
Abstract
The efficiency of gene repair by homologous recombination in the liver is enhanced by CRISP/Cas9 incision near the mutation. In this study, we explored interventions designed to further enhance in vivo hepatocyte gene repair in a model of hereditary tyrosinemia. A two-AAV system was employed: one virus carried a Staphylococcus pyogenes Cas9 (SpCas9) expression cassette and the other harbored a U6 promoter-driven sgRNA and a fragment of fumarylacetoacetate hydrolase (Fah) genomic DNA as the homologous recombination donor. In neonatal mice, a gene correction frequency of ∼10.8% of hepatocytes was achieved. The efficiency in adult mice was significantly lower at ∼1.6%. To determine whether hepatocyte replication could enhance the targeting frequency, cell division was induced with thyroid hormone T3. This more than doubled the gene correction efficiency to 3.5% (p < 0.005). To determine whether SpCas9 delivery was rate limiting, the gene repair AAV was administered to SpCas9 transgenic mice. However, this did not significantly enhance gene repair. Finally, we tested whether the Fanconi anemia (FA) DNA repair pathway was important in hepatocyte gene repair. Gene correction frequencies were significantly lower in neonatal mice lacking the FA complementation group A (Fanca) gene. Taken together, we conclude that pharmacological induction of hepatocyte replication along with manipulation of DNA repair pathways could be a useful strategy for enhancing in vivo gene correction.
Introduction
The most common current approach to gene therapy of inherited liver disease involves AAV-mediated episomal gene addition. 1,2 Although promising, this method is limited by the lack of permanence in dividing cells, reducing its utility in settings of liver growth and regeneration. Homologous recombination-mediated targeted gene repair has also been achieved using AAV, but suffers from inefficiency. Our laboratory has demonstrated that gene targeting mediated by recombinant AAV can successfully correct the point mutation in the Fah5981SB mouse model 3 of human liver disease hereditary tyrosinemia type I (HT1). 4,5
HT1 is a severe autosomal recessive metabolic disease caused by accumulation of hepatotoxic metabolites due to mutations in the fumarylacetoacetate hydrolase (FAH) gene. Treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), an inhibitor that blocks upstream of FAH, prevents metabolite accumulation, and rescues the phenotype. 6 The Fah5981SB mouse model (herein described as Fah−/− ) carries a G-to-A transition that causes loss of exon 8, a subsequent frameshift in the resulting mRNA, and a severe reduction in Fah enzymatic activity, and represents an acute form of human HT1. 3 By delivering a homology containing AAV vector, we achieved in vivo gene correction frequencies of 0.1% to 1% in Fah−/− neonatal mice and even lower correction levels in adults. 4,5 This low frequency of gene correction was subtherapeutic due to inefficient homologous recombination. We subsequently used the CRISPR-associated RNA-guided endonuclease Staphylococcus pyogenes Cas9 (SpCas9) to promote homologous recombination using single-stranded oligonucleotides as donors. 7 Although improved, the gene correction frequency remained subtherapeutic at <1% in adults.
Using the same model system, others then used AAV as a homology donor while delivering Cas9 with an RNA nanoparticle. 8 This approach resulted in mutation repair frequencies of up to ∼6%, even in adults. Others employed a dual AAV system (one vector expressing Staphylococcus aureus Cas9 (saCas9) and the other containing homology donor) in a mouse model of ornithine transcarbamylase (OTC) deficiency. 9 Gene repair frequencies of 7–20% were seen in neonates, whereas much lower (0.3–2.1%) was observed in adults. In addition, hepatotoxicity was found in adults.
In this study, we wished to explore the use of a dual vector system using SpCas9 to optimize gene repair efficiency in vivo. We tested whether the level of Cas9 expression was rate limiting, whether cell replication could enhance efficiency, and whether the Fanconi anemia (FA) pathway is important for AAV-mediated homologous recombination repair in the liver in vivo.
Materials and Methods
Mice
Fah5981SB
mice were maintained under C57 strain background and were supplied with 32 mg/L NTBC in drinking water.
3
To generate Fanca−/−Fah−/−
mice, heterozygotes were crossed inter se to generate double mutant mice and littermate controls. Cas9-transgenic mice were initially purchased from The Jackson Laboratory (Bar Harbor, ME) and backcrossed fully to the C57 strain background. All mice used were either neonates (newborn mice within 48 h after birth) or 8-week-old adults. T3 diet was manufactured at the TestDiet (Richmond, IA) by mixing thyroid hormone T3 (3,3,5-Triiodo-
Plasmid construction
The Fah-targeting sgRNA was designed using the online CRISPR design tool. 10 Oligonucleotides for three sgRNA designs were ordered from Integrated DNA Technologies (52241; Coralville, IA), annealed, and cloned into pX330 plasmids (Addgene plasmid 42230). 11 The resultant plasmids were used to transfect the human liver cell line Huh7 cells (purchased from ATCC) and the indel frequencies were evaluated by Tide analysis 3 days after transfection. 12 The most active sgRNA was chosen and cloned into pAAV2 plasmid under the control of U6 promoter. This sgRNA recognized the DNA sequence 5′-ATCTAGATGACCCGATGCTC-3′ upstream of a PAM sequence TGG in exon 8 of mouse Fah gene. A 3.6 kb DNA fragment that flanks the target site was also cloned into this plasmid. The detailed information about the resultant plasmid is shown in Fig. 1A.

AAV vector design and the timeline of the experiments.
AAV purification and titering
Recombinant AAV2/8 virus was produced at the Molecular Virology Core at the Oregon National Primate Research Center, OHSU. In brief, the virus was made through the standard triple plasmid co-transfection method and purified with Iodixanol Gradient Ultracentrifugation. 13 AAV titers were determination by dot-blot hybridization and the virus purity was assessed by silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Liver immunohistochemistry and antibodies
Four weeks after the initial virus administration, liver samples were harvested and fixed in 10% phosphate-buffered formalin. Liver sections were stained with either rabbit polyclonal anti-Fah serum or hematoxylin and eosin (H&E) as reported previously. 5
For the BrdU assay, an additional DNA hydrolysis and permeabilization steps were performed after fixation to allow the anti-BrdU antibody (Abcam, Cat no. ab6326) access to the BrdU within the DNA.
Statistical analysis
Statistical analyses were done using Prism 7.0 software (GraphPad Software, CA). Two-tailed unpaired Student's t-tests were performed to calculate p-values. A p-value <0.05 was considered as statistically significant.
RESULTS
Vector design and experimental scheme
A dual AAV approach was used to correct the point mutation of Fah−/− mice. One virus, AAV-sgRNA-repair, delivered a U6-driven sgRNA targeting a 20 nt DNA sequence 145 nt away from the point mutation site and a 3.6 kb genomic DNA fragment flanking the target site that could serve as the DNA donor for homologous recombination. The second virus, AAV-Cas9, carried a SpCas9 expression cassette. Since many AAV vectors show a broad tissue tropism, a liver-specific gene regulatory cassette was used to achieve restricted liver specific of Cas9. 14 The gene repair scheme is depicted in Fig. 1. The CRISPR-Cas9 will generate a DNA double-stranded break near the mutation site specifically in hepatocytes, which will then promote either nonhomologous end-joining (NHEJ) or homologous recombination events. However, since the AAV-delivered DNA repair donor does not provide a promoter, only homologous recombination events can lead to gene correction and restore Fah gene expression.
Gene correction in neonatal mice
The dual AAV-mediated gene repair system was first tested in neonatal Fah−/− mice. A mix of AAV-sgRNA-repair (8 × 1010 vg per mouse) and AAV-Cas9 (4 × 1010 vg per mouse) viruses was injected into newborn Fah−/− mice through the facial vein. The same dose of either virus was used in parallel single-virus control experiments. To prevent an overestimation of the gene correction frequency due to selective growth of gene corrected hepatocytes, all breeders and breastfeeding mice were provided with 32 mg/L NTBC in the drinking water. The experimental animals grew normally and no adverse health issues were observed during the 1-month period after AAV administration. No hepatocyte toxicity, from either Cas9 or AAV, was visible by H&E staining of the liver samples (not shown). Four weeks after the initial virus administration, the livers of the mice receiving both AAV viruses displayed 10.8% (95% confidence interval [CI]: 6.7–14.8%) positive Fah-expressing hepatocytes (Fig. 2A, C). In stark contrast, the livers of the mice receiving AAV-sgRNA-repair alone showed only rare clusters of Fah-positive hepatocytes. The livers of the mice receiving only AAV-Cas9 did not show any Fah-positive hepatocytes. These results indicate that CRISPR/Cas9-generated DNA double-strand breaks markedly increased the efficiency of homologous recombination-mediated mutation correction.
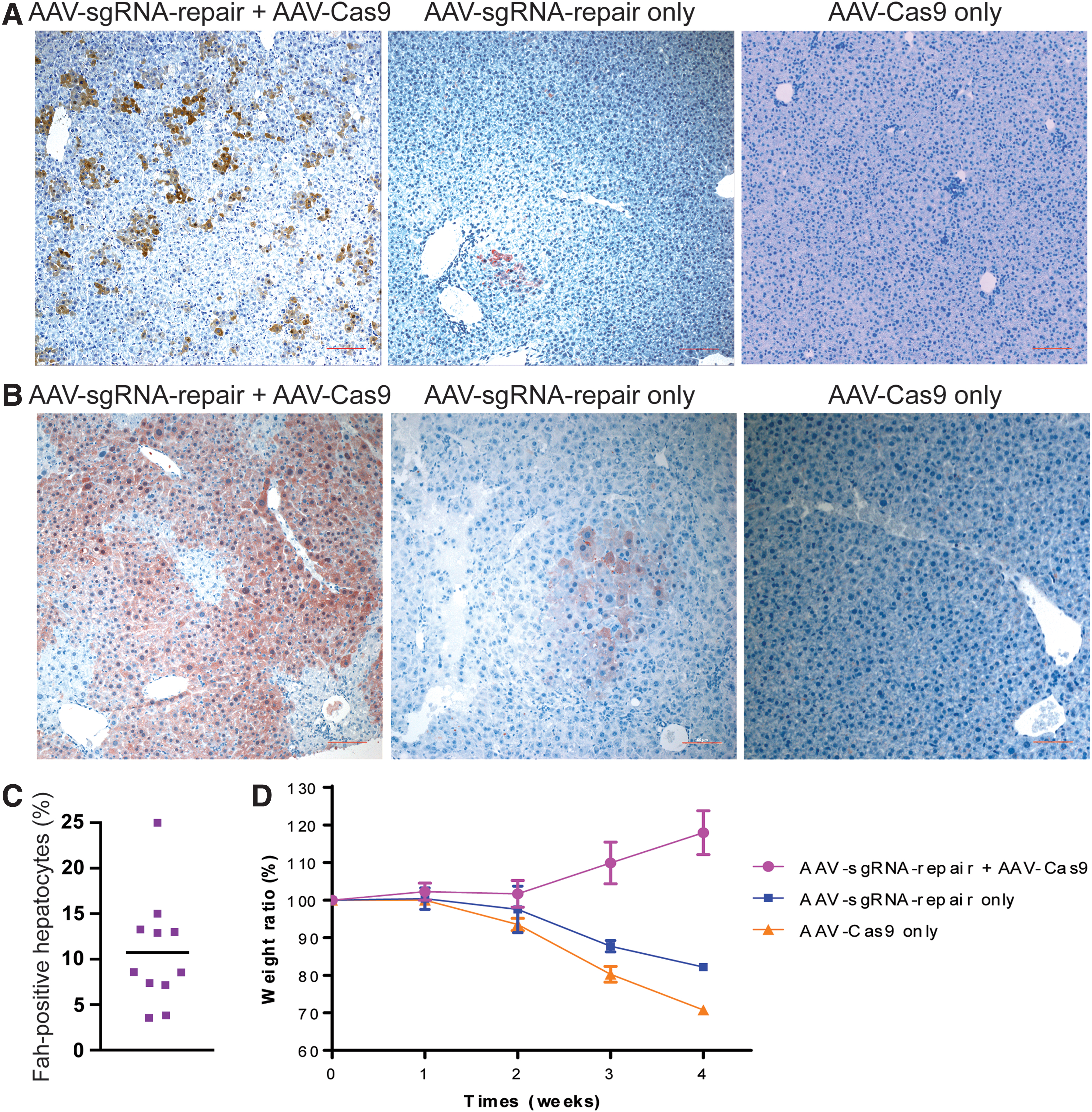
Gene correction in neonatal Fah−/−
mice.
For a small cohort of experimental mice, NTBC was removed from the drinking water 4 weeks after the initial AAV administration and monitored for 4 more weeks. The mice receiving the combination of AAV-sgRNA-repair and AAV-Cas9 viruses showed steady growth. At harvest, >80% of the hepatocytes had positive Fah staining (Fig. 2B). As we had seen before, the mice receiving AAV-sgRNA-repair virus alone suffered from an initial weight loss before expanding the Fah positive hepatocyte population due to the selection pressure induced by NTBC withdrawal (Fig. 2D). The mice receiving the AAV-Cas9 virus alone never recovered from the initial weight loss and died soon after NTBC withdrawal. These results indicate that the gene repair by our dual virus system was sufficient to support a full functional correction of Fah deficiency hepatocytes.
Gene correction in adult mice
Next we tested this AAV-mediated gene repair system in adult Fah−/− mice. A clinically relevant dose of 4 × 1011 vg/mouse of AAV-sgRNA-repair virus and 2 × 1011 vg/mouse of AAV-Cas9 virus was injected intravenously into 8-week-old Fah−/− mice, along with single virus injections in parallel control experiments. The mice receiving AAV-sgRNA-repair alone did not show any Fah-positive hepatocytes, indicating very low homologous recombination levels in adult mice. However, the mice receiving both AAV-sgRNA-repair and AAV-Cas9 viruses showed 1.6% (95% CI: 1.0–2.3%) Fah-positive hepatocytes in the liver (Fig. 3A). This low but solid gene correction frequency was dependent on the presence of both AAV-sgRNA-repair and AAV-Cas9 viruses.
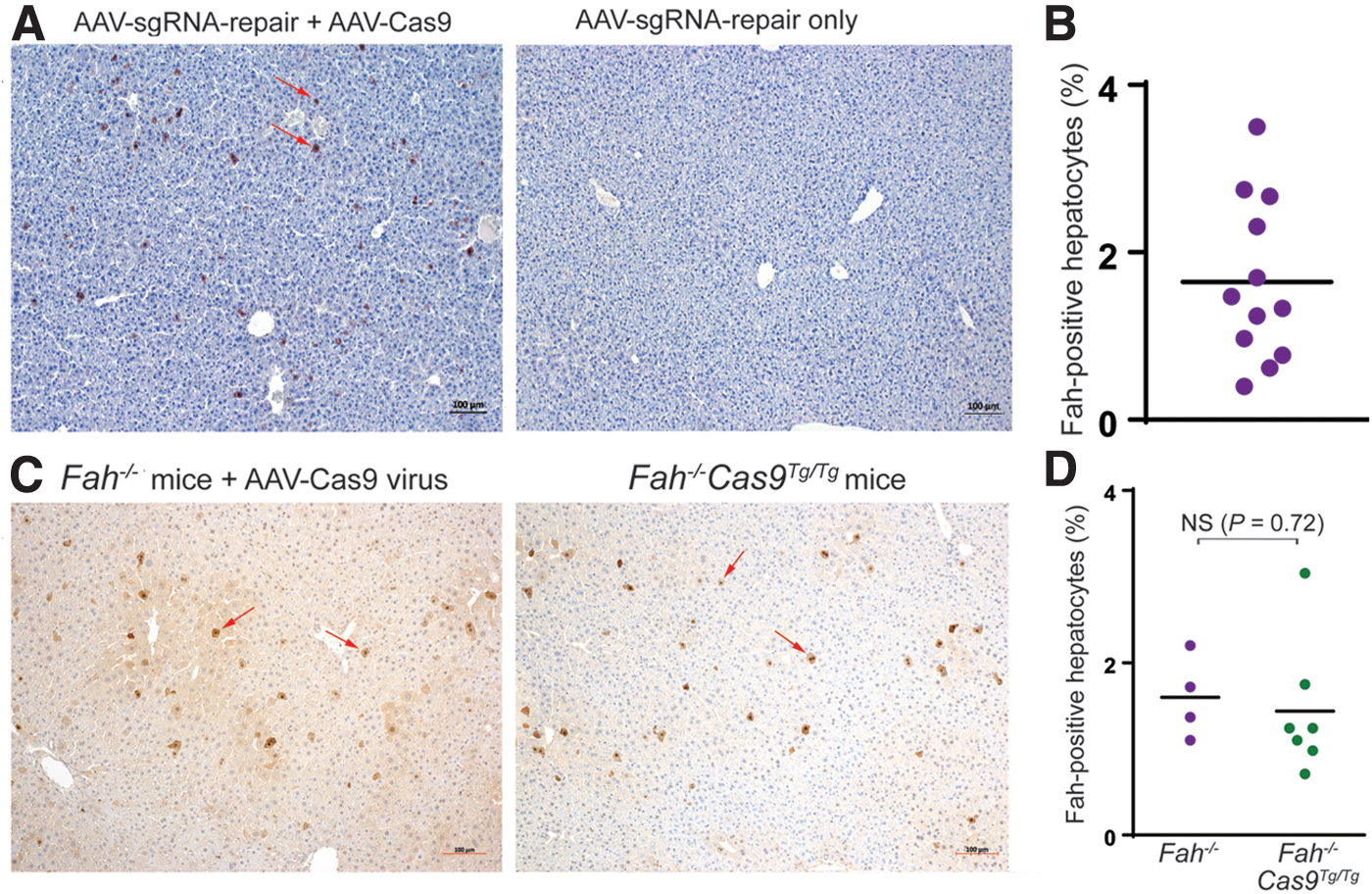
Gene correction in adult Fah−/−
mice.
Several explanations exist for why the same system worked well for neonates but poorly for adults. One possibility was that the AAV virus dose used was not sufficient for adult mice. Considering that the target cell needs to receive both AAV-sgRNA-repair and AAV-Cas9 viruses at the same time to have its mutation corrected, an insufficient virus dose would certainly affect the gene correction efficiency. Since the ratio of AAV-sgRNA-repair virus to AAV-Cas9 virus was 2:1 in our two virus system, AAV-Cas9 virus was more likely to be the limiting factor. To test this possibility, we utilized the Cas9-transgenic mouse model in which Cas9 is expressed constitutively in all the tissues. Fah−/− mice were crossed with Cas9-transgenic mice to generate the Fah−/−Cas9Tg/Tg strain. 4 × 1011 vg/mouse of AAV-sgRNA-repair virus was injected into 8-week-old Fah−/−Cas9Tg/Tg and Fah−/− mice, with the latter also receiving an injection of 2 × 1011 vg/mouse of AAV-Cas9 virus. Four weeks after the virus administration, the gene correction efficiency was measured by anti-Fah staining. As shown in Fig. 3C, despite having abundant Cas9 activity, the gene correction frequency of the Cas9 transgenic mice was the same (p = 0.72) as in Fah−/− mice receiving both viruses (Fig. 3D). This finding indicates that Cas9 expression was not a rate-limiting factor in the dual vector approach.
Proliferation is the key for gene correction in adult hepatocytes
It is well known that adult hepatocytes rarely divide, whereas hepatocytes in neonatal mice are proliferating. 15 Since homologous recombination depends on cell division and occurs mostly in late S phase and G2 phase of the cell cycle after DNA replication in eukaryotes, 16 we postulated that the higher gene correction frequency in younger animals might be caused by more active hepatocyte replication. Therefore, we sought to determine whether promoting hepatocyte proliferation could boost homologous recombination (HR)-mediated gene correction in adult mice. To do this, mice were given a diet containing tri-iodothyronine (T3). T3 is essential for normal organ growth and known to induce hepatocyte proliferation in vivo in rodents. 17 BrdU was delivered in the drinking water in vivo to label proliferating cells. Consistent with the previous report, 10 days after being fed with T3-supplemented chow, the mice showed increased hepatocyte proliferation, evidenced by substantially enhanced BrdU-positive immunostaining in the liver (Fig. 4A). As a comparison, the mice fed with the placebo diet showed very few BrdU-positive hepatocytes. No adverse effects were seen in T3-treated mice and the mice also displayed a normal H&E staining of liver sections (not shown).

Gene correction with induced liver regeneration.
A cohort of Fah−/− mice was fed either T3-supplemented chow or placebo diet and injected with 4 × 1011 vg/mouse of AAV-sgRNA-repair virus and 2 × 1011 vg/mouse of AAV-Cas9 virus 5 days after the initiation of T3-diet intervention. This special diet intervention was maintained for 2 more weeks before switching back to the normal rodent chow. When harvested 4 weeks after the virus administration, the mice receiving this 19-day T3 diet intervention showed an average of 3.5% (95% CI: 2.5–4.5%) Fah-positive hepatocytes in the livers, significantly more (p < 0.005) than the average of 1.6% Fah-positive hepatocytes observed in the mice receiving the placebo diet (Fig. 4B, C). These results indicate that a temporary boost of hepatocyte proliferation by T3 promotes the efficiency of gene correction in hepatocytes.
The FA pathway promotes AAV-mediated gene correction
The FA pathway promotes the use of homologous recombination to repair DNA double-strand breaks by suppressing NHEJ. Recent study has shown that the FA pathway is required for Cas9-mediated single-strand template repair. 18 To further understand whether the status of FA pathway function affects AAV-mediated CRISPR-Cas9 genome editing, intercrossed Fah−/− mice were intercrossed with Fanca−/− mutants to produce double-knockout Fah−/−Fanca−/− mice. A low-dose mixture of 5 × 1010 vg/mouse of AAV-sgRNA-repair virus and 2.5 × 1010 vg/mouse of AAV-Cas9 virus was injected into a cohort of neonatal Fah−/−Fanca−/− mice and their littermate Fah−/−Fanca+/+ controls. Four weeks later, at harvest, Fah−/−Fanca +/+ mice showed a modest gene correction, with 5.2% of hepatocytes positive for anti-Fah immunostaining (Fig. 5A, C). As a comparison, the gene correction efficiency in Fah−/−Fanca−/− mice was significantly (p < 0.05) reduced to 2.1% of the hepatocytes (Fig. 5B, C). These results indicate that the FA pathway was not essential for the AAV-mediated CRISPR-Cas9 genome editing but did facilitate this process.

The impact of the FA pathway on AAV-mediated gene correction in neonatal mice.
DISCUSSION
Gene repair of disease-causing mutations by homologous recombination has obvious conceptual advantages over gene addition strategies. 4,7,9,19 Gene correction will persist for the life of the cell, result in proper regulation of the disease gene, and avoid insertional mutagenesis. However, homologous recombination is inefficient in mammalian cells and, therefore, strategies to enhance gene repair frequencies have been investigated by several research groups. 4,20 The liver is a major organ for intermediary metabolism and many genetic diseases are caused by defects in hepatocytes. For this reason, liver-directed gene therapy has been of significant interest. 1 The choice of the donor DNA represents one approach to enhancing HR efficiency and it has been found that AAV a single-stranded DNA virus works very well, particularly in vivo. 21,22 In rodents, rAAV can mediate readily measurable rates of HR in hepatocytes without any additional manipulations. 5,19 The rates can be as high as 1/200 cells in neonates, but are much lower in adults (<1/5,000). 5 However, this number remains well below the frequency required for therapeutic benefit in any known genetic disease. It is well known that double-strand breaks in the target sequence dramatically enhance homologous recombination. 7,8,23,24 We and others, therefore, used the CRISPR/cas9 system to create DNA double-strand breaks near the disease-causing mutation. Our group has used a point mutation in the Fah gene, a model for the human genetic disease hereditary tyrosinemia, for gene repair experiments. Indeed, CRISPR/Cas9-mediated strand breaks significantly enhanced the frequency of gene correction. 7
In our first experiments, a CRISPR/Cas9 expression plasmid was administered by hydrodynamic injection, along with single-stranded oligonucleotides as a donor template.
7
Gene correction frequencies of 0.4% were seen in adult mice, substantially improved compared with results obtained without the induction of double-strand breaks. However, this level of gene correction was still subtherapeutic. Subsequently, gene repair frequencies of ∼6% were found in adult Fah−/−
mice, when the donor template and gRNA were delivered by rAAV and CRISPR/Cas9 was administered as nanoparticles.
8
We wished to compare this result to a dual rAAV system, similar to that used by others to treat OTC deficiency.
9
In contrast to the OTC gene repair system, we utilized SpCas9, whereas they used the S. aureus enzyme.
9
The approach of co-injecting multiple rAAV vectors to achieve a double-strand break and enhanced homologous recombination has also been used to target therapeutic cDNA to a safe-harbor locus.
25,26
Using zinc-finger nucleases to induce the DSB, this method has even been used in a human clinical trial (
Our dual vector approach was fairly efficient in neonatal mice with an average of ∼11% (range 4–25%). The high inter-mouse variability was probably caused by the technical challenges of facial vein injection. Nonetheless, this level of gene correction would be in the therapeutic range for many disorders, especially secreted enzyme deficiencies. In adults the average frequency was 1.6% (range 0.3–3.8%), lower than the 6% reported using nanoparticles to deliver cas9 in the same model system. We, therefore, wondered whether Cas9 expression was rate limiting and whether the dual virus system was suboptimal in terms of Cas9 levels or the timing of its expression. Cas9 is constitutively expressed in all cells in Cas9 transgenic mice. Interestingly, gene repair frequencies were not increased at all when the rAAV harboring the gRNA and donor homology was given to Fah−/−Cas9Tg/Tg mice. This suggests that suboptimal Cas9 expression was not the reason for the relatively lower gene repair frequency observed in our dual vector experiments.
RNA nanoparticles can have hepatotoxicity 27 and we, therefore, wondered whether injury-induced liver regeneration could potentially explain the higher gene repair frequency in the study by Yin et al. 8 Although no overt liver toxicity was reported by Yin et al., the Fah+ hepatocytes seen in their study were frequently seen in small clusters of 2 or 3 cells, suggesting potential cell division of gene repaired cells. Similarly, it is well known that hepatocytes are actively dividing in the growing liver of neonates 15,28 and hepatocyte replication, therefore, could also explain the higher repair frequency in younger animals. We directly tested this hypothesis by pharmacological induction of liver regeneration using the potent mitogen T3. Indeed, the gene repair frequency was doubled on average by this manipulation, reaching up to 7% in some animals. Pharmacological and temporary induction of liver regeneration may, therefore, be a viable strategy to enhance hepatocyte gene repair in a clinical setting. In fact, T3 analogs with hepatocyte specificity have been used in rodents to induce liver regeneration 29 and are in clinical trials for hyperlipidemia in humans. 30
We also investigated whether the FA DNA repair pathway 31,32 plays a role in hepatocyte gene repair. The FA pathway has been found to be a critical player in single-strand oligonucleotide-mediated homologous recombination. 18 Specifically, the FA pathway was shown to be required for single-stranded template repair (SSTR). Loss of the FA pathway function reduced gene editing at double-strand breaks in favor of NHEJ. In this study, we found a modest but significant effect on rAAV-mediated recombination. This finding is consistent with the hypothesis that rAAV-mediated gene repair uses SSTR. In previous study, we showed that inhibition of NHEJ enhances AAV-mediated gene repair. 4,33 Together these findings suggest that pharmacological stimulation of the FA pathway, particularly in combination with blockage of NHEJ, could be used to improve gene repair in hepatocytes.
Footnotes
Acknowledgment
We appreciate the help with AAV virus preparation from the Molecular Virology Core at the Oregon National Primate Research Center, OHSU.
Author Disclosure
M.G. has a significant financial interest in Yecuris and Ambys Medicines, companies that may have commercial interest in the results of this research and technology (Fah mouse).
Funding Information
This study was supported by CA190144 (to M.G.).