear that the lack of CFTR in the airways leads to impaired mucociliary clearance caused by poor hydration of airway surface liquid (ASL) and that the resultant viscous mucus secretions provide the environment for bacterial colonization. However, a significant knowledge gap that remains for CF lung gene therapy is a clearer understanding of the target cell types required for complementation of disease. CFTR expression in the lung is highly regulated at the cellular level. 27 –30 For example, submucosal glands (SMGs) embedded beneath the airways express CFTR 27 and CFTR-dependent secretions from these structures are important in innate immunity in the airway. 31,32 If these structures play a role in CF pathogenesis, as research in CF animal models suggests, 33 strategies for CFTR delivery to these structures, which are relatively inaccessible from the airway lumen, may require unique vector systems.
An additional potential unique cellular target includes the newly characterized pulmonary ionocyte, 29,30 which expresses the majority of CFTR in the conducting proximal airways and SMG ducts. Ionocytes compose only ∼1–2% of cells on the surface of the large airways and these cells have a very unique cellular anatomy and composite of channels that suggests they are involved in pH regulation and ion transport. Whether ionocytes are a required cell type for complementation of CF lung disease remains unknown, but based on the composition of other channels that are co-expressed with CFTR in these cells, their discovery raises the question as to whether certain functions of CFTR are cell-intrinsic. 34 CFTR also physically interacts with other ion channels, such as the epithelial sodium channel (ENaC), 35 to regulate ion and fluid movement across airway epithelia. 36,37 These studies emphasize that certain functions of CFTR may be dependent on the cellular composite of other channels that control ion movement.
Our current understanding of how CFTR maintains airway clearance and innate immunity in a multicellular airway epithelium is based predominantly on the notion that CFTR functions similarly in any columnar cell type (i.e., cells with an apical and basolateral membrane) to move anions across the epithelium. This notion is supported by the finding that CFTR gene replacement using many types of viral vectors, including rAAV, 15,16,38 human bocavirus type 1 (HBoV1), 39 lentivirus, 40,41 helper-dependent adenovirus (HDAd), 42 and human parainfluenza virus, 43 can complement CFTR-mediated Cl− currents after transduction of bronchial airway cultures derived from humans with CF that are grown at an air-liquid interface (ALI). If one accepts that this wide array of viral vectors likely has differing cellular specificities for transduction of airway cell types, then the cellular partitioning of CFTR expression in vivo is likely not an obstacle for CF lung gene therapy if Cl− transport is the primary driver of disease.
Puzzling paradoxes, however, exist in translating in vitro findings to the in vivo setting in terms of the cellular specificity of CFTR function. For example, forced expression of CFTR in polarized human CF ALI cultures using human parainfluenza virus, which is believed to primarily target ciliated cells, can correct defects in Cl− transport and mucociliary clearance. 43 However, studies in transgenic mice overexpressing CFTR under the control of the Foxj1 promoter (which drives expression specifically in ciliated cells) on a CFTR-knockout (KO) background failed to rescue CFTR-dependent nasal potential difference despite high levels of CFTR mRNA and protein expression. 44 These contradictory findings raise two important questions: Is the ciliated cell an effective target for CF gene therapy? Has the heavy reliance on Cl− transport as an endpoint for efficacy in the development of CF gene therapies led to oversimplified thinking about disease complementation in vivo, ignoring the fact that airway fluid dynamics and pH, mucus secretion and clearance, and innate immunity are regulated by specific cell populations, including those in the surface airway epithelium (SAE) and SMGs? 6,31 –33,45 –48 Addressing this in vivo biology has historically been difficult, because mice do not develop lung disease in the absence of CFTR 49 –52 and this species also lacks SMGs in the cartilaginous airways. 53 –55
Armed with next-generation viral vector systems and an improved understanding of viral transduction biology in the airway epithelia, the field is now well positioned with effective tools to deliver CFTR to the lung. However, remaining unknowns about CF lung pathogenesis, and how various CFTR-expressing cell types coordinate lung clearance and innate immunity, present potential hurdles for gene therapy. New CF animal models have begun to pave the way to address these challenges and will continue to play an important role in the preclinical development of CF lung gene therapies. This article will review these knowledge gaps, discuss opportunities to address them, and describe their potential impact for future CF lung gene therapy approaches.
Animal Models of CF
After being solely dependent on CF mouse models for 20 years, the past decade has positioned the field with a Noah's Ark of CF animal models (Table 1). Each of these models was initially characterized in the CFTR-KO state. The most well-studied larger animal CF models include the ferret and pig, and each gives rise to the spectrum of multiorgan disease seen in humans with CF. Although CF mice and rats do not develop spontaneous lung infections, the CF rat does develop defects in tracheal mucociliary clearance with age that are associated with SMG hypertrophy. 56 Relatively new CF rabbit and sheep models have yet to be fully characterized for their lung phenotype in the adult state. At birth, all CF models lack gross pathology in the lung, and in the case of CF ferrets and pigs, disease in the lung develops with age. Given the focus of this review on the lung, we limit discussion to the CF ferret and pig models for which the lung phenotype has been studied in more depth.
CF animal models
CF, cystic fibrosis.
Despite the lack of gross pathology in the lungs of newborn CF ferrets and pigs, both species demonstrate dysregulated inflammatory responses after the first exposure to bacteria 84,85 and an impaired ability of the lung to eradicate bacteria. 74,85 Within weeks to months after birth, both CFTR-KO ferrets and pigs spontaneously acquire bacterial infections in the lung. 63,74 Lung disease in both CF ferret and pig models is characterized by excessive mucus that obstructs the airways, 60,61,74 impaired mucociliary clearance, 33,85 defective SMG secretions, 63,86 and polymicrobial bacterial infections. 61,74 CF ferrets are extremely sensitive to lung infections early in life before full development of airway SMGs at about 1 month of age 63 and thus require aggressive antibiotic treatment to survive. 61 When symptomatically treated with antibiotics, CFTR-KO ferrets live longer (average of 105 ± 27 days), but they still succumb to bacterial colonization of the lung. 61 When aggressively treated with three antibiotics from birth, CFTR-KO ferrets lived an average of 1,143 ± 77 days (N = 4 females and N = 3 males) and although they do not acquire bacterial lung infection, they still develop structural and mucoinflammatory lung disease. 60 The average lifespan was also not significantly different between females (1,144 ± 136 days) and males (1,174 ± 54 days). These findings have led to the concept that there are two separate components of CF lung pathogenesis involving defects in innate immunity and inflammation caused by abnormal mucus and obstruction. 87
Both pig and ferret CFTR-KO models have severe gastrointestinal defects before and after birth that has made them particularly difficult to rear; this has limited their utility as preclinical models for gene therapy. For example, all CFTR-KO pigs are born with meconium ileus (in utero obstruction of the intestine) 71 and significant exocrine pancreatic disease that is initiated in utero. 77 Similarly, a large fraction (80%) of CFTR-KO ferrets have meconium ileus 63 and their exocrine pancreatic disease progresses rapidly after birth. 59,62 Although meconium ileus in the CFTR-KO pig can be treated surgically, 74 this is not possible in CFTR-KO ferrets due to size.
For these reasons, second-generation gut-corrected CFTR-KO ferrets 63 and pigs 88 have been generated, which express CFTR under the direction of the intestinal fatty acid-binding protein (FABPI) promoter. Although the gut-corrected CF pig model has shown some protection from meconium ileus, 88,89 the similar model in ferret did not increase the ease of rearing despite protection from meconium ileus (unpublished). For these reasons, a third-generation CF ferret model was engineered to harbor a CFTR-G551D mutation (CFTR G551D) that is responsive to the CFTR modulator drug VX-770. 66
When pregnant female ferrets (jills) harboring CFTR G551D/G551D kits are given VX-770 at embryonic day 28, the CF kits born are protected from pancreatitis and meconium ileus, and when the drug is continued postnatally, the CF kits have normal growth and survival rates. CFTR G551D/KO kits are only partially protected by in utero VX-770 therapy due to reduced (∼50%) expression from the CFTR G551D allele caused by a neomycin expression cassette in the intron that was used to generate the model. Termination of VX-770 at any age reactivates disease in the pancreas, intestine, and lung 66 ; however, when this is done before 2 weeks of age, there is high mortality due to both lung and intestinal pathology. When VX-770 is terminated at 2–3 weeks of age, terminal lung disease with bacterial colonization typically occurs in the majority of animals within 1–3 months. This VX-770-responsive model will greatly expand the utility of the CF ferret for studying disease pathophysiology and the preclinical testing of gene therapies that can either prevent or reverse CF lung disease. 90
With the advent of CRISPR/Cas9 gene editing in zygotes, several new ferret models are emerging. For example, a ROSA-26 knock-in has been generated that expresses a CreERT2-responsive LoxP-Tomato-stop-LoxP-EGFP (ROSA-TG) reporter that will enable lineage tracing of stem cells in ferrets. 91 Such a ROSA-TG model will also be useful for evaluating gene transfer from Cre-expressing vector systems. Several Cre-driver ferrets have also been generated to enable lineage tracing of stem cells and the conditional deletion of CFTR in a cell-type specific fashion (unpublished). These models will assist in dissecting CF pathogenesis and the cellular targets for gene therapies.
Cellular Targets in the Lung for CF Gene Therapy
The conducting airways are believed to be the predominant target for CF gene therapy and are composed of several unique domains that house a variety of CFTR-expressing cell types important for coordinating innate immunity and clearance in the lung. Recent single-cell RNA sequencing (scRNAseq) data have significantly enhanced the ability to classify various cell types in the human conducting airways. 29,30,92,93 In humans, the proximal cartilaginous airways (trachea and bronchi) are composed of the SAE and SMGs. The SAE comprises primarily basal cells, intermediate basal cells (transitional states of basal cell differentiation to secretory and ciliated cells), secretory (goblet) cells, and ciliated cells, but they also contain less abundant neuroendocrine cells, brush cells, and ionocytes (Fig. 1A). 30,94 –96 SMGs are found in the submucosa of the cartilaginous airways and secrete mucus and fluid that are rich in antibacterial factors. 31,32 These structures contain a network of interconnected serous acini and mucous tubules that are surrounded by myoepithelial cells and are secreted into collecting ducts composed of simple columnar cells and ionocytes. 27,95 –97 Secretions exit SMGs through ciliated ducts that have a similar cellular composition to the SAE but are enriched with ionocytes. Terminal bronchioles contain basal cells, secretory club cells, ciliated cells, neuroendocrine cells, and fewer ionocytes, whereas respiratory bronchioles contain primarily club cells. 94 –97

Cellular expression patterns of CFTR in the proximal human airway.
CFTR is expressed in several of these cell types at widely divergent levels and this has created controversy over what cell types contribute to CF lung pathogenesis and have the ability to restore lung function after gene therapy. 34 For example, ciliated cells have long been considered to be a predominant CFTR-expressing cell type involved in CF pathogenesis and a target for CF gene therapy. 43,98 However, some studies have failed to detect both CFTR protein and mRNA in this cell type. 27,30 Recently, scRNAseq on human and mouse proximal airway cell types 29,30 has demonstrated that the majority of CFTR mRNA in the proximal airway is present within ionocytes, and that ciliated cells express very little in only a small fraction of cells (Fig. 1B, C). scRNAseq has also demonstrated that a subset of intermediate basal cell precursors of secretory cells and differentiated secretory cells express low levels of CFTR in greater numbers than ciliated cells (Fig. 1B, C). 29,30
CFTR mRNA and protein expression in intermediate basal cells has been previously shown in human bronchus, 27 but the functional significance remains unknown. Notably, after tracheal injury in mice, cycling basal cells increase transcription of CFTR, 30 suggesting that CFTR may play a role in basal cell differentiation to secretory cells and/or ionocytes (Fig. 1D). Ionocytes represent only ∼1–2% of proximal conducting airway epithelial cells and were first identified as “jackpot” CFTR-expressing cells enriched in gland ducts and the SAE surround these gland ducts. 27 Other cell types known to express CFTR include serous cells of SMGs 27,99 and club secretory cells, which demonstrate high levels of expression in the respiratory bronchioles. 28,99
The Pulmonary Ionocyte
scRNAseq of human proximal airway epithelial cells demonstrates that 65% of ionocytes express CFTR, whereas only 1.4% of ciliated cells have detectable CFTR mRNA at levels that are 100-fold lower than ionocytes. 100 Further, 67% of all CFTR mRNA in the large airways comes from ionocytes. 100 These findings are similar to mouse tracheal epithelia cells, where ionocytes express 54% of all CFTR transcripts, whereas ciliated cells express only 1.5%. 29 Ciliated cells constitute ∼60–70% of all columnar cell types in the normal human proximal conducting airways 101 and only polarized columnar cell types with an apical membrane can contribute to ion movement across an epithelium.
These findings raise an important question about how various cell types in the airway coordinate CFTR-dependent anion movement to control ASL volume, airway pH, innate immunity, and clearance. Do cell-autonomous functions of CFTR exist that are coordinated by other cell-specific apical and basolateral channels that collectively control ion gradients and membrane potential required for the movement of ions and fluid across the epithelium? The answer to this question has important implications for gene therapy. In this regard, pulmonary ionocytes are enriched for a number of specialized ion channels and V-ATPases 29,30 that could impart cell-specific CFTR functions as demonstrated in other systems where ionocytes participate in acid-base regulation and salt/water balance. 102 –106
Consistent with this notion, the abundance of ionocytes in polarized cultures of human bronchial epithelium grown at an ALI correlates with cAMP-inducible Cl− current, 30 and the elimination of ionocytes from mouse tracheal epithelial ALI cultures increases the depth of the ASL, the viscosity of the ASL mucus, and also alters the ciliary beat frequency. 29 The expression of V-ATPases, which transport H+ within ionocytes of the inner ear, kidney, and epididymis, is driven by Foxi1, 107 a transcription factor required for ionocyte specification in mouse, human, and zebrafish. 29,30,106 Studies of ionocyte function in the epithelia of amphibians and fish 102 –106 have implicated three classes that are involved in acid-base regulation and osmoregulation, and one of these is enriched for CFTR. 34,105 Inhibiting Foxi1 in the mucociliary skin epithelium of frogs, which ablates ionocytes, leads to altered ciliary beat frequency 106 —a finding similar to that of Foxi1-KO mouse tracheal cultures. 29 Given the importance of fluid and anion transport, as well as pH, in maintaining mucociliary transport (MCT) and innate immunity in the airway, 2,69,72,75 the known functions of ionocytes in other systems appear to be well aligned with CF phenotypes. Thus, determining whether ionocytes play a role in CF lung pathogenesis is needed to understand whether this cell type will be a required target for gene therapy.
Potential Mechanisms for Defected pH and Hydration in the CF Airway
The maintenance of an ASL layer with well-hydrated mucin is critical for effective MCT. In the absence of CFTR, ion and fluid transport across the airway epithelium is significantly impaired and this leads to dehydration of the ASL and impaired MCT. 6,36 Dehydration of ASL in the CF airway is driven by ENaC, a serine protease-activated and pH-sensitive ENaC that absorbs Na+ from the ASL and thus also drives fluid absorption. 108 –111 CFTR conducts both Cl− and HCO3 −, and defective HCO3 − secretion in CF airway epithelia reduces the pH of the ASL. 69,72,112 Given that serine proteases in the ASL are more active at a lower pH, the loss of CFTR-dependent HCO3 − secretion has been proposed to be responsible for hyperactivity of ENaC in CF and dehydration of the ASL. 111 CFTR has also been proposed to inhibit ENaC activity through direct physical interactions. 113
Distinguishing these two mechanisms of CFTR-dependent ENaC regulation are noncell autonomous and cell-autonomous modes of action (i.e., whether or not CFTR has to be expressed in the same cell as ENaC). Determining these modes of actions has a direct implication for CF gene therapy approaches and the level of cellular specificity required for CFTR expression to properly regulate ENaC. Defective CFTR-dependent ion transport has also been shown to affect fluid absorption, with CF airway epithelia absorbing fluid more slowly than non-CF epithelia after a small volume challenge. 114 The mechanism responsible for this defect includes reduced protease-dependent activation of ENaC in the absence of CFTR.
There are many potential mechanisms that may explain why CFTR expression is highly regulated at the cellular level in the airway. We will focus our discussion on two potential models that may control pH-dependent fluid movement in the airway and how they could be particularly important for designing gene therapy approaches (Fig. 2). These proposed mechanisms are based on known channels in the airway epithelium that participate in transepithelial ion and fluid transport together with CFTR 36,115 –117 and on scRNAseq data on the cell types that express CFTR. 29,30 For simplicity, we have focused on the apical channels that may participate in ion transport and fluid movement at the airway surface, but we recognize that the activity of basolateral channels and the partitioning of their expression in various cell types of the airway is equally important in establishing electrical and chemical driving forces for channel activation. 36,115,116 Both models propose that ionocytes participate in regulating ASL pH through CFTR-dependent HCO3 − secretion to control ENaC activity. These two models also assume that fluid absorption and secretion are opposing forces that maintain ASL height at steady state in a normal airway epithelium. 118
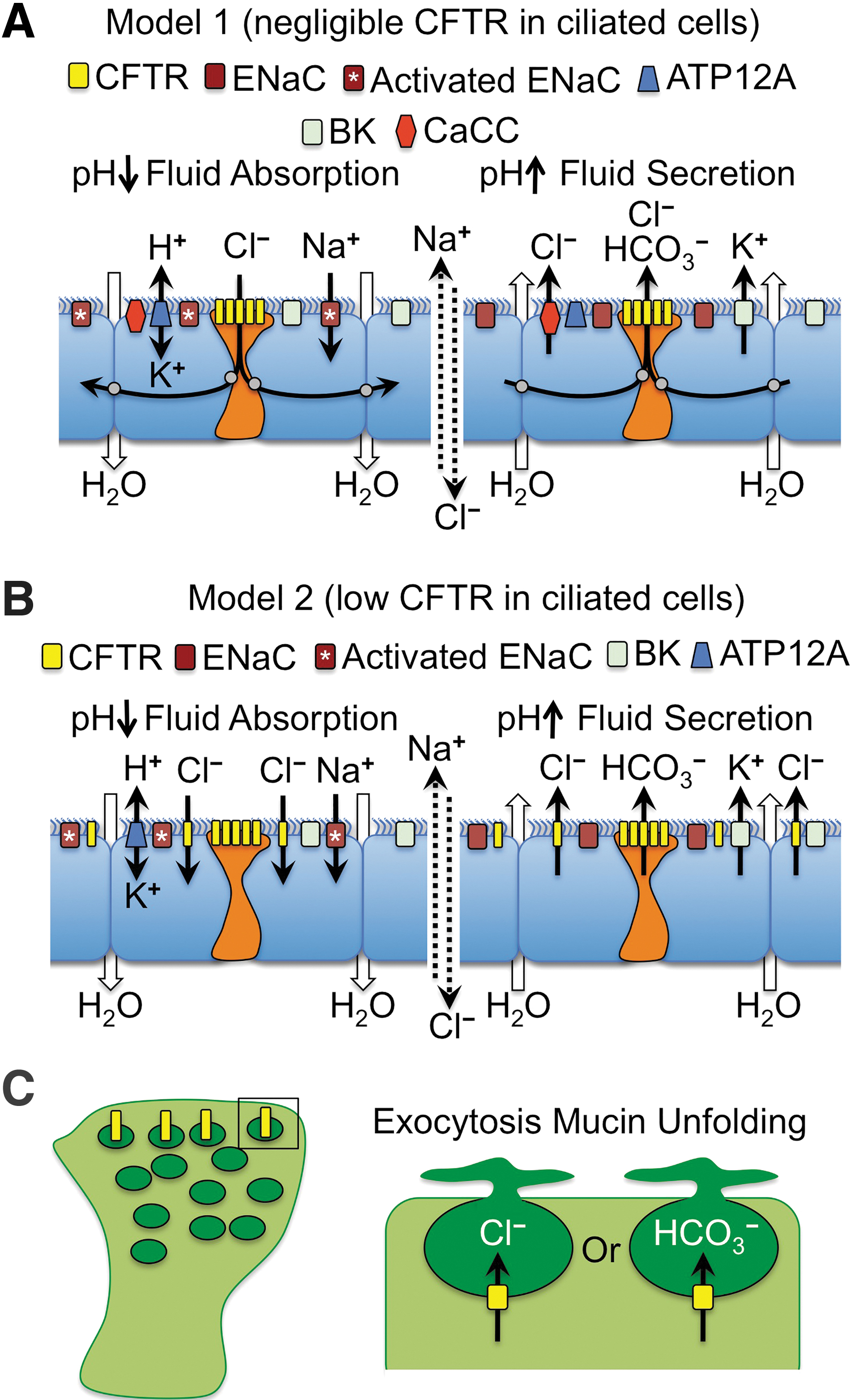
Models for the multicellular integration of CFTR function in ion and fluid movement across the proximal airway epithelium.
The first model proposes that fluid absorption and secretion defects in CF are associated with CFTR defects in ionocytes (Fig. 2A). Under conditions of fluid absorption (Fig. 2A, left panel), this mechanism proposes that the nongastric H+/K+ ATPase (ATP12A) drives acidification of the ASL 73 and the pH-dependent proteolytic activation of ENaC. Active Na+ absorption by ENaC creates an electrical driving force for Cl− absorption through ionocyte CFTR. We propose that CFTR mediates Cl− absorption since fluid absorption after a small volume challenge is impaired in CF ALI cultures. 114 Since ionocytes are at such low abundance in the airway, we propose that they may be electrically coupled with ciliated cells through gap junctions that are capable of passing Cl− between cells, as previously suggested. 119 Under conditions of fluid secretion (Fig. 2A, right panel), CFTR-dependent HCO3 − secretion by ionocytes drives alkalinization of the ASL and inactivation of ENaC, as previously suggested. 111 Cl− secretion could occur through ionocyte CFTR and/or the calcium-activated chloride channel (CaCC), 36,115,116 whereas K+ secretion by the voltage-dependent K+ channels (BK) provides the counter ion for the movement of salt and fluid into the airway lumen. 117 Importantly, when BK channels are inhibited in polarized airway epithelium, through either channel blockers or knockdown of the BK alpha subunit (KCNMA1), ASL dehydrates similarly to CF cultures. 117
An alternative second model proposes that low-level CFTR expression in ciliated cells is functionally relevant to fluid homeostasis at the airway surface (Fig. 2B). Similar to Model 1, this model proposes that fluid absorption is driven by ATP12A-dependent acidification of the ASL and activation of ENaC to absorb Na+ (Fig. 2B, left panel). However, this model proposes that the overall low level of CFTR in ciliated cells collectively absorbs the predominance of Cl− and that ionocytes primarily function to secrete HCO3 −, which is inactive during fluid absorption conditions. When fluid absorption passes the optimal ASL height and becomes slightly dehydrated, Model 2 proposes that ionocytes are activated to secrete HCO3 −, potentially though mechanical forces, 36 and the ASL pH rises leading to inactivation of ENaC (Fig. 2B, right panel). Cl− and K+ secretion through ciliated cell CFTR and BK channels leads to salt and fluid movement into the airways, raising ASL volume.
Although the two models just mentioned were chosen as provocative examples representing two cell types with extremely different levels of CFTR expression (ionocytes>>ciliated cells), secretory cells may also participate in transepithelial Cl− movement as they express CFTR at higher levels than ciliated cells based on scRNAseq. Secretory cells in the cartilaginous airways (i.e., goblet cells) also differ in their biology to those in the bronchioles (i.e., club cells) and thus CFTR may play different roles in these two types of secretory cells. For example, vesicular CFTR expression in goblet cells may play a role in exocytosis of mucin granules and the proper hydration and unfolding of mucin (Fig. 2C). By contrast, given that ionocyte abundance appears to decrease in the distal airways, CFTR expression at the apical membrane in club cells may play an important role in pH regulation and hydration of the ASL. Further, scRNAseq 120 and in situ hybridization 28 demonstrated that alveolar Type II (AT2) cells also express CFTR. Given their large numbers, AT2 cells collectively express the greatest total amount of CFTR in the lung. Although CFTR within AT2 cells is known to control fluid absorption in the newborn lung and in pulmonary edema, 121 –123 whether AT2 cells contribute to CF lung pathogenesis remains largely unknown.
Although these are only two potential models and many other possibilities exist that can explain how cell type-specific CFTR expression could impact innate immunity and clearance in the airway, they emphasize several key points regarding complementation of CF airway defects by gene therapy. For example, will it be necessary to express or correct CFTR within ionocytes to obtain proper airway pH and thus also regulation of ENaC? Can ciliated cells adopt ionocyte-like functions if CFTR is overexpressed using gene therapy? Or do ciliated cells not have the proper channel composition to properly regulate HCO3 − secretion even if high levels of CFTR are expressed. Further, if CFTR plays a role in mucin secretion as a Cl− and/or HCO3 − channel within mucin granules, complementation of this cell autonomous function would likely require CFTR expression within goblet and/or club cells.
CF Gene Therapy in Ferret and Pig Models of CF
Given the robust lung phenotype in CF ferrets and pigs, these models provide opportunities for the preclinical testing of gene therapy vector systems. Several recombinant vector systems have been evaluated in both species, including lentivirus, HDAd, bocavirus (BoV), and AAV. Each vector system has advantages and disadvantages. For example, lentiviruses are integrating viruses and thus provide longer-term persistence, whereas HDAd, BoV, and AAV have episomal genomes that dilute with cell division. AAV vectors suffer from packaging limitations, which are significant when delivering the large CFTR cDNA, whereas a related parvovirus vector using HBoV1 capsids has increased packaging capacity to enable the production of virus with the intact CFTR cDNA and a strong promoter. HDAd vectors have a very large packaging capacity, but carry significantly more foreign protein into cells, which may enhance immunogenicity. Recently, HDAd and rAAV vectors have been adapted to allow for integration of their genomes by using the PiggyBac transposase. Each of these vector systems and their application in ferret and pig models is discussed in more detail later.
rAAV vectors
The only viral requirements for rAAV vectors are two 145-nucleotide ITRs. However, the coding sequence of full-length CFTR is 4430 nucleotides. With the addition of the ITRs, the recombinant AAV genome exceeds its native size of 4,680 nucleotides, leaving little room for a promoter and polyadenylation sequence. Although rAAV vectors can effectively package up to 4,900 nucleotides, vector genomes containing CFTR greater than this size incur small deletions within the ends of the genome. 16 Two approaches have been used to enhance the capacity of rAAV vectors to deliver CFTR. The first approach created CFTR minigenes that produced channels with similar functional characteristics to full-length CFTR. 17,124 One of the better-characterized CFTR minigenes included a 156-nucleotide deletion in the CFTR regulatory (R) domain and was capable of rescuing intestinal obstruction in CF mice. 125
The second approach to circumvent packaging limitations has been to reduce the size of promoters that drive CFTR expression and the use of small synthetic poly-A sequences. 15,16,18,126 Promoters that can be packaged with a shortened CFTR minigene have included a minimal human cytomegalovirus (CMV) promoter containing core elements from the CMV immediate early promotor 18 and thymidine kinase 126 promoters/enhancers as well as synthetic promoter/enhancer elements. 15,16 Using the latter approach, a synthetic enhancer-element screen identified a 100-nucleotide sequence (F5) that gave expression from an 83-nucleotide synthetic promoter (tg83) at levels equivalent to the CMV/beta-actin promoter/enhancer. 16 When the F5tg83 promoter was used in conjunction with the CFTR-ΔR cDNA, the rAAV-CFTR-ΔR vector gave near normal levels of CFTR-mediated Cl− current in human CF airway epithelia.
Additional advances in rAAV transduction of airway epithelia have stemmed from the identification of alternative capsids that have high tropism for the apical surface of human airway epithelia 38,127,128 and an improved understanding that AAV encounters an intracellular proteasome-dependent block during movement to the nucleus 21,22,129 that can be overcome by co-administration of proteasome inhibitors. 23,130 Directed evolution with an AAV2/AAV5 capsid library after multiple rounds of apical infection of ALI cultures has generated a novel AAV capsid variant (AAV2.5T) that is highly tropic for the apical surface of well-differentiated human airway epithelia in vitro. 38
Collectively, these advances in rAAV vector systems have solved major obstacles encountered in the first rAAV2 CF clinical trials, including: (1) expression using small highly active promoters, (2) packaging capacity using a CFTR minigene, and (3) tropism to the airway surface using evolved capsids. In addition, the apical postentry barriers limiting productive rAAV transduction of polarized airway epithelium can be largely overcome by transient inhibition of the proteasome during or after the infection period.
Several of these rAAV vector systems have been tested in ferret and pig models and have demonstrated additional barriers to translating studies from human airway epithelium in vitro to animal models in vivo. For example, rAAV1 is highly effective at transducing well-differentiated human and ferret airway epithelia in the presence of proteasome inhibitors. 127,131 rAAV1 can also transduce the neonatal ferret lung. 24 However, when ferrets reach 1 month of age, rAAV1 transduction of the lung declines significantly due to airway secretions that inhibit postentry processing of the virus, which is refractory to proteasome inhibition. 24 Similarly, AAV2.5T, which is highly effective at transducing well-differentiated human airway epithelium in vitro, 38 does not transduce well-differentiated pig airway epithelium in vitro or in vivo. For these reasons, the group evolved an rAAV vector in pig airway in vivo, 132 using a shuffled capsid library generated from AAV-1, -2, -4, -5, -6, -8, and -9 cap genes, 133 an error-prone AAV2 library, 134 and a diversified AAV2 capsid library generated by peptide replacement at four capsid surface loops. 135 The isolated virus, called AAVH22, was highly tropic for pig airways. 132 Further, when AAVH22 was used to package the F5tg83-pCFTR-ΔR expression cassette, it was effective at correcting Cl− currents, ASL pH, and bacterial killing in CF pigs. 132 More recently, a system was developed to enhance the persistence of rAAV-CFTR genomes by incorporating PiggyBac transposase binding sites into the AAV vector and delivering a second AAV vector carrying the PiggyBac transposase. 136 Although the complete system has yet to be tested in vivo, the CFTR-containing vector complemented disease endpoints when aerosolized in the lungs of CF pigs.
HBoV1 vectors
HBoV1 is an autonomously replicating human parvovirus commonly associated with acute respiratory tract infections in infants and young children. 137,138 Unique to HBoV1 is its ability to reinfect children after seroconversion from a primary infection. 139 HBoV1 has a genome size of 5,543 nucleotides that is considerably larger than the 4,700-nucleotide AAV genome. This provided opportunities to engineer a recombinant HBoV1 vector that could carry the full-length CFTR cDNA driven by a strong promoter. To this end, methods were developed to package the rAAV2 genome into the HBoV1 capsid. The resultant rAAV2/HBoV1 hybrid vector was highly tropic for well-differentiated human airway epithelia after apical infection; however, proteasome inhibitors were still required for maximal transduction. 39 This vector system has the capacity to package viral genomes up to 5,800 nucleotides and thus may also be useful for gene editing approaches that require a larger payload and RNA-guided nuclease. Further, improved packaging systems are now enabling production of rAAV2/HBoV1 on par with that of rAAV2. 140
Humans are the only known host for HBoV1 respiratory tract infections and this presents challenges for preclinical animal studies using the rAAV2/HBoV1-based vector systems. However, studies evaluating rAAV2/HBoV1 transduction in the ferret lung have proved promising and suggest that the CF ferret model could potentially be used to develop rAAV2/HBoV1 as a gene therapy vehicle. 141 This study in wild-type ferrets demonstrated that repeat rAAV2/HBoV1 dosing to the lungs of 7-day- and 29-day-old animals was effective. Further, ferrets exposed to the virus at 7 days of age gave rise to fivefold higher levels of transduction after reinfection at 29 days of age, as compared with that observed in naive infected 29-day-old animals. The ability of HBoV1 capsids to reinfect ferrets is reminiscent of findings in young children. 139
Lentiviral vectors
Unlike rAAV and rAAV/HBoV1 vectors, lentiviral vectors have the advantage of integrating into the target cell genome and thus may persist longer if stem or progenitor cells are transduced. A wide array of lentiviruses have been proposed for gene therapy of CF lung disease, including: human immunodeficiency virus, 142 –144 simian immunodeficiency virus, 41,145 feline immunodeficiency virus (FIV), 40,146,147 and equine infectious anemia virus (EIAV). 148,149 Although these lentiviruses are not typically tropic for the airway epithelium, advances in pseudotyping the lentiviral envelope proteins have allowed for the development of efficient vector systems. For example, pseudotyping EIAV with influenza virus hemagglutinin 148,149 and FIV with baculovirus GP64146 produces recombinant lentiviruses with high-level tropism for the apical surface of the airway. Improvements to the airway tropism of lentiviruses have also been made by using libraries of GP64 envelope mutants. 150
Preclinical testing of lentiviral vectors has also been performed in pigs and ferrets. For example, recombinant EIAV pseudotyped with influenza A virus subtype H7 hemagglutinin highly transduces ferret bronchial and bronchiolar airway epithelium. 151 FIV pseudotyped with GP64 also efficiently transduces ferret bronchiolar epithelium. 151 Although these studies in ferrets have thus far only evaluated lentiviral transduction using reporter genes, in vivo studies in CF pigs have evaluated lentiviral-mediated CFTR delivery and the complementation of CFTR defects in excised tissues. 152 These studies have demonstrated complementation of transepithelial cAMP-stimulated current, ASL pH, and bacterial killing defects observed in CF pig airways.
HDAd and Piggybac/adenovirus vectors
The first CF gene therapy trials targeting the nasal and lung epithelium were performed by using E1-deleted recombinant adenoviral vectors. 153 –160 CFTR gene transfer in these studies was low and the approach using first-generation E1-deleted adenovirus was subsequently abandoned due to concerns about safety caused by innate and acquired immunity against viral genes that remained within the vector. More recently, HDAd has been developed for CF lung gene therapy. HD-Ad has the advantage that all virally encoded genes are deleted from the vector. Although deletion of viral genes reduces adaptive T cell responses against virally infected cells, incoming viral capsid proteins can still mount a CD8+ T cell response through the presentation of viral capsid epitopes by infected dendritic cells. 161 This limitation is universal to all protein capsid viruses (including rAAV), but since the adenovirus capsid is larger than AAV, the acquired immune responses to HD-Ad will likely be more severe. HD-Ad vectors have been tested in both pigs and ferrets and demonstrated to be highly efficient in gene transfer to the airways when tight junctions are disrupted with lysophosphatidylcholine to expose the basolateral coxsackievirus and adenovirus receptor for infection. 151,162
Additional modifications to adenoviral vector systems have included the use of transposase-mediated integration of a transgene cassette. Using the DNA transposon piggyBac, a hybrid piggyBac/adenovirus vector can highly transduce the large and small conducting airways of pigs with a GFP reporter cassette when lysophosphatidylcholine is co-administered. 163 Basal cells, a known stem/progenitor cell in the conducting airways, were also highly transduced, suggesting that this vector system could potentially provide long-term persistence when a transposase is co-delivered. Further, the delivery of an adenoviral vector expressing CFTR to CF pig airways corrected anion channel activity, ASL pH, and bacterial killing defects observed in the CF pig airways. 163 Future work is needed to demonstrate the utility of this vector system to integrate CFTR.
Conclusions and Future Prospects
Thirty-six CF gene therapy clinical studies or trials have occurred worldwide in the 30 years since the CFTR gene was identified. 164,165 Of these, only two clinical trials were designed and powered with a sufficient number of patients to assess efficacy by using pulmonary function tests, including rAAV213 and nonviral/plasmid. 166 This review has focused on viral vectors for CF lung gene therapy and the role that larger CF animal models have in the development of these approaches.
Regardless of the vector used for CF lung gene therapy, there are shared knowledge gaps. Larger CF animal models that faithfully reproduce the human CF lung phenotype will play an important role addressing these questions. For example, will it be necessary to reconstitute the native cellular CFTR expression patterns in the lung to delay disease progression? What are the pathophysiologically most important cellular targets in the CF lung? Are SMGs required gene therapy targets for the treatment of CF lung disease? Recent advances in the ability to genetically engineer ferrets that are capable of conditional inactivation or reactivation of CFTR in a cell-specific manner will aid in understanding the cellular targets that are most critical for CF lung gene therapy.
Although not discussed in this review, the immune response to viral vector administration to the lung will likely become a major focus of research over the next decade as gene therapy trials for CF commence. For example, lessons from systemic delivery of AAV vectors have demonstrated that both humoral and cellular immunity against viral antigens can limit vector readministration and persistence of transgene expression, respectively. Solutions to these immunologic barriers may include transient immunosuppression and tolerization strategies using engineered host T cells. Larger animal models, which more accurately reflect the immune responses of humans and the CF disease state, will likely also have a significant role in developing these targeted immunosuppression strategies.
Lastly, the development of viral vector systems that perform similarly in humans and the CF animal models used to test efficacy of gene therapies may require alternative approaches, such as interspecies-directed evolution. The landscape of gene therapy is rapidly changing with the development of new gene editing tools and such an approach targeted at airway stem cells would solve potential technical hurdles if CFTR expression needs to be regulated at the cellular level for therapeutic efficacy. Although these newer technologies are not discussed in this review, they also rely heavily on the development of effective gene delivery systems. Thus, successful CFTR gene replacement therapies will likely inform next-generation gene editing approaches. Despite these challenges, there is tremendous excitement for CF lung gene therapy and the prospects of a mutation-agnostic cure for all CF patients.
Footnotes
Acknowledgments
The authors gratefully thank Dr. Jennifer Barr for editorial assistance in the preparation of this review.
Author Disclosure
Z.Y. and J.F.E. receive income for consulting with Spirovant Science. J.F.E. has sponsored research with Vertex Pharmaceuticals and Spirovant Science.
Funding Information
This work was supported by grants from the NIH (HL051670 and DK047967 to J.F.E.), Cystic Fibrosis Foundation (to J.F.E. and Z.Y.), the University of Iowa Center for Gene Therapy (DK054759 to J.F.E.), and the Roy J. Carver Chair in Molecular Medicine (to J.F.E.).