
Abstract
Oncolytic adenoviruses (OAds) are promising agents for cancer therapy, representing a novel therapeutic strategy for pancreatic ductal adenocarcinoma (PDAC). However, there are challenges associated with the successful use of an OAd alone, involving the security of the viral vector and screening of an effective antitumor gene. In the present study, a novel OAd CD55-ST13-tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) was constructed in which the dual therapeutic genes ST13 and TRAIL were inserted, featuring the carcinoembryonic antigen (CEA) as a promoter to control E1A and deletion of the 55 kDa E1B gene. ST13, known as a colorectal cancer suppressor gene, exhibited lower expression in PDAC than in tumor-adjacent tissues and was associated with poor prognosis in PDAC patients. In vitro studies demonstrated that CD55-ST13-TRAIL was effective in promoting the expression of ST13 and TRAIL in CEA-positive pancreatic cancer cells. Moreover, CD55-ST13-TRAIL exhibited a synergistic effect toward tumor cell death compared with CD55-ST13 alone or CD55-TRAIL alone, and inhibited tumor cell proliferation and induced cell apoptosis dependent on caspase pathways in PDAC cells. Furthermore, xenograft experiments in a mouse model indicated that CD55-ST13-TRAIL significantly inhibited tumor growth and improved the survival of animals with xenografts. The findings demonstrate that oncolytic virotherapy under the control of the promoter CEA enables safe and efficient treatment of PDAC, and suggest that it represents a promising candidate for the treatment of metastatic diseases.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) represents the fourth leading cause of cancer death worldwide, the high mortality rates closely paralleling the incidence rate among solid tumors. 1 Since the pancreas is anatomically inaccessible and because no specific symptoms are observed in the early stages of PDAC, the low rates of survival are largely attributed it being first identified in advanced PDAC patients with metastasis and tumor infiltration into normal tissues. 2 Despite substantial progress in PDAC therapy having been achieved over the past several decades, limitations in chemotherapy and radiotherapy, in addition to those of surgical resection, have been experienced. 3 –5 Thus, early-stage detection methods and effective treatment strategies are urgently required to combat this disease. With the development of genetic engineering, oncolytic virotherapy has become a novel strategy for cancer therapy and has attracted increasing interest from both researchers and clinicians. 6
Oncolytic adenoviruses (OAd) have emerged as promising antitumor agents because they can selectively replicate in malignant tumors, and lyze tumor cells in a targeted manner without eliciting significant damage to normal cells. 7,8 As the most common vectors of gene therapy, adenoviruses can display tumor-selective tropism through genetic modification. 9 The use of tumor-specific promoters to control essential viral genes can achieve the targeting of tumors combined with viral propagation, ensuring that oncolytic virotherapy is both safe and effective. Previous studies by our and other groups have demonstrated that a panel of tissue- or tumor-specific promoters can be used to regulate OAd gene expression, such as the survivin promoter that targets solid tumors, 10 the GP73 promoter that targets liver cancer, 11 the carcinoembryonic antigen (CEA) promoter that targets colorectal cancer, 12 and the tyrosinase promoter that targets melanoma. 13 It is worth mentioning that CEA promoter-regulated OAd was initially used to target pancreatic cancer and demonstrated significant inhibition of tumor growth, 14 suggesting that CEA is an alternative candidate target for PDAC therapy.
The choice of antitumor gene for targeting plays a vital role in successful cancer gene therapy. From in-depth studies of the genetic changes in different tumors, we have identified numerous candidate genes for cancer-targeting gene virotherapy (CTGVT). 15 For example, we previously developed an OAd expressing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and obtained a significant antitumor effect in colorectal cancer. 16 TRAIL is a member of the tumor necrosis factor superfamily and activates the extrinsic apoptosis pathway in cancer cells while sparing the surrounding healthy cells. 17,18 TRAIL is a ligand that induces apoptosis or necroptosis by binding to transmembrane death receptors that promote the formation of death-inducing signaling and activation of the caspase pathway, leading to apoptotic tumor cell death. 19 –21 However, malignant tumor cells may produce resistance to TRAIL therapy. 22 Thus, effective therapeutic methods are required to combine with TRAIL to improve the targeted therapy effect or reverse the resistance to TRAIL.
Recently, dual-gene CTGVT (CTGVT-DG) has been proposed as a promising antitumor strategy. 23 However, the appropriate combination of dual antitumor genes is critical to the success of the CTGVT. A novel human adapter protein, ST13, also known as suppression of tumorigenicity-13, is frequently downregulated in tumors. 24 Furthermore, the biological function of heat shock protein-70 (Hsp70) is that of a molecular chaperone in cancer. Hsp70 has been shown to be expressed in the cytoplasm and nuclei of tumor cells, and to have involvement in tumor cell apoptosis and immune synergism. 25 –27 ST13 has been identified both as a cofactor and as Hsp70 interacting protein, in addition to possibly acting as a chaperone of Hsp70. 28 In a previous study, we established that ST13 expression in colorectal cancer tissue was significantly lower than in tissue adjacent to the tumor, and overexpression of ST13 induced apoptosis in colorectal carcinoma cells. 29 However, the evaluation of ST13 expression and its role in PDAC, or the potential synergistic antitumor effect of ST13 and TRAIL remains incompletely understood.
In the present study, we first measured ST13 expression in PDAC tissues and analyzed the correlation between ST13 expression, clinical pathology, and patient survival in PDAC. We used a CEA promoter to construct the OAd CD55-ST13-TRAIL that carried dual genes ST13 and TRAIL linked by Ile-Glu-Thr-Asp (IETD), which suggested a strong antitumor targeting capability and a synergistic antitumor effect in PDAC. We also found that CD55-ST13-TRAIL induced cancer cell-selective apoptosis and suppressed cancer cell proliferation through the expression of TRAIL and ST13 via a signaling pathway that triggered caspase and the epithelial/mesenchymal transition (EMT). CD55-ST13-TRAIL significantly promoted PDAC xenograft tumor regression in immunodeficient mice, and so may represent an effective and safe cancer gene therapy for PDAC.
MATERIALS AND METHODS
Patients and tissue sample
Samples were obtained from a total of 205 patients who had undergone surgical resection in the Department of Surgery, Zhejiang Provincial People's Hospital, Hangzhou, China, from March 2011 to August 2017. No patients received chemotherapy, radiotherapy, or other antitumor therapy before surgery. Each tissue sample was frozen immediately in liquid nitrogen following surgical resection and diagnosed by the Pathology Department of Zhejiang Provincial People's Hospital. The tissues were analyzed using tissue microarrays (TMAs), which were constructed by Shanghai Biochip (Shanghai, China).
The patient cohort consisted of 164 males and 38 females, aged from 32 to 91 years with a median age of 61.5 years. Tumors were staged according to the American Joint Committee on Cancer tumor-node-metastasis staging criteria. There were 120 patients at stages I–III and 85 stage IV patients. Survival time was calculated from the date of surgery to follow-up examination or mortality, and analyzed overall survival rate of patients. The present study was approved by the Zhejiang Provincial People's Hospital Ethics Committee. Informed consent was provided by every patient.
Immunohistochemistry assay and evaluation
TMA sections were processed by immunohistochemical (IHC) staining, using a Histostain-Plus IHC kit (Invitrogen), in accordance with the manufacturer's protocol. First, two TMA sections were baked at 70°C for 2 h, then deparaffinized and dehydrated using xylene and an increasing gradient of alcohol concentrations, respectively. For antigen retrieval, a high-pressure cooker was used to boil samples at 120°C with TE buffer for 3 min, which were subsequently treated with 3% hydrogen peroxide for 15 min to inhibit endogenous peroxidase activity and then incubated with 10% normal goat serum for 20 min to reduce background nonspecific binding. The TMA sections were then incubated with the rabbit anti-ST13 monoclonal antibody (Santa Cruz) overnight at 4°C. The sections were washed three times with phosphate-buffered saline (PBS), and then incubated with the biotin-labeled secondary antibody at room temperature for 15 min and streptavidin/peroxidase at room temperature for 15 min. Finally, a DAB kit was used to develop the color until a brown staining was clearly observed in the TMAs, which were then counterstained with hematoxylin for 3–5 min, dehydrated with ethyl alcohol for 5 min, cleared with xylene for 10 min, and then mounted with gelatin resin.
Stained slides were observed using light microscopy by expert pathologists who scored the sections for staining intensity and the proportion of the section that was stained. Staining intensity was graded as follows: 0 = negative; 1 = weak; 2 = moderate; and 3 = strong. Positive staining percentage of cells was scored as follows: 0: <5% cells stained: 1: 6–25% cells stained; 2: 26–50% cells stained; and 3: >51% cells stained. Scores of staining intensity and the proportion of stained cells were multiplied to further calculate a staining index score: a score ≤3 was defined as low expression and a score of ≥4 was defined as high expression.
Cell lines and cell culture
The human PDAC cell lines SW1990, PANC-1, and Bxpc-3, the normal human pancreatic ductal epithelial cell line hTERT-HPNE, and HEK293 human embryonic kidney cells were acquired from the Shanghai Cell Collection (Shanghai, China). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., MA), supplemented with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin at 37°C in a 5% CO2 humidified incubator.
Construction of the recombinant OAd
A schematic representation of the process for OAd construction is displayed in Fig. 2A. All OAd vectors were based on pSD55 and generated using a combination of traditional pShuttle vectors and CMV expression cassette-mediated cloning. pSD55 is an E1B 55-kDa-deleted adenovirus vector containing the E1A gene necessary for selective replication in cancer cells. The CEA promoter was subcloned into pSD55 to form pSD55-CEA in the present study. To link therapeutic gene expression with viral replication, ST13 and TRAIL expression cassettes were separately inserted to obtain pSD55-CEA-E1A-ST13 and pSD55-CEA-E1A-TRAIL. In addition, the vectors pSD55-CEA-E1A-EGFP and pSD55-CEA-E1A-ST13-IETD-TRAIL were constructed using a method similar to that described above. The sequence of each recombinant plasmid was verified by restriction enzyme digestion, PCR, and DNA sequencing. The OAds CD55-ST13, CD55-TRAIL, and CD55-ST13-TRAIL were generated by homologous recombination in Escherichia coli strain BJ-5183 using the shuttle plasmids described above and the adenovirus packaging backbone plasmid pAdeasy-1. The packaged recombinant plasmids were then transfected into HEK293 cells to form recombinant OAd using Lipofectamine 3000 (Thermo Fisher Scientific). Finally, all recombinant OAds were amplified in HEK293 cells, purified using cesium chloride density gradient centrifugation after which the titer was measured.
RNA extraction and quantitative real time-PCR
Cells were lyzed using TRIzol reagent (Invitrogen) to extract total RNA, in accordance with the manufacturer's instructions. Following spectrophotometric quantification, cDNA was synthesized using a PrimeScript first-strand cDNA synthesis kit (TaKaRa) in accordance with its instructions. Quantitative real time-PCR (RT-PCR) was conducted using a SYBR green reagent in a Step One Plus Real-time PCR System (Bio-Rad, USA) in accordance with the manufacturer's protocol. GAPDH mRNA was used for normalization. The primer sequences of CEA for PCR were: forward: 5′-GCTTGTGGCAGCTACACTCTG-3′; reverse: 5′-GTTGATGAAACCTGTCGTCCC-3′. Target gene expression for CEA was calculated using the comparative delta CT method. Each quantitative PCR was performed in triplicate and repeated independently three times.
Cell viability and colony formation assay
Cell viability was measured using an MTS assay (Sigma-Aldrich, St Louis, MO). Briefly, Bxpc-3, PANC-1, and SW1990 cells (5 × 103 cells/well) were seeded into 96-well plates infected with recombinant adenovirus and incubated at 37°C for 1–4 consecutive days. At the same time of each day, 20 μL of MTS (Promega, China) was added to each well, which contained 200 μL of DMEM. After incubation for 4 h, absorbance at 490 nm of wells was measured using a microplate reader.
For the colony formation assay, SW1990 and PANC-1 transfected cells at a density of 200 cells/mL were seeded into 6-cm-diameter dishes. After 14 days of culture, surviving colonies were fixed with absolute ethanol (Sangon, China) and stained with crystal violet, after which the colonies were counted using light microscopy. Colony formation assays were performed in triplicate.
Combination index analysis
Cell viability was examined using an MTS assay, as described above. The combination index (CI) value was analyzed in PANC-1 cells using CalcuSyn software, which is calculated using the following formula: CI = (D)S/(Dx)S + (D)T/(Dx)T + (D)S(D)T/(Dx)S(Dx)T, where (D)S and (D)T represent concentrations of drugs S or T in a combination that inhibits cell growth, and (Dx)S and (Dx)T are concentrations of single drugs S or T that inhibit cell growth. The nature of the drug interaction is synergistic if CI <1 and antagonistic if CI >1.
Flow cytometry and apoptotic cell staining assay
For the cell apoptosis assay, an FITC Annexin V and propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA) was used, in accordance with the manufacturer's protocol. Forty-eight hours after infection, the cells were digested and washed twice with PBS. The cells were resuspended in 500 μL aliquots of binding buffer and stained with Annexin V FITC/PI. The cells were then immediately analyzed using fluorescence-activated cell sorting.
Cells were seeded into six-well plates then stained with CD55-ST13 (5 multiplicity of infection [MOI]), CD55-TRAIL (5 MOI), or CD55-ST13-TRAIL (5 MOI). All cells were incubated with Hoechst 33342 (Beyotime Institute of Biotechnology, Nantong, China) for 10 min after 48 h, then washed twice with PBS, and subsequently observed using a fluorescence microscope (Nikon).
Western blotting
Protein was extracted using the RIPA buffer (Beyotime Institute of Biotechnology), as described previously. 30 Protein concentrations were measured using a BCA protein assay kit (Thermo Fisher Scientific). The proteins were then heat-inactivated at 100°C for 5 min and then separated using 10–15% SDS-PAGE. They were transferred onto a PVDF membrane, blocked in 5% nonfat dried milk at room temperature for 2 h, and then incubated in fresh blocking buffer with primary antibodies overnight at 4°C. The membranes were washed and then incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. The membranes were washed in Tris-buffered saline three times and then visualized with an Immobilon Western Chemiluminescent HRP substrate (Merck Millipore, Germany) using a GelDoc XR System (Bio-Rad). Antibodies were purchased from Abcam (United Kingdom) against E1A (No. ab204123) and from Cell Signaling Technology (Danvers, MA) against Bcl-2 (No. 15071), caspase-8 (No. 9746), caspase-9 (No. 9502), caspase-3 (No. 9662), TRAIL (No. 3219), BAX (No. 5023), XIAP (No. 14334), and β-actin (No. 4970).
Animal experiments
All animal experiments were consistent with procedures and regulations approved by the U.S. Department of Agriculture and the National Institutes of Health. BALB/c nude mice (3–5 weeks old) were purchased from Zhejiang Chinese Medical University, into which PANC-1 cells were injected subcutaneously to form tumor xenografts that were utilized for in vivo studies. Based on the previous research findings of our group, OAd doses of 2 × 109 pfu were selected for intratumoral injection and were divided into three injections. After the first injection, tumor size was measured every 5 days and the tumor volume calculated.
For analysis of viral replication and accumulation in vivo, mice were sacrificed after treatment with a recombinant adenovirus or PBS, and the tumor tissues, liver, and kidneys from each mouse harvested, fixed in 5% paraformaldehyde for 48 h, and then embedded in paraffin. The tissue samples were sliced into 4–5-μm-thick sections. IHC staining was performed using an anti-E1A antibody, in accordance with an IHC protocol. Finally, the sections were counterstained with hematoxylin and eosin (H&E) and observed using microscopy.
Statistical analysis
Statistical analysis was performed using SPSS (version 17.0), GraphPad Prism (version 7.0), Origin (version 8.5), and Photoshop (version 6.0) software. All experiments were repeated three times. Data are expressed as mean ± SD (in vitro) or mean ± SEM (in vivo). A Student's t-test was used to test differences between groups. p < 0.05 was considered significantly different.
RESULTS
Expression of ST13 in PDAC tissue correlated with clinicopathologic parameters
To investigate the expression of ST13 in PDAC, we conducted IHC analysis of ST13 in PDAC and adjacent normal tissues. The results indicated that ST13 was mostly expressed in the cytoplasm of PDAC or normal tissues (Fig. 1B). Furthermore, the expression of ST13 was at a low level in PDAC tissues compared with adjacent tissues. As shown in Table 1, ST13 expression was correlated with the clinical variables of PDAC patients, demonstrating a negative correlation between ST13 expression with cancer metastasis (p = 0.042), microvascular invasion (p = 0.046), CEA (p = 0.016), and cancer status (p < 0.001). Meanwhile, Kaplan–Meier plots were used to analyze the relationship between survival rate and ST13 expression in PDAC patients within 5 years in accordance with the follow-up results. These revealed that reduced expression of ST13 was associated with poor prognosis in PDAC patients.

Low ST13 expression predicted shorter survival.

Characterization of oncolytic adenovirus CD55-ST13-TRAIL.
Relationship between ST13 and clinicopathological characteristics in pancreatic ductal adenocarcinoma patients
Bold indicates statistically significant p values (<0.05).
CEA, carcinoembryonic antigen.
Construction and characteristics of OAd CD55-ST13-TRAIL
The pancreatic cancer-specific promoter CEA was used to promote the oncolytic virus to specifically target pancreatic cancer. First, we measured the expression of CEA in PDAC cells using RT-PCR. Compared with normal pancreatic cells, CEA was found to be highly expressed in PDAC cells (Fig. 2D). Based on the selective replication capability of the oncolytic virus, we constructed a novel OAd that harbored the dual therapeutic genes ST13 and TRAIL (Fig. 2A). To verify the infection efficiency of CD55-EGFP in PDAC cells, we measured the expression of enhanced green fluorescent protein (EGFP) using fluorescence microscopy. The results indicate that greater EGFP expression was observed in PDAC cells after infection with CD55-EGFP after 48 h (Fig. 2E, F), but almost no expression in normal pancreatic cells (data not shown). Similarly, expression of adenovirus proteins E1A, ST13, and TRAIL was observed in PANC-1 cells infected with OAd CD55-ST13, CD55-TRAIL, and CD55-ST13-TRAIL at the indicated MOI, respectively. It revealed that the various OAds were able to efficiently mediate the expression of viral E1A and therapeutic genes ST13 and TRAIL in PDAC cells (Fig. 2B, C).
OAd CD55-ST13-TRAIL exerts synergistic inhibition of PDAC cell growth
To investigate the antitumor effect of CD55-ST13-TRAIL in vitro, PDAC cell lines Bxpc-3, PANC-1, and SW1990, and normal pancreatic hTERT-HPNE cells were infected with CD55-ST13, CD55-TRAIL, or CD55-ST13-TRAIL at different concentrations for 48 h, after which cell viability was analyzed using an MTS assay. The results indicated that CD55-ST13-TRAIL significantly inhibited cell growth in PDAC cells compared with other treatment groups in a dose-dependent manner, and displayed few side effects toward the normal pancreatic cells (Fig. 3A). In addition, colony formation analysis was used to evaluate the cell proliferation capability of PDAC cells following various treatments. The results indicated that CD55-ST13-TRAIL significantly inhibited the colony formation capability of SW1990 and PANC-1 cells compared with CD55-ST13 or CD55-TRAIL (Fig. 3B, C), suggesting that CD55-ST13-TRAIL was able to inhibit proliferation of PDAC cells.
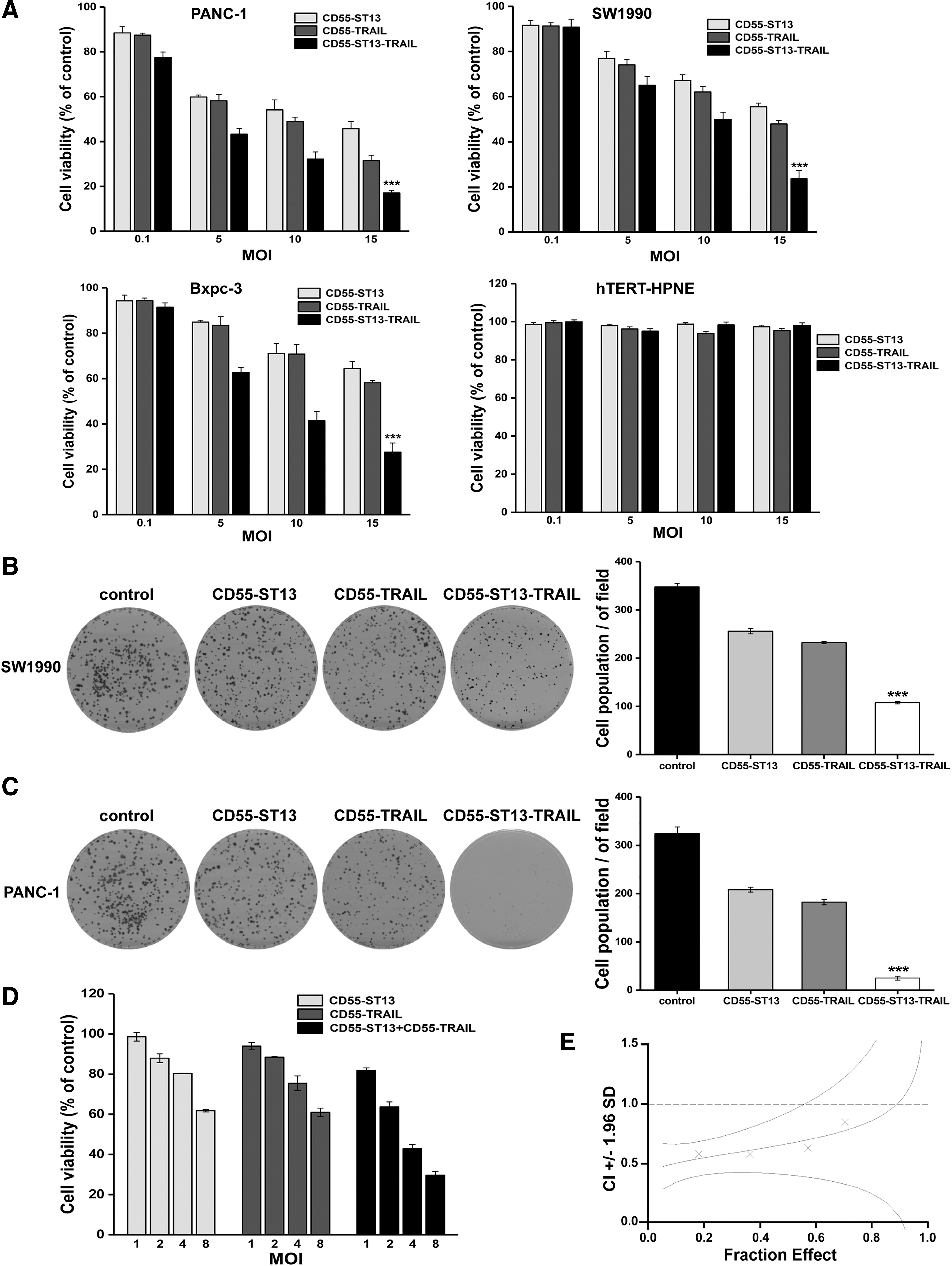
Oncolytic adenovirus CD55-ST13-TRAIL suppresses tumor cell proliferation.
To further confirm whether the combination of CD55-ST13 and CD55-TRAIL had a synergistic antitumor effect, we analyzed their therapeutic potential using CalcuSyn software. PANC-1 cells were treated with CD55-ST13, CD55-TRAIL, or a combination of both at different concentrations (Fig. 3D). As shown in Fig. 3E, CI values are represented by X-marks. As can be seen, combined treatment of CD55-ST13 and CD55-TRAIL resulted in CI values <1, indicating a synergistic effect.
OAd CD55-ST13-TRAIL selectively induces cell apoptosis in PDAC cells
In an attempt to determine whether the antitumor effect of CD55-ST13-TRAIL was mediated by an enhancement of cancer cell apoptosis, a flow cytometry assay was first used to examine its apoptotic effects in PANC-1 cells. As shown in Fig. 4A, we used CD55-ST13, CD55-TRAIL, or CD55-ST13-TRAIL infected PDAC cells, and the results showed that CD55-ST13-TRAIL treatment increased the proportion of apoptotic cells at 31.4% compared with 15.2% for CD55-ST13 alone and 24.8% for CD55-TRAIL alone. Morphological changes in the apoptosis of PANC-1 cells were verified using a Hoechst33342 assay. The result demonstrated that significant chromatin condensation and formation of apoptotic bodies were observed in PANC-1 cells, especially those treated with CD55-ST13-TRAIL (Fig. 4C). However, almost no apoptotic cells or cells undergoing characteristic changes were observed in the normal cell population (Fig. 4C). Thus, these data suggest that the combination of ST13 and TRAIL significantly induced apoptosis in PDAC cells.
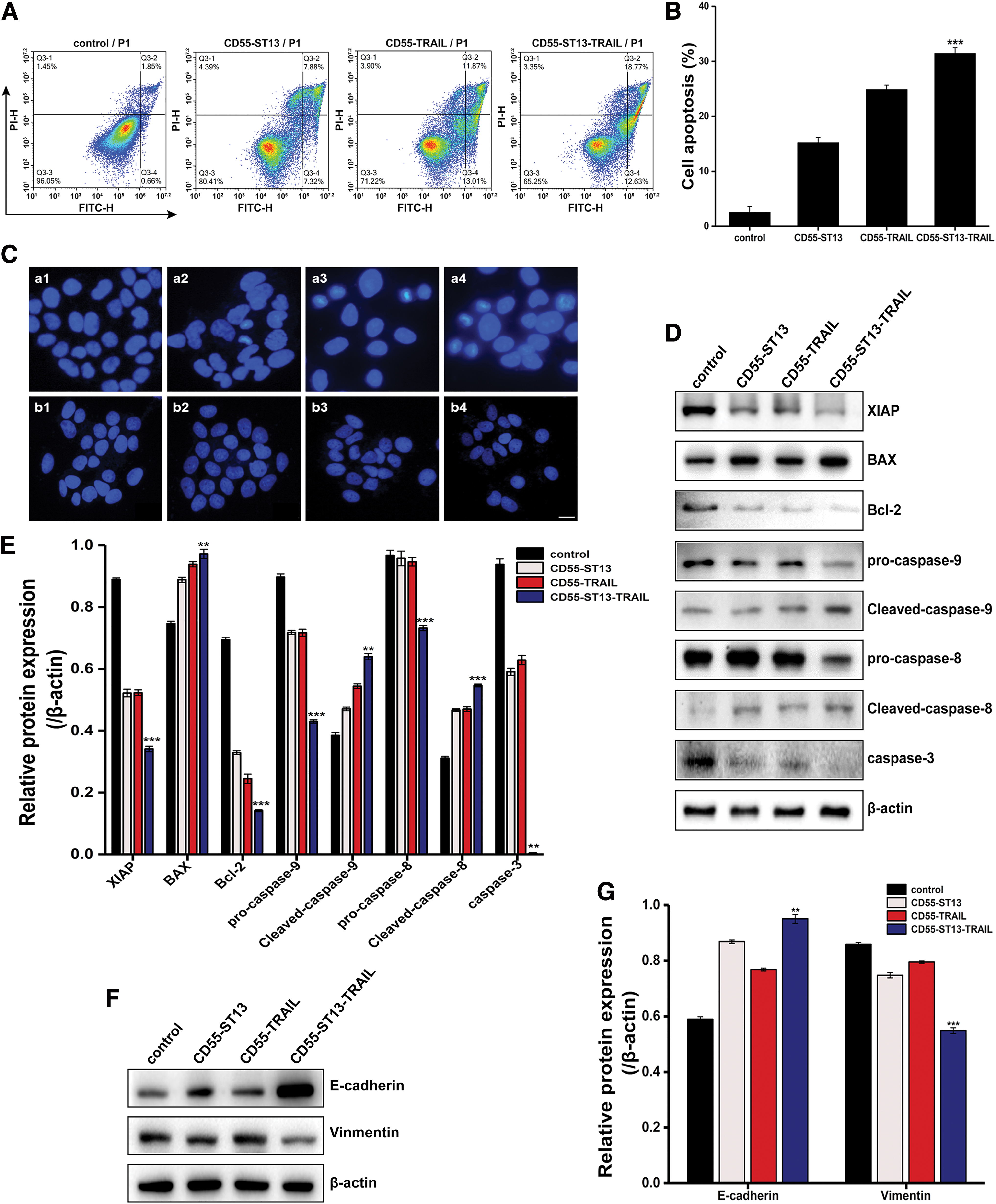
Oncolytic adenovirus CD55-ST13-TRAIL induces apoptosis in PANC-1 cells.
To further explore the potential mechanism of CD55-ST13-TRAIL-induced apoptosis, we measured the expression of the caspase-related pathway in PDAC cells. As shown in Fig. 4D, CD55-ST13-TRAIL treatment markedly increased the expression of BAX, cleaved caspase-8, and cleaved caspase-9 in the PDAC cell lines compared with other treatments. Nevertheless, the expression of XIAP, Bcl-2, procaspase-8, procaspase-9, and caspase-3 decreased in the CD55-ST13-TRAIL group. In addition, it was found that CD55-ST13-TRAIL decreased the expression of E-cadherin and increased the expression of vimentin in PDAC cells (Fig. 4E). These findings revealed that the induction of apoptosis and inhibition of metastasis by CD55-ST13-TRAIL may depend on EMT and the caspase pathway.
OAd CD55-ST13-TRAIL causes tumor regression in vivo
To evaluate whether CD55-ST13-TRAIL enhances antitumor efficacy in vivo, we first established PANC-1 tumor xenografts in an athymic nude mice model (Fig. 5A). Neither CD55-ST13 nor CD55-TRAIL monotherapy was able to suppress tumor growth more than CD55-ST13-TRAIL therapy. Notably, treatment with CD55-ST13-TRAIL significantly suppressed tumor growth and achieved a greater degree of tumor regression (Fig. 5C). Furthermore, the survival rate was analyzed in mice administered with CD55-ST13, CD55-TRAIL, and CD55-ST13-TRAIL. Compared with CD55-ST13 or CD55-TRAIL treatment, the survival of mice injected with CD55-ST13-TRAIL was significantly improved (Fig. 5D). In addition, adenovirus E1A expression was measured by IHC analysis in tumor tissues, which indicated that E1A was expressed in tumor tissues treated with CD55-ST13, CD55-TRAIL, and CD55-ST13-TRAIL (Fig. 5B). Importantly, no significant toxicity was observed in the liver, kidneys, or spleen by H&E staining, indicating that the OAd did not cause damage to these tissues (Fig. 5B). The results suggest that CD55-ST13-TRAIL is safe and can selectively inhibit PDAC cell growth.

Antitumor efficacy of oncolytic adenovirus CD55-ST13-TRAIL in xenografts. Mice were treated with intratumoral injection of PBS, CD55-ST13, CD55-TRAIL, or CD55-ST13-TRAIL.
DISCUSSION
PDAC is a common malignant tumor worldwide. 31 –34 Although different approaches are offered for the treatment of PDAC, all deliver unsatisfactory results, characterized by high levels of tumor metastasis and poor prognosis. Thus, there is an urgent need to develop novel therapeutic strategies that have high efficacy with low adverse effects for targeting malignant tumors, especially metastatic tumors. The limitations of traditional gene therapy include a short half-life in vivo combined with poor tumor-targeting efficacy. 35 CTGVT strategies can enhance therapy efficacy and have wide preclinical application in the treatment of malignant tumors. 36 –39 Currently, oncolytic adenoviral vector-based gene therapy has become an effective approach for targeting refractory cancer. A therapeutic gene is inserted into an oncolytic virus and is highly expressed in the tumor, but not in normal tissues due to virus-specific replication. 40 TRAIL has been shown to be a promising drug candidate for the treatment of a variety of cancers in current preclinical and clinical studies. However, there are no current clinical trials of TRAIL for PDAC therapy.
Thus, we have attempted to use the TRAIL gene to further enhance the oncolytic efficacy of adenovirus therapy in PDAC, in addition to enabling selective TRAIL expression and continuously induced PDAC cell apoptosis by viral replication. However, TRAIL-based therapy has a number of disadvantages, such as resistance to TRAIL monotherapy, and the TRAIL receptor acts as an important determinant of tumor-stroma-interplay during PDAC metastasis, playing a role in the promotion of metastasis in PDAC. 41 In this regard, based on CTGVT-DG strategies, a dual-regulated OAd was designed to overcome the limitations of single-gene therapy. Selection of the appropriate dual therapeutic genes to synergistically target the tumor cells is critical to the success of the CTGVT-DG strategy.
Until now, multiple studies have demonstrated that ST13 plays an important role in tumor progression. In the present study, ST13 was found to be poorly expressed in PDAC tissues and to be associated with cancer metastasis, CEA expression, and microvascular invasion. We also found that the survival rate of PDAC patients with high ST13 expression was greater than those with low ST13 expression. These results suggest that ST13 may act as a tumor suppressor in PDAC, and downregulation of ST13 may be a predictor of poor prognosis in PDAC patients. Moreover, ST13 promotes Hsp70 expression in vivo, enhances the immunogenicity of tumor cells, which induces an immune response, and promotes the interaction of tumor cells with NK cells. 28 Similarly, it is known that OAds generate a strong immune response, activating an inflammatory response at sites of tumors, thereby promoting uninfected cells to become infected by the adenovirus. 42 Thus, an ST13-armed OAd may acquire a better therapeutic efficacy in immunogenic mice in clinical applications. Notably, not only did overexpression of Hsp70 not influence TRAIL-induced apoptosis, it also increased the stability of TRAIL-R1 and TRAIL-R2 during ligand binding, leading to enhanced extrinsic apoptosis signaling. 43 Together, the results confirm that ST13 and TRAIL may be appropriate dual therapeutic genes for the CTGVT-DG strategy.
In the present study, we constructed the novel OAd CD55-ST13-TRAIL that was regulated by the promoter CEA, in which a four-peptide sequence encoding a caspase-8 cleavage site (IETD) mediated double gene expression. In addition, we deleted a 55 kDa gene in the E1B region, which allowed the virus to specifically replicate only in the tumor. Importantly, the novel OAd increased the expression and release of the ST13 and TRAIL genes in PDAC cells. Our studies found that CD55-ST13-TRAIL could selectively infect and kill PDAC cells, exerting significant oncolytic activity against PDAC cells both in vitro and in vivo, combined with few side effects in normal cells. Furthermore, CD55-ST13-TRAIL exhibited a higher antitumor activity toward tumor cells compared with gene monotherapy. Therefore, these results revealed that the linker IETD-mediated cleavage site did not affect ST13 or TRAIL function and that the dual therapeutic genes exhibited a synergistic effect in inhibition of the growth of PDAC cells. However, the potential antitumor mechanism of CD55-ST13-TRAIL remains unclear and needs to be explored.
In general, PDAC cells display high rates of metastasis with low levels of apoptosis, possibly involving EMT and apoptotic processes. 44 EMT is a crucial process for distant metastasis of PDAC cells in which epithelial cells lose intercellular adhesion and cellular polarity. 45,46 In the present study, considering that ST13 is related to cancer metastasis, we speculate that ST13 possibly induces EMT. To test this hypothesis, we measured the expression of related proteins by Western blot analysis. Our studies indicated that CD55-ST13-TRAIL upregulated the expression of E-cadherin and downregulated vimentin expression. The results indicate that CD55-ST13-TRAIL may inhibit metastasis in PDAC cells via regulation of the EMT pathway, although additional investigation is required to confirm this assertion. In addition, we explored whether CD55-ST13-TRAIL plays a key role in cell death and maintenance of mitochondrial membrane integrity via the intrinsic caspase pathway and Bcl-2 family members, including pro-/antiapoptotic proteins. The results demonstrate that CD55-ST13-TRAIL induced increased BAX and XIAP expression, which initiated apoptosis through mitochondrial cytochrome c release into the cytoplasm, activating the proapoptotic caspase cascade. 47 These findings indicate that the OAd CD55-ST13-TRAIL enhanced the antitumor effect by the inhibition of metastasis and promotion of apoptosis. However, no matter what the safety of CD55-ST13-TRAIL construct, the nonspecific toxicity and safety of oncolytic virotherapy in future clinical application should be a concern.
Recently, inhibitors of immune checkpoint molecules such as PD-1, PD-L1, and CTLA4 have been shown to substantially improve the treatment of solid tumors. Thus, a rational combination of oncolytic virotherapy and immune therapy could result in synergistic therapeutic efficacy of both potential anticancer strategies, even in advanced cancers. 48 In addition, the small-molecular reagents sodium butyrate, 49 N,N-dimethyl-D-erythro-sphingosine, 50 and mycophenolic acid 51 have demonstrated antitumor potential, and so these may be effective when used as a combination regimen with an OAd. Aberrant signaling pathways are involved in carcinogenic processes, such as RTK and FAK-p53-MDM2 signaling. 52,53 Thus, molecular targeted inhibitors against these aberrant signals also may deliver a new combination strategy with oncolytic virotherapy for future PDAC treatment.
In summary, the novel construct of the OAd CD55-ST13-TRAIL suppresses PDAC cell proliferation and induces the apoptosis of PDAC cells by activation of the caspase pathway. Therefore, the present study may provide a new predictive indicator and therapeutic strategy for PDAC treatment.
Footnotes
ACKNOWLEDGMENT
We thank Prof. Zheng Shu for kind suggestions and modification to this article.
ETHICAL APPROVAL
This research study complied with all applicable international, national, and/or institutional guidelines for the care and use of animals.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Natural Science Foundation of Zhejiang Province of China (No. LY18C070002), the National Nature Science Foundation of China (No. 81803069), and the 521 Talent Project of Zhejiang Sci-Tech University.