
Abstract
Recombinant adeno-associated viral (rAAV) vector-based gene therapy has been adapted for use in more than 100 clinical trials. This is mainly because of its excellent safety profile, ability to target a wide range of tissues, stable transgene expression, and significant clinical benefit. However, the major challenge is to produce a high-titer, high-potency vector to achieve a better therapeutic effect. Even though the three plasmid-based transient transfection method is currently being used for AAV production in many clinical trials, there are complications associated with scalability and it is not cost-effective. Other methods require either large-scale production of two herpes simplex viruses, rHSV-RepCap and rHSV-GOI (gene of interest), with high titers, or a stable cell line with high titer wild-type adenovirus infection. Both of these options make the process even more complex. To address this issue, we have developed a stable cell line-based production with the use of only one rHSV-RepCap virus. Using this new methodology in small-scale production, we achieved ∼1–6 E + 04 vg/cell of AAV9 in the top producer clones. Large-scale production in 10-CS (10-Cell Stack) of one of the top producing clones resulted in ∼1–2 E + 13 vg/10-CS with 50% of full capsid ratio after purification. This method could potentially be adapted to suspension cells. The major advantage of this novel methodology is that by using the rHSV-RepCap virus, high titer AAV can be produced with any GOI containing a stable adherent or suspension producer cell line. The use of this AAV production platform could be beneficial for the treatment of many diseases.
Introduction
Adeno-
The broadly accepted standard rAAV production method involves using adherent HEK293 cells and transfecting them with two plasmids (gene of interest [GOI], RepCap with helper) or three plasmids (GOI, RepCap, Helper) by using calcium phosphate or polyethylenimine (PEI). 8,19 However, lot-to-lot variations and scalability problems were observed in viral vector production by transfection methods. 20
The other attractive large-scale AAV production method involves using herpes simplex virus-1 (HSV-1) or baculovirus production in insect cells. 21 –23 Also, in their study, Conway et al. showed that a proviral HEK293 cell line, GFP-92, up on infection with rHSV1 vector engineered to express AAV2 rep and cap genes can produce scalable AAV-GFP. 21 Another approach involves using stably transfected producer cell lines (PCL) infected with wt adenovirus. 24 –27 These helper virus-based production methods require viral clearance steps in the purification process. 10,26,28,29
The AAV production platform using HSV-1 virus utilizes two engineered replication-deficient d27.1 HSV-1 vectors, one carrying AAV RepCap and the other with the GOI -AAV ITR expression cassette. 21,30 –32 The main drawback of this existing herpes simplex system is the labor and cost involved with scalability of the production of the d27.1 HSV-1 vectors. These viral vectors were produced by using adherent vero (V27) cells in a fixed bed reactor system. 33 However, producing two HSV viruses simultaneously is a labor-intensive and expensive process even if the packed bed bioreactor system can be scaled up to 2,000 L. Nevertheless, the V27 cell line is the one widely used for HSV production; choosing any cell line that complements the d27.1 mutation in the KOS strain of HSV would be suitable for amplifying the helper rHSV. This will lead to a path of new research findings for the easier and feasible production of HSV viruses using different cell lines.
For production, either HEK293 or BHK cell lines were co-infected with both HSV1 viruses to produce AAV. The replication-deficient d27.1 HSV1 was (with deletion in ICP27 gene) produced by using V27 cells for this study to improve safety and yield. 21,31 The HSV particles were reduced (14.04 log 10 reduction in HSV) in the final AAV product by downstream processing, which ensured safety and very low-risk adverse effects. 33 –36 Even though this method is currently being used in many clinical trials, there are limitations associated with this method.
The increasing clinical demand for high yields of AAV vectors also requires a large-scale production of both HSV-1 viruses when using this platform. Advancements in this HSV-based platform will be crucial for the successful production for AAV for gene therapy. One of the approaches we are reporting here would be the development of a stable 293-derived PCL transfected with a plasmid containing rAAV vector genome along with a drug selection marker. Such a platform would require infection with just one type of ICP27-deletion mutant HSV vector (RepCap) (Fig. 1). A similar approach was reported by Toublanc et al., where a UL30 gene-deletion mutant HSV vector was used. The advantage of this system is that instead of two HSV vectors only one HSV vector will be required, thereby reducing the production cost for AAV for gene therapy. 37 This new method will be beneficial in minimizing the duration of HSV virus production, because only one virus is required. This will lead to a higher quality AAV final product with greater HSV clearance compared with previous methods.
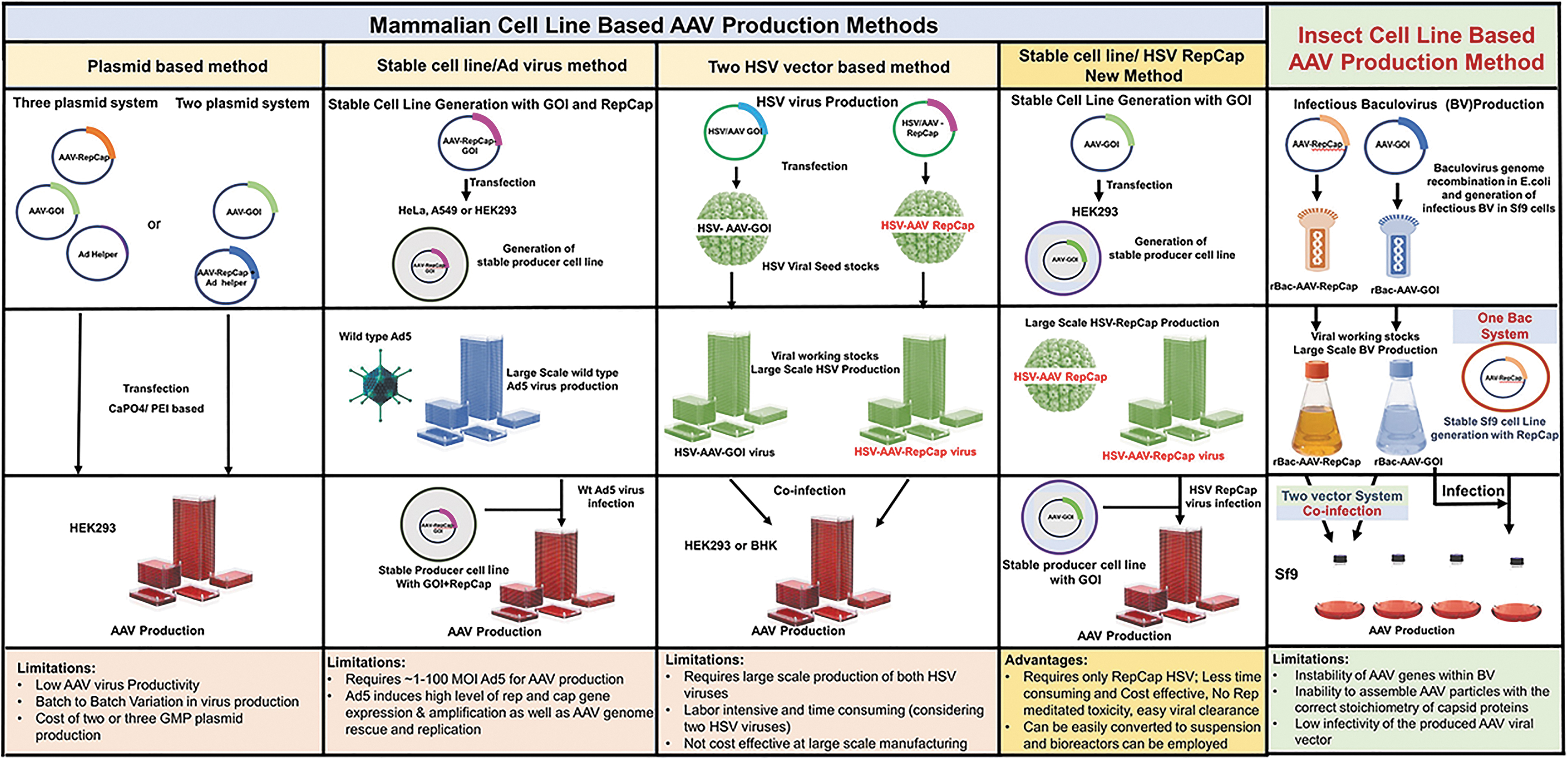
Schematic representation of simplified work flow for the manufacturing of AAV vectors using different platforms. Due to the limitations with the existing methods, as an alternative the stable cell line production and infection with rHSV-RepCap virus method was illustrated as a promising novel approach. AAV, adeno-associated virus; GOI, gene of interest; HSV, herpes simplex virus.
Materials and Methods
Cells, plasmids, and virus
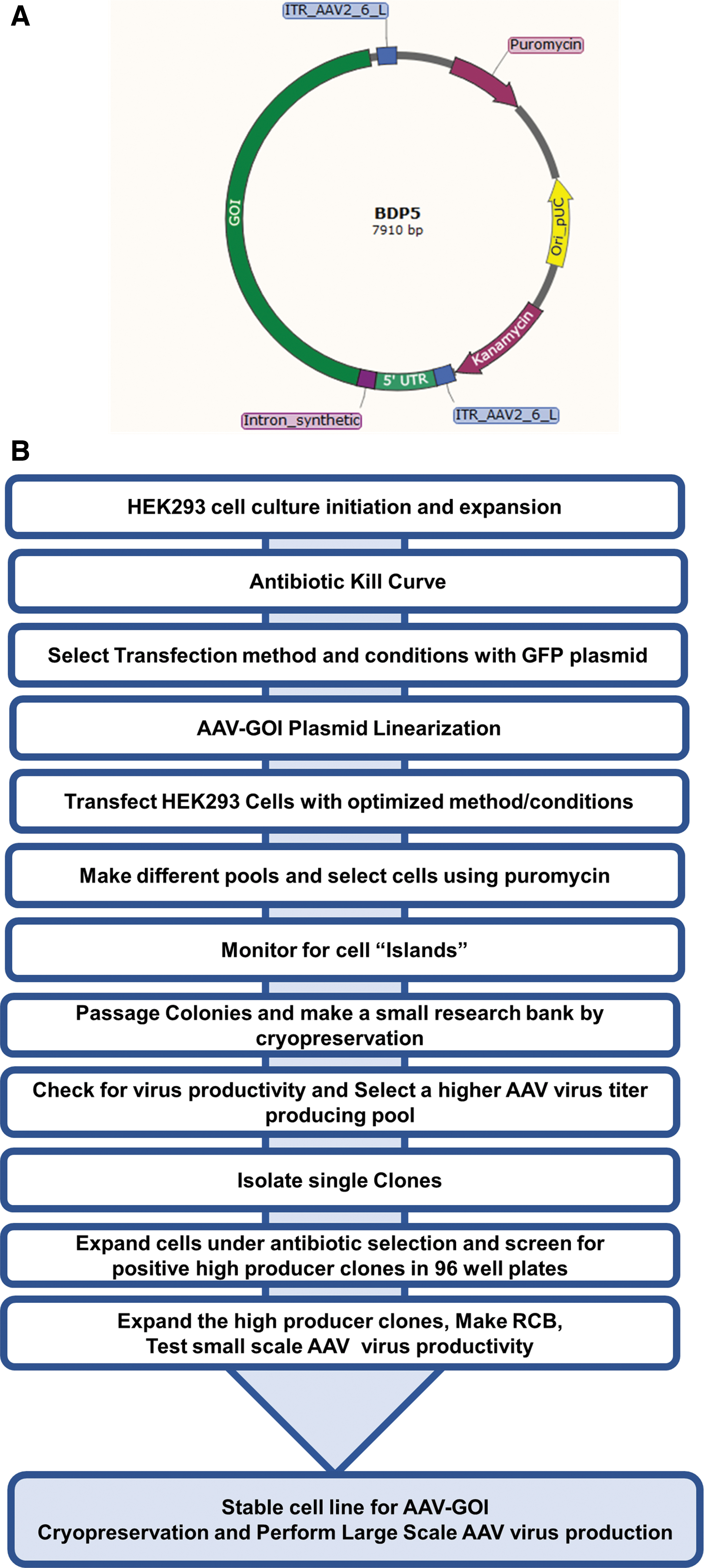
Adherent HEK293 cells (originally ATCC; F-11984) were propagated in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) with 10% fetal bovine serum (FBS from HyClone™). V27 cell lines were obtained from the University of Florida (Lot 091613TC), and a research cell bank (RCB) was made by culturing the cells in DMEM with 5% FBS. The cell culture flasks and cell stacks used were manufactured by Corning®. The BDP5-GOI plasmid was designed by BDP and synthesized by DNA2.0 (ATUM) (Fig. 2A). The HSV RepCap virus seed stock was from Solid Biosciences Inc., the HSV RepCap virus was produced by using the V27 cell line, and infectious viral particles were determined by plaque assay. The HSV virus working stocks and large-scale production were carried out by following the method of Adamson-Small. 38
(
Antibiotic kill curve generation and transfection efficiency testing for HEK293 cells
The day before the experiment, 0.8E+06 cells were seeded in a six-well plate containing DMEM +10% FBS medium. For the puromycin kill curve study, various concentrations of puromycin were added to the medium containing cells. The cells were propagated for 5 days, replacing the selective medium as needed. Each day, the cell viability was examined by using an automated cell counter. The most appropriate selective puromycin concentration required to kill the HEK293 cells was determined by the kill curve plot.
For the transfection efficiency study, seven different transfection reagents (Turbofect [ThermoScientific], Liopfectamine 3000 [ThermoScientific], Calcium Phosphate [Sigma], PEI [Sigma], PEI-Pro [Polyplus], Dreamfect [OZ biosciences], and Fugene [Promega]) were tested according to the respective manufacturer/available protocols. The pVectOZ-GFP plasmid was purchased from OZ Biosciences, and 1 μg of this plasmid was transfected with the respective transfection reagents for this study. After 48 h post-transfection, bright field and green fluorescent protein (GFP) fluorescence images were taken by a fluorescence microscope and the transfection efficiency was calculated. Transfection efficiency was calculated as the percentage of transfected cells by counting transfected cells holding the GFP signal as well as total cells from bright-field images recorded after 48 h.
Generation and selection of the AAV9-GOI stable producer cells
Briefly, the HEK293 cells were transfected with a plasmid that contained the GOI and puromycin drug resistance gene. The plasmid was linearized by restriction digestion with the PvuI enzyme (New England Biolabs). About 10 μg of the linearized plasmid was transfected with Turbofect reagent (Thermofischer scientific) according to the manufacturer's instructions. After 72 h, the transfected cells were divided into 16 different pools and stable pools were selected with puromycin (2 μg/mL) concentration. After selection, the stable pools were expanded up to T225 flasks to make a mini-RCB using Cryostor CS10 (BioLife Solutions) as the freezing medium. The stable pools were tested for their AAV productivity by using HSV Rep2Cap9 virus using small-scale six-well plate experiments. The top producer pool was selected for single-cell cloning. For single-cell cloning, the cells from the top producer pools were randomly sorted by using FACS ARIA III. The single cells were grown under antibiotic selection. After the complete growth of clones in 96-well plates, the clones were transferred to 24-well plates. At this stage, the expansion of single-cell clones and screening for AAV productivity were similar to that described earlier for the stable pool generation and selection. For large-scale AAV production, the high producer clone was expanded from T25 to T225, 1-CS and up to 10-CS by using trypsinization and seeding. The AAV production was performed as described later (Fig. 2B).
AAV production, harvesting, and lysis
The HEK293 stable producer cells containing the GOI were infected when the confluency reached greater than 80%. The cells were counted on the day of infection to calculate the amount of HSV RepCap virus to be added to the fresh medium for 12 multiplicity of infection (MOI). The cells were harvested after 48–72 h postinfection, centrifuged at 2,600 RPM for 20 min at 4°C, and finally washed with phosphate buffered saline (PBS). The cell pellets were stored frozen at −80°C until further processing. For cell lysis, the harvested cell pellet was thawed, lysis buffer (20 mM Tris, 150 mM NaCl, 0.5% OPE, pH.8.0) was added to the cell pellet, and the mixture was freeze-thawed three times by using a dry ice/ethanol bath. The lysed cell pellet was centrifuged at 2,600 RPM for 15 min at 4°C. The supernatant was tested for virus productivity by qPCR. For large-scale production, the cells were lysed, benzonase treated, and microfluidized before purification.
AAV purification
The harvested cell pellets from 10-CS were removed from −80°C and thawed at 37°C. The pellet was resuspended in 50 mL of 20 mM Tris, 150 mM NaCl, 0.5% OPE (Octyl Phenol Ethoxylate; J.T.Baker), pH 8.0 buffer. The salt active nuclease (50 U/mL; Arcticzymes) final concentration was formulated in 1 mM MgCl2. This was added to the cells with lysis buffer and incubated at 37°C for an hour with intermittent mixing. To overcome the virus aggregation problems in large-scale production and purification, a high-pressure homogenizer (Microfluidics) was used to disrupt the cells after lysis. The microfluidizer was operated as per the user manual for the Model HC-5000. The nuclease-treated harvest was transferred to a 500 mL sterile bag attached to the product inlet line of the homogenizer. An empty 500 mL bag was attached to the receiving end (outlet) of the homogenizer. The sample was passed through the homogenizer chamber at 4,000–5,000 PSI pressure until the feed bag was almost drained. Additional buffer (20 mM Tris-HCL, 50 mM NaCl, 0.1% OPE pH 7.6) was added to the feed bag, and the process was repeated to chase the rest of the product from the homogenizer chamber. The outlet receiving bag was disconnected. The homogenized lysate was centrifuged at 1,500 RPM for 20 min, and the supernatant was carefully transferred to a PETG bottle without disturbing the pellet. Poros CaptureSelect AAV9 affinity resin (Thermo Fisher) was packed in a column and equilibrated with a five-column volume of 25 mM Tris, 25 mM NaCl pH 7.2. The supernatant from above was loaded onto the equilibrated column. The column was washed with a 10-column volume of 25 mM Tris, 25 mM NaCl, pH 7.2 buffer. The AAV9 main peak was eluted by using 25 mM Tris, 25 mM NaCl, pH 3.0 buffer. The eluted virus fractions were immediately neutralized by using Tris buffer (pH 10) to pH 7.2. The Poros CaptureSelect AAV9 main peak fractions (9 and 10) were concentrated and diafiltered into PBS, pH 7.2 buffer by using a UF filter (Spectrum Filter, MPES filter 100 KD; 75 cm2). The overall yield was about 35–60%. The purified and concentrated virus sample was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver stained to check the purity. For western blotting, samples were transferred on to polyvinylidene fluoride membranes (Thermo Fischer); AAV cap proteins were detected by using an anti-AAV VP1/VP2/VP3 monoclonal antibody (B1-mouse monoclonal; Cat no:61084) at a 1:100 dilution and a secondary detection antibody was used at 1:2,000 dilution.
qPCR analysis of vector genomes
A 150 μL aliquot of each sample was combined with 20 μL of 1 × PBS, 20 μL 10 × DNase I Reaction Buffer (Thermo Scientific), and 10 μL of DNaseI, RNase-free (Thermo Scientific). The samples were incubated at 37°C for 30 min to facilitate digestion of nonencapsulated DNA, and at 65°C for 10 min to inactivate the DNase enzyme. The entire contents of each DNase reaction (200 μL) were extracted by using the Qiagen QIAamp DNA Mini Kit according to the manufacturer's instructions. AAV9 genomes were detected and quantified by qPCR with the following primer/probe set: GOI Forward Primer: 5′-CCAAGTGCAACATCTGCAAAG-3′, GOI Reverse Primer: 5′-TCTGGCAGATATCGTAGTTGAAGTG-3′ GOI Probe: 5′-FAM/TGCCCCATC/ZEN/ATCGGCTTCCG/3IABkFQ-3′. An aliquot of a GOI plasmid was diluted over a series of 10-fold dilutions to establish a standard curve for quantification. Ten microliter aliquots of sample, GOI plasmid dilution or 1 × TE (No Template Control) were tested in 30 μL reactions. Thermal cycling was performed on an Applied Biosystems 7900HT Sequence Detection System with the following temperature profile: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min.
Empty versus full capsid ratio analysis
The empty versus full capsid ratio in the final purified product was calculated by using size exclusion chromatography (SEC)-UV-MALS-dRI analysis. The SEC-UV-MALS-dRI analysis is based on the method developed by Wyatt Technology (Michelle Chen, PhD and Anatolii Purchel, PhD, Wyatt Technology Corporation. AN1617: Quantifying attributes of AAV gene therapy vectors by SEC-UV-MALS-dRI). This method utilized a triple detection system consisting of an Agilent HP1100 UV-Vis detector measuring at wavelengths 260 and 280 nm, Wyatt Technology's miniDAWN TREOS II multi-angle light scattering (MALS) detector with a Wyatt QELS embedded online dynamic light scattering (DLS) module and a Wyatt Optilab T-rEX differential refractive index (dRI) detector. The Empty (no DNA payload) and Full Capsid, AAV9-CMV-GFP (a single – stranded DNA of full-length payload) controls used in the assay were at 2.03E+13 and 4E+13 vg/mL, respectively and were received from Virovek Inc. Chromatographic separation was achieved with a Wyatt WTC-050S5 column (7.8 × 300 mm) and the corresponding guard column. The mobile phase, 1 × PBS (Lonza), was run at a flow rate of 0.5 mL/min. The duration of each run was for 45 min. Data from the MALS, DLS, and dRI detectors were acquired and processed by using the ASTRA® software.
Results and Discussion
Selection of antibiotic concentration and comparison of different transfection methods for efficacy determination using HEK293 cells
To generate a fully transfected stable cell population, it is very important to determine the optimal puromycin concentration required to eliminate the non-transfected cells. This is accomplished by performing a puromycin dose-response kill curve using HEK293 cells. Determining the selection capacity of puromycin will lead to a higher success rate in the development of stable PCL. 39,40 In the current study, HEK293 cells were grown in five different concentrations of puromycin (2, 4, 6, 8, and 10 μg/mL) containing medium for 5 days. The viability of the cells was estimated by using an automated cell counter. The kill curve was generated by plotting the viable cells versus days after adding the various puromycin (antibiotic) concentrations. It was determined from the curve that 2 μg/mL of puromycin would be the optimal dose for HEK293 cells (Fig. 3A).

A major challenge for the generation of stable cell lines is low transfection efficiency. Stable expression of a gene in a particular cell line can be influenced by the transfection method used. The choice of transfection reagent for stable cell line production is a critical factor that is often overlooked. Based on the source of HEK293 cells, the cells may respond differently to a given transfection reagent or method. Therefore, selecting the proper method is necessary to maximize the results. To identify the best method, the present study comparatively analyzed the transfection efficiency of seven different transfection systems (Turbofect [ThermoScientific], Liopfectamine 3000 [ThermoScientific], Calcium Phosphate [Sigma], PEI [Sigma], PEI-Pro [Polyplus], Dreamfect [OZ biosciences], and Fugene [Promega]) in HEK293 cells. Transfection was assessed through expression of GFP (pVectOZ-GFP plasmid). Forty-eight hours after transfection, the expression of GFP was detected by fluorescence microscopy and compared with the total cells to calculate the transfection efficiency. Figure 3B provides the summary of the transfection study. Several researchers have shown that lipofectamine transfection works well for HEK293 cells 41 –43 ; however, the data in Fig. 3B indicate that in our study Turbofect transfects the HEK293 cells with >80% efficiency. Hence, for this study Turbofect has been evaluated as a suitable gene transfer system for the HEK293 cells for the production of stable producer cells with GOI.
Developing a novel methodology using stable PCL with GOI and HSV RepCap virus for the production of AAV
Generation of stable pools containing GOI
Stable HEK293 producer cells containing the GOI were generated by transfecting the cells with linearized plasmid with GOI and puromycin resistance using the Turbofect reagent. Seventy-two hours post-transfection, the cells were split into 16 mini pools in six-well plates and selected for resistance to puromycin (2 μg/mL). The medium was replaced once every 3 days with fresh puromycin until all the un-transfected cells were killed by puromycin selection. In the transfected cells, multiple colonies/islands of cells started growing after selection. This resulted in stable pool generation. After the complete cell death in the non-transfected control cells, the selected stable pools were grown in the antibiotic selection media to achieve >70% outgrowth. The stable pools were expanded from T25, to T75, and then T225 flasks. A small-scale RCB (5 vials) for the stable pools was generated.
Identification of producer clones making high-titer AAV using RepCap HSV virus infection strategy
To determine the AAV productivity in the stable-GOI-HEK293 pools, one of the pools cells was infected with different MOI (2, 6, 12, 18, and 24) of HSV RepCap virus for 72 h in DMEM with 10% FBS infection media. Based on the results, 12 MOI was chosen for determining the productivity of all the pools. After 72 h postinfection with HSV RepCap virus infection, the stable cell pools that produced AAV were harvested by centrifugation at 1,200 RPM, 15 min at 4°C. The cell pellet was lysed, and the AAV titer was determined via qPCR estimation. Among the 16 pools tested, Pool 14 produced high-titer virus (243 vg/cell) compared with other pools. One of the alternate methods to improve the virus productivity in the stable cell line pool is to double transfect the cell lines and perform selection at high antibiotic pressure. To test this, Pools 10, 11, and 14 (high producer pools) were further transfected with 10, 20, 30, 40, and 50 μg of GOI plasmid per million cells (second transfection). The transfection concentrations above 20 μg of GOI plasmid resulted in cell death. The 10 and 20 μg transfected cell pools were cultured and selected by using 3, 6, and 10 μg of higher antibiotic pressure. When these cells were tested for virus productivity, even though their DNA copy number increased compared with single transfection, it did not result in higher virus productivity compared with single transfected high producer pools (data not shown).
Generation of stable cell line from the high-titer AAV-producing single-cell clones
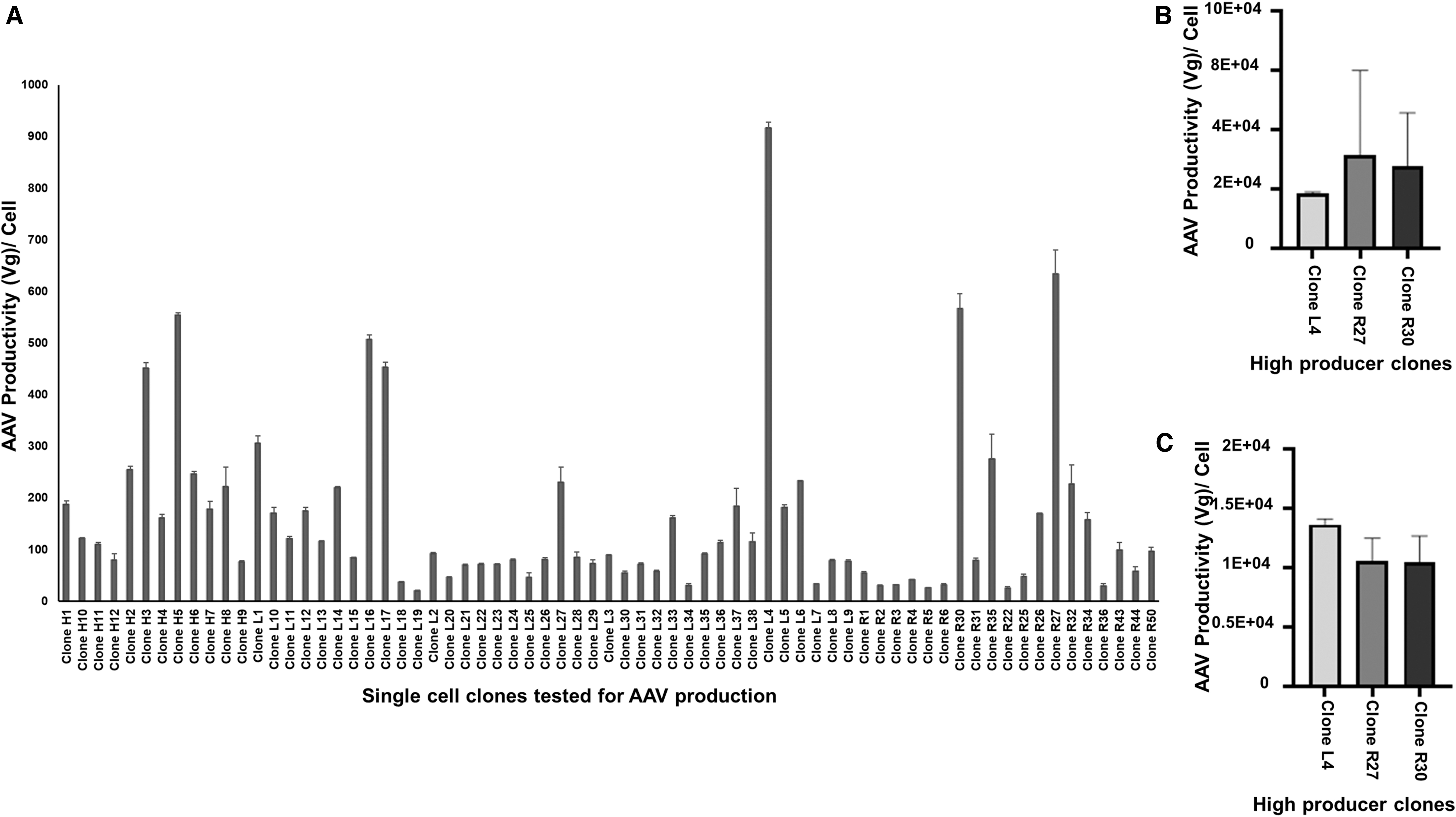
Among the top producing pool of cells, Stable pool 14, was selected for single-cell cloning. For the single-cell cloning, the cells from Pool 14 were sorted randomly by using FACS ARIA III sorter in 20 × 96 well plates. The cells were grown in puromycin selection media and inspected every day to identify the cells containing one colony. Single-cell clones were maintained in 96-well plates until the colony size was sufficient to allow transfer to 24-well plates in DMEM with 10%FBS with 2 μg/mL puromycin selection. Among the expanded clones, 70 clones were tested for AAV productivity by using 12 MOI of HSV Rep2Cap9 virus infection for 72 h in DMEM with10% FBS media. The AAV productivity was tested in each clone by the same qPCR method described earlier (Fig. 4A). The top producing AAV9 stable clone (L4) was found to have ∼1E+03 vg/cell. The other high producer clones were R27 (0.63E+03 vg/cell) and R30 (0.57E+03 vg/cell). The L4, R27, and R30 clones were expanded in six-well plates and eventually scaled up to T75 flasks, where a small RCB was made. The AAV productivity in these clones was tested in small-scale experiments by using six-well plates and different media conditions. Under the optimized conditions (12 MOI HSV RepCap infection in DMEM with 2% FBS, and harvesting at 48 h), the top producer single-cell clones (L4, R27, R30) produced ∼1-6E+04 vg/cell of AAV9 virus in small-scale six-well plate experiments (Fig. 4B). This is a higher productivity for AAV using the HEK293 PCL with HSV RepCap infection, compared with other plasmid-based transfection methods. 44,45 To improve the AAV productivity further, the concentration of HSV RepCap virus infection was increased from 12 MOI to 24 MOI in a small-scale study using optimized conditions for Clones L4, R27, and R32. The results indicated that the increase in the HSV RepCap virus concentration did not improve the virus productivity (Fig. 4C). After successfully setting up this novel production methodology, it was applied to the manufacturing of AAV on a larger scale. This methodology was scaled up by using 1-CS and 10-CS, which produced 1E+12 vg/1-CS and ∼1-2E+13 vg/10-CS (Fig. 5A) of AAV9 viral vector. This process can be adapted from stable adherent cell lines to suspension cell lines, which could require less HSV RepCap virus. 32 HEK293 cells were adapted to suspension culture in multiple studies. Tsao et al. have developed a process for adaptation of the adherent HEK293 cell line into a serum-free suspension medium through gradual serum weaning. 46 Grieger et al. have successfully adapted an adherent HEK293 cell line from a qualified clinical master cell bank to grow in animal component-free suspension conditions. 47 Similarly, the adherent producer cells can also be adapted to serum-free suspension cells. It is possible that these changes may result in higher cell density/mL but a decrease in production levels in suspension cells in some cases. But it is reasonable to assume that if the suspension adaptation is started with multiple high PCL, this process could increase the chance of getting a high producer suspension cell line. The AAV vector produced from large-scale manufacturing was purified by using Poros CaptureSelect AAV9 affinity chromatography (Fig. 5B). The final purified and concentrated AAV product was separated on an SDS-PAGE and silver stained. The purity of the AAV fractions was confirmed by the presence of VP1, VP2, and VP3 proteins (1:1:10 ratio) (Fig. 5C). A western blot with anti-VP1+VP2+VP3 monoclonal antibody also confirmed the AAV capsid proteins in the final product, which is shown in Fig. 5D. Another critical quality attribute to determine the rAAV vector quality is to determine the empty versus full capsid ratio in the final product. To achieve this, a simple and robust direct SEC method with UV-MALS-dRI detection was utilized (Wyatt Technology). The results demonstrated that this stable cell line-based method has improved vector quality and reduced empty capsids when compared with both transient-based and two HSV-based AAV production methods. The empty versus full capsid ratio for the purified final product was found to be 50:50 (Fig. 5E). Adamson-small et al. have reported 16% full capsids for the transient transfection-based method and 28% full capsids for two HSV infection-based AAV production methods. 38 The proportion of the full capsid using this stable cell line-based novel method is about 50% (vector size ∼4.3 Kb). This is in agreement with Burnham et al., who in their analytical ultracentrifugation approach showed that the stable cell line-based AAV production method produced a higher percentage of full capsids compared with that of the vectors produced by the transient transfection method. 48

Recently, Adamson-Small et al. showed that increasing the sodium chloride concentration in the medium at the time of rAAV production process increased AAV titers up to four- to six-fold when using the HSV infection method in HEK293 cells. 32 In addition, they have shown that converting an adherent platform to a suspension platform with serum-free media increased the AAV production by greater than three-fold in large-scale manufacturing. 32 However, their approach used a two HSV vector-based methodology. Producing two HSV vectors at large scale is a time-consuming, labor-intensive, high-cost process. Adapting these measures in a novel stable cell line with one HSV methodology may lead to a scalable, high-titer, high-quality AAV production. A scalable production of AAV2-GFP with a proviral cell line GFP-92 with single rHSV1 was explored by Conway et al. 21 Once the producer cells are developed, they can be used continuously for AAV production. 27 Ye et al. have published a research paper on the clearance of HSV virus during purification of AAV from the two HSV-based AAV production processes. 49 Given the previously established protocol for HSV virus clearance, it is a very easy process to completely clear the HSV virus from a final product produced using this new method. 36
Together, these data establish a user-friendly novel process that can be used to produce high-titer AAV with commercial feasibility. This study constitutes a step forward toward an improved scalable AAV production method aiming at clinical supply and expanding gene therapy applications. This alternative methodology can be easily utilized by current HSV-based AAV manufacturers for gene therapy clinical trials to minimize the labor, time, and cost.
Conclusions
AAV vectors are excellent tools to promote gene transfer and stable long-term gene expression in human gene therapy studies. 50,51 For large-scale AAV production, a stable PCL is desirable since it allows scalable and cost-effective viral production with increased reproducibility and safety. 52 However, producing Rep-Cap containing stable cell lines is very challenging because of the toxicity associated with the expression of Rep proteins. 53 Using either the two HSV-based vector system, 31,33,49,54 or the stable producer with Ad5 infection 26,27,55,56 for AAV production has major setbacks and limitations for large-scale production. The two HSV-based production system requires large-scale production of both HSVs, which is labor intensive, time consuming and imposes limitations on feasible scale. Similarly, the adenovirus system requires large-scale virus production and the establishment of downstream processes to remove wtAd5 for viral clearance from the final product. Here, we described a new AAV production method by generating a stable cell line with GOI and infecting with HSV RepCap virus. In this method, the RepCap is supplied by the HSV virus, so that the Rep mediated toxicity concerns are eliminated. In addition, developing a HSV RepCap virus platform will be very useful to extend this new methodology in many current high-titer requiring gene therapy trials with the generation of a stable GOI cell line. In this study, a single plasmid containing the GOI and a puromycin selectable marker was stably transfected into HEK293 adherent cells. Single-cell clones were infected with HSV Rep2Cap9 virus for AAV production. In small-scale studies, the top producer single cell clones produced ∼1-6E+04 vg/cell of the AAV9 virus. This is a high productivity for AAV production using an HEK293 PCL with HSV Rep2Cap9 infection when compared with other plasmid-based transfection methods. This novel methodology was scaled up to 1-CS and 10-CS production of AAV9 virus, which produced 1E+12 vg/1-CS and 1-2E+13 vg/10-CS. The final purified rAAV particles contained 50% full capsids. This method can potentially be converted to a suspension platform and may be able to achieve higher viral production. Together, these data establish a user-friendly novel process that can be used to produce high-titer AAV. This method offers manufacturing advantages, because production of only one HSV vector is required. Further, this method potentially can also be adapted to suspension culture and large-scale bioreactor-based production of AAV vectors can be tested to enable preclinical and clinical studies.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This research was supported by the Intramural research program of The National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH).