
Abstract
Adeno-associated viral (AAV) vectors represent an ideal vehicle for human gene transfer. One advantage to the AAV vector system is the availability of multiple naturally occurring serotypes that provide selective tropisms for various target cells. Strategies to enhance the properties of the natural AAV isolates have been developed and can be divided into two approaches, rational design or directed evolution. The rational design approach utilizes knowledge of AAV capsids to make targeted changes to the capsid to alter transduction efficiency or specificity, while the directed evolution approach does not require a priori knowledge of capsid structure and includes random mutagenesis, capsid shuffling, or random peptide insertion. In this study, we describe the generation of novel variants for both AAV2 and AAV5 using a rational design approach and knowledge of AAV receptor binding, surface charge, and AAV capsid protein posttranslational modifications. The novel AAV2 and AAV5 variants demonstrate improved transduction properties in both the mouse retina and cornea. The translational fidelity of the novel AAV2 variant was confirmed in the context of the nonhuman primate (NHP) retina, whereas a NHP tissue explant model was established to allow the rapid assessment of translational fidelity between species for the AAV5 variants. The capsid-modified AAV2 and AAV5 variants described in this study have novel attributes that will add to the efficacy and specificity of their potential use in gene therapy for a range of human ocular diseases.
Introduction
The successful clinical application of adeno-associated viral (AAV) gene therapy vectors for ocular disease has been demonstrated and ocular gene therapy products are now a reality. Notably, the first human gene therapy drug to be approved by the Food and Drug Administration, and to enter the U.S. market, was Luxturna, a treatment for an inherited retinal dystrophy, Leber's congenital amaurosis 2. 1,2 Numerous ongoing clinical and nonclinical studies further underscore the utility of the AAV vector system as an effective gene delivery vehicle for the eye, and the availability of a variety of naturally occurring serotypes, displaying various retinal cell tropisms, are considered effective tools to target therapeutic genes to the retina. One disadvantage to the first-generation capsids, in the context of posterior retinal gene therapy, is that their delivery requires an invasive subretinal surgery, thus to address the need for a less-invasive delivery route, research is focused on developing AAV vectors that can transduce outer retina (i.e., photoreceptors [PR]/retinal pigment epithelium [RPE]) following intravitreal injection. 3,4 Moreover, the delivery of genes to the anterior chamber of the eye, specifically corneal endothelial cells, would likewise benefit from a less-invasive surgical procedure than intracameral delivery 5 or corneal puncture. 6 Notably, both intravitreal and subretinal delivery of AAV vectors are associated with a dose-dependent inflammation, 4,7 thus an important advancement to engineering capsids with improved efficiency, is the ability to address immunogenicity and dosage challenges without compromising efficacy.
Strategies to identify next-generation capsids can be divided broadly into two approaches, rational design 8 and directed evolution, 9 with the latter approach involving iterative screening of complex AAV capsid libraries to identify capsids with desired attributes. Additionally, the directed evolution approach does not require any knowledge of underlying structure–function relationships in the capsid proteins. In contrast, rational design requires knowledge of the AAV capsid structure and function, and progress using this strategy has been facilitated by the use of X-ray crystallography and cryoelectron microscopy, to solve the structures of first-generation AAVs. 10 –16 Structure knowledge provides a basis for developing customized capsids for specific targeting of AAV vectors. The interaction of domains on the surface of the AAV capsid with specific cellular receptors dictates the tissue tropism for the various AAV serotypes, with different AAV serotypes utilizing unique cellular receptors. For example, the primary receptor for AAV2 is heparan sulfate proteoglycan (HSPG), 17 whereas the receptors for AAV5 have been identified as α-2-3-N-linked sialic acid and platelet-derived growth factor receptor. 18,19 Previously, we and others, described AAV2 variants devoid of canonical heparan sulfate (HS)-binding residues. These AAV2 variants were shown to mediate very efficient photoreceptor transduction when delivered subretinally, a tropism that is in vast contrast to that observed with the parental, heparan sulfate-binding AAV2, which shows limited tropism for photoreceptor cells with subretinal delivery. 20,21 Thus, knowledge of structural determinants of receptor binding and tropism, provide insights for modifying the capsid to generate variants with desired transduction profiles.
The AAV capsid is composed of the three structural proteins viral protein (VP)1, VP2, and VP3, which are expressed from the same open reading frame in an approximate stoichiometry of 1:1:10, with an alternative start codon for VP2 and VP3. 22 –24 The capsid proteins share most of their amino acid sequence; VP1 and VP2 differ from VP3 by a shared N-terminal extension of ∼65 amino acids, depending on the serotype, with VP1 containing an additional ∼135 unique amino acids, a region known as VP1-unique (VP1u). 10,22 –24 VP1 is required for viral infectivity, in part due to the presence of a highly conserved, N-terminal phospholipase A2 (PLA2) homology domain (amino acids 44–138) that is buried within the capsid interior but becomes externalized through pores found at the fivefold symmetry axis following a conformational change in the acidic endosomal compartment. 25 –27 While VP2 is dispensable for capsid assembly and infectivity of the virus, any deletion or mutation in VP1 that results in loss of the phospholipase A2 (PLA2) catalytic domain and its activity, results in a significantly reduced AAV infectivity. 25 –27 In addition, it has been postulated that other signals affecting infectivity might be located on the VP1-unique region of AAV2, which has been reported for several autonomous parvovirus capsid proteins. For example, it has been demonstrated that AAV2 VP1u has protease activity and that the protease site overlaps, in part, with the PLA2 domain in AAV2 VP1u. 28 Previously, we reported on the development of a direct liquid chromatography/mass spectrometry (LC/MS) intact protein analysis to characterize viral capsid proteins. 29 Using this method, the complete characterization of the constituent viral capsid proteins of several AAV vectors, including their sequences and posttranslational modifications (PTMs), including acetylation and deamidation were reported. 29 Notably, the N-termini of all VPs of the six serotypes analyzed (AAV1, 2, 5, 7, 9, and rh10), was confirmed to start at one residue after the predicted N-termini based on DNA sequences, with one exception the VP3 of AAV7. Additionally, the VP1 and VP3 of AAV serotypes 1, 2, 5, 7, 9, and rh10, were shown to contain an N-terminal acetylation. Although N-terminal acetylation of proteins is a widely known phenomenon, the biological significance of N-terminal acetylation of viral capsid proteins is not well understood. One likely hypothesis we proposed, was a potential link between N-terminal acetylation of VP1 and VP3 and viral capsid degradation and uncoating before nuclear entry. 29
AAV-mediated transduction of multiple retinal cell types in rodent models of inherited retinal disease, after either subretinal or intravitreal injection, is well reported. 30,31 Additionally, the efficiency and tropism of different AAV serotypes toward photoreceptor cells, have been evaluated in other animal model species, including pigs, 32 dogs, 33,34 and nonhuman primates (NHPs). 35,36 One limitation of the direct application of results from animal studies is that viral transduction can vary widely across species. Intravitreal injection in the rodent retina, in contrast to subretinal delivery, 37 results in robust transduction of retinal ganglion cells, the inner nuclear layer as well as Muller glia (Internal data and Refs. 20,38 ). However, although some retinal ganglion cells can be transduced in NHPs by intravitreally injected AAV2-based vectors, the area of transduction is restricted primarily to a ring of retinal ganglion cells around the fovea, with no Muller glia cell transduction. 39 The absence of a cone-enriched fovea in the rat and mouse, further highlights the limitations of using rodent retina to model human retinal disease and the need to confirm translatability of AAV transduction patterns to a larger mammalian retina. Large mammal model availability can be limited, so to assess translatability of AAV transduction patterns, the use of ex vivo mammalian retinal explants is being explored. 40 The merits of using an organotypic explant approach to test the viral transduction efficiency and tropism of different AAV serotypes in human retina and RPE choroid has been demonstrated, 40 and while there was evidence of variability in outer nuclear layer (ONL) transduction efficiency across different donor retinas, this approach provides a reliable method to determine how the different AAV capsids transduce a primate retina.
In this study, we describe the use of a rational design approach to engineer AAV capsids with improved transduction for photoreceptors and corneal endothelial cells. The translational fidelity of our novel heparan sulfate binding knockout (HBKO) variant, AAV2-HBKO, in the NHP retina was confirmed, moreover, the use of an organotypic NHP retinal explant model, to confirm translation of novel AAV5 variants between species, was evaluated. The novel AAV variants described in this study will provide additional tools for gene therapy strategies in the treatment of human blindness.
Materials and Methods
AAV vector production and purification
AAV vectors were produced using the transient triple transfection method as previously described. 41 Briefly, HEK293 cells were transfected using polyethyleneimine, and a 1:1:1 ratio of three plasmids (AAV vector, AAV rep/cap, and Ad helper plasmid). In the case of some AAV production campaigns, the Ad helper functions were replaced by using wtAd5 at a multiplicity of infection (MOI) of 10. The vector plasmid contains the vector genome (vg) chicken β-actin hybrid promoter (CBA)-enhanced green fluorescent protein (eGFP) and ITR sequences from AAV2. eGFP expression is driven by the cytomegalovirus enhancer, CBA promoter as described. 42 The AAV rep/cap helpers contained rep sequences from AAV2 and serotype-specific capsid sequences with the nomenclature, rep2/cap2, rep2/cap5, rep2/cap7, etc. The pAd helper used was pHelper (Stratagene/Agilent Technologies, Santa Clara, CA). AAV vectors were purified by affinity column chromatography (AVB Sepharose High-Performance medium; GE Healthcare) as previously described. 41 The fractional content of empty and genome-containing capsids was assessed by analytical ultracentrifugation, as previously described. 43
Analyzing recombinant AAV vector purity using SYPRO Ruby Protein Gel Stain
Samples from purified vector were loaded onto a NuPage 4–12% Bis-Tris gel (Invitrogen). Typically, 1–5 × 1010 vgs of purified vector was analyzed. The gel was stained with SYPRO Ruby Protein Gel Stain (Life Technologies).
LC/MS intact protein analysis
As previously described, 29 AAV virions was first concentrated with an Amicon Ultra centrifugal filter (0.5 mL, 10 kDa molecular weight cut off [MWCO]) and then washed with 25 mM Tris pH 7.1 three times. The concentrated AAV virions were denatured with 10% acetic acid vortexed and further diluted with an equal volume of High Performance Liquid Chromatography water. The final acetic acid concentration was 5%. Fifty microliters of AAV solution (∼2–5 μg of proteins) was injected to Acquity Ultra-Performance Liquid Chromatography (UPLC) coupled with a Xevo G2-XS qTOF MS instrument (Waters, Milford, MA). The separations were performed on a Ethylene Bridged Hybrid (BEH) C8 column (2.1 × 100 mm) at a flow rate at 250 μL/min. Mobile phase A and mobile phase B were 0.1% formic acid in water and acetonitrile, respectively. The final gradient for C8 column was as follows: from 10% B to 20% B over 6 min, from 20% B to 30% B over 10 min, then from 30% to 38% B over 40 min. The capillary voltage and sampling cone voltage of the mass spectrometer were set at 3.5 kV and 45 V, respectively. The mass spectra were acquired in positive sensitivity mode over m/z 500–4,000. MaxEnt1 in MassLynx software version 4.1 was used for protein deconvolution.
Enzymatic digestions of AAV1 and AAV2 VPs
AAV2-eGFP generated from triple transfection as well as producer cell line (PCL) process 41 (∼10 μg capsid protein) was first concentrated using Amicon Ultra centrifugal filters (10 kDa MWCO), denatured with 6 M Guanidine-HCl, and 50 mM Tris at pH 8.5. The proteins were reduced with 5 mM Dithiothreitol at 60°C for 30 min in darkness and then alkylated with 15 mM iodoacetamide at room temperature for 30 min. The samples were buffer exchanged into 25 mM Tris pH 7.1 for digestion using Bio-Spin® 30 Tris column. After buffer exchange, the samples were split into two aliquots. Each aliquot was digested with trypsin at 1:25 or Asp-N at 1:50 enzyme: protein ratio (w/w) for 2 h at 37°C, respectively.
UPLC/MS/MS peptide mapping
The protein digests were analyzed by UPLC/MS/MSE using an Acquity UPLC-Xevo G2-XS qTOF MS system (Waters). The separation was achieved using a BEH300 C18 column (2.1 × 150 mm) with a linear gradient from 2% to 40% B (0.1% formic acid in acetonitrile) over 68 min at a flow rate of 250 μL/min. For MS, the capillary voltage and sampling cone voltage were set 3.0 kV and 30 V, respectively. The mass spectra were acquired in the positive sensitivity MSE mode in the m/z range of 500–2,000.
Determination of deamidation levels in AAV VPs
The extracted ion chromatograms of peptides and their corresponding deamidated species were used for calculation of deamidation levels. 29
Generation of AAV capsid variants
Deamidation variants N57D and G58D are based on pim45BD-cap2, an AAV helper plasmid that contains both rep and cap sequences from AAV2. Fragments containing the designated mutations were synthesized (Genscript) and subcloned into pim45BD-cap2. Mutations were verified by DNA sequencing (Genewiz). Acetylation variants were constructed as above, using pHLP19-cap5.2, a helper plasmid that contains rep from AAV2 and cap from AAV5, as the parental plasmid. AAV2-HBKO and AAV5 arginine mutant capsid plasmids were generated by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies), according to the manufacturer's protocol. The pIM45BD plasmid was used to generate AAV2-HBKO using a polymerase chain reaction (PCR) mutagenesis primer designed to alter the codons encoding arginine 585 and 588 on VP3 to alanine. The sequence of the mutagenic primer used to generate the R585A and R588A mutations was: TATCTACCAACCTCCAGGCAGGCAACGCACAAGCAGCTACCGCAG. The AAV5-G474R, AAV5-N564R, and AAV5-N573R mutants were generated using the AAV5 rep/cap plasmid (pHLPcap5.2) as a template and PCR mutagenic primers designed to change the respective arginines to alanines. The sequence of the mutagenesis primer used to introduce the G474R mutation was CCAGGTTCCAGCGCTGGGTTCGGCC. The sequence of the mutagenesis primer used to introduce the N564R mutation was CCGCGTGGCGTACCGCGTCGGCGGGCAG. The sequence of the mutagenesis primer used to introduce the N573R mutation was CAGTGGTGGAGCTCTGTCTGTTGGTGGCCATCTG. All mutations were confirmed by DNA sequencing.
Infectious AAV particle titration
Titration of infectious AAV particles by quantitative PCR (qPCR) was performed using the endpoint dilution assay as described previously. 44 Tissue culture infective dose (TCID)50 titers were calculated from two independent assays and results are shown in Supplementary Table S1. Briefly, HeLa RC32 cells (ATCC® CRL-2972™) harboring integrated copies of both rep and cap genes, were infected with 10-fold dilutions of the AAV vector preparations, in the presence of wild-type Ad5 (MOI 10). Infectious titer was calculated as IU/mL using qPCR assay targeting the bovine growth hormone poly A sequence encoded in the vg. Additionally, titration of infectious AAV particles was also assessed by transgene expression. 44 HeLa cells were infected with serial dilutions of AAV vectors. Analysis of GFP expression by fluorescence microscopy of cells was performed with the following limit of quantitation (LOQ) and limit of detection (LOD) and results are shown in Supplementary Table S1.
LOQ: Detectable signal in wells but not quantifiable (too few wells positive).
LOD: No detectable signal in any wells (<1,000 IU/mL).
NHP studies
The NHP study was performed at Covance Laboratories, Inc. (Madison, WI). Four adult male cynomolgus monkeys (Macaca fascicularis) were screened for neutralizing antibodies against AAV5 and AAV2 before surgery as previously described. 35 Animals considered seronegative (serum titers ≤1:4) were included in the study. Cynomolgus monkeys were assigned to two groups and were administered either AAV2-HBKO-eGFP or AAV5-eGFP. Animals were dosed through subretinal injection to both eyes (OU) once on day 1 of the dosing phase at a volume of 120 μL/eye. The diluent was Alcon® BSS® with 0.014% polysorbate 20. Dosing syringes (1.0 mL Luer Lok™ Becton Dickinson Product 309628 or equivalent) were filled and affixed to a Dutch Ophthalmic Research Center (DORC) 23-gauge needle with an extendible 41-gauge subretinal injection needle on the day of dosing using aseptic procedures under a laminar flow hood. Injections were administered within 30 min of filling the DORC injector. Dosing was done performed in OU of each animal. Animals were anesthetized with intramuscular injections of atropine (0.01 mg/kg), ketamine (2–10 mg/kg), and dexmedetomidine (0.25 mg/kg). Following the procedure, the anesthesia was reversed with atipamezole (0.25 mg/kg). The anesthesia regimen was adjusted based on the responsiveness of animals and according to Covance veterinary staff. Eyes were cleaned with an ∼1% povidone iodine solution (prepared with sterile saline and 5% povidone iodine) and rinsed with sterile saline. An ∼2.5% povidone iodine solution was used at the dose site before injection. The injection was performed following a study-specific procedure, briefly described as follows.
Pupils were dilated with a topical mydriatic agent. The DORC disposable dual-bore injection needle (23-gauge) was introduced directly through the sclera in the superior temporal quadrant of the globe ∼3 mm posterior to the corneal limbus and moved through the vitreous under visual control using a surgical microscope viewing through a dilated pupil, with a modified fundus-viewing lens placed on the cornea. The 41-gauge cannula tip was advanced from the 23-gauge needle and gently touched the retinal surface. The dose was injected through the neural retina into the subretinal space, resulting in a subretinal bleb. The 41-gauge cannula tip was retracted, and the 23-gauge needle was withdrawn. A topical antibiotic and steroid ointment (Neo-poly-dex) was instilled in each eye following all other postdose ocular procedures. The injection occurred in a small portion of the retina in the mid-arcade region.
Fundus autofluorescence imaging
The fundus autofluorescence (FAF) imaging was done once during the predose phase and during weeks 2, 4, and 6 of the dosing phase. Animals were anesthetized (remained under anesthesia used for dosing) for FAF imaging. Eyes were dilated with a mydriatic agent (1% tropicamide). FAF images were taken of each eye to include the subretinal dose sites and the fovea. Images were taken with a Heidelberg SPECTRALIS® instrument.
Tissue processing of NHP retina
On day 43 of the dosing phase, all animals, having been fasted overnight, were anesthetized with sodium pentobarbital, exsanguinated, and necropsied. The eyes from each animal were injected with and submerged in chilled 4% paraformaldehyde (PFA) and stored in a refrigerator, set to maintain 2–8°C, for 48–72 h. The eyes were then embedded in paraffin, sectioned, and slides were prepared for immunohistochemistry (IHC) analysis. From the temporal calotte, 20 serial sections were taken from the fovea. In addition, 20 serial sections were taken every 250 μm for a total of eight steps from the remaining temporal calotte. One slide from the fovea and one slide from each of the eight steps of the temporal calotte were stained with Hematoxylin and Eosin.
Immunohistochemistry analysis
A single slide through the fovea of each eye was examined by bright field microscopy. The presence or absence of eGFP expressing photoreceptors was noted at the time of observation. The antibody that identified eGFP was tagged with a chromogen that produced a brown precipitate. The antibodies that identified rhodopsin were tagged with a chromogen that produced a red precipitate. The presence or absence of rod photoreceptors that expressed eGFP was noted. Selected representative images were recorded. To assess the eGFP expression, we performed immunohistochemistry. Briefly, sections were washed with xylene three times for 5 min each. Sections were rehydrated by washing in graded series of alcohol, rehydrated in distilled water. Following antigen retrieval, endogenous peroxidase activity was blocked with hydrogen peroxide and nonspecific protein binding with normal goat serum. Sections were incubated with eGFP and rhodopsin antibodies. After washing, sections incubated with ChromoPlex 1 Dual Detection for bond (Leica, Wetzlar, Germany) were used for the visualization of dual histochemical staining, according to the manufacturer's instructions. Sections were counterstained with Hematoxylin, which were then dehydrated with ethanol before mounting. Paraffin-embedded sections were deparaffinized and rehydrated in a graded ethanol. Antigen retrieval was performed, and sections were blocked with protein block serum-free reagent (Dako). Sections were then incubated with mouse anti-eGFP and were incubated overnight at 4°C and then washed, and incubated with anti-mouse Alexa Fluor 488 secondary antibody. The sections were washed in phosphate-buffered saline (PBS), and coverslipped using mounting solution and imaged using microscope.
Immunofluorescence
The remaining slide was labeled for eGFP and 4′,6-diamidino-2-phenylindole (DAPI) (immunofluorescence). The slides were stained for the immunofluorescence detection of eGFP and costained with DAPI to visualize the nuclei.
Photoreceptor cell counting and quantification of transduction
The step-sectioned slide set from each eye was reviewed and the slide that contained the fovea was selected for analysis. The photoreceptor layer was imaged from one border of the subretinal bleb to the other. Morphometric analysis was performed using NIH ImageJ (version 1.49t) to determine the percentage of photoreceptors expressing the eGFP transgene within the borders of the subretinal bleb. Color balance command was used to adjust the brightness and eliminated background fluorescence. The surface area of ONL was measured with measuring command. Then we used thresholding to create a mask of GFP-expressing cell bodies and measured surface area. The percentage of transduced photoreceptors was determined using the formula: Percentage of Transduced Photoreceptors = PR transduced / PR total × 100. The result from each microscope image was plotted. The total percentage was calculated by taking the sum of the areas of the PR transduced and PR total using the above formula.
Mouse studies
All animal studies were performed according to a detailed protocol, approved by Sanofi's Internal Animal Care Use Committee. At postnatal day P45, mice were placed under general anesthesia with intraperitoneal injection of ketamine (90 mg/kg)/xylazine (9 mg/kg). Pupils were dilated with topical application of 1% tropicamide (Akorn Pharmaceuticals, Lake Forest, IL). To perform subretinal injections, aliquots of AAV were thawed on ice, fluorescein (AK-FLUOR, 10%—Akorn Pharmaceuticals) was added to the viral preparation to aid in visibility of AAV delivery. Under visualization with an operating surgical microscope (PSMT5N; World Precision Instruments, Sarasota, FL), an incision was made through the sclera immediately posterior to the nasal limbus using a beveled 30-gauge needle. Through this incision, a 35-gauge blunt needle (NF35BL; World Precision Instruments), which is mounted in to the SilFlex tubing connected to NanoFil syringe, was introduced into the subretinal space. Viral suspension was injected over 20 s into the subretinal space using a programmable microsyringe pump with foot pedal control (UMP3, UltraMicroPump; World Precision Instruments). The needle was held in place for at least 20 s after completion of the injection. Fundus and OCT examination was performed following injection to visualize the location of injection and thus ensure that AAV vectors were injected into the subretinal space. A 3.5% Akten (lidocaine hydrochloride ophthalmic gel; Akorn Pharmaceuticals) Ophthalmic gel was applied to the cornea as a topical anesthetic. A small amount of neomycin/polymyxin B/dexamethasone ophthalmic ointment (Alcon Laboratories, Inc., Fort Worth, TX) was spread over the eye before placing the animal in a 37°C incubator to recover from anesthesia. For intravitreal injections, mice were anesthetized, and eyes were dilated same as described above. One microliter of AAV suspension was injected into the vitreous through sclera, which is 1 mm from the limbus using 35G beveled-tip needle attached to a 10-μL Nanofil syringe through SilFlex tubing (World Precision Instruments). OCT was performed immediately to check for any retinal damage following injections. A small amount of Akten and triple antibiotic ophthalmic ointment was applied on the eye before placing at 37°C.
Quantification of recombinant AAV transduction in mouse retinal lysates
Following euthanasia, eyes were collected, frozen immediately on dry ice, and stored at −80°C for future dissection. All dissection steps were performed under a dissecting microscope using cold instruments, while maintaining the eye in a frozen state during dissection. The anterior segment was removed using a razor, excess tissue was removed from the back of the eye if needed, and the lens were then removed. Retinal lysates were generated by placing the frozen vitreous humor, retina, and eye cup in 200 μL of cell lysis buffer from the GFP Enzyme-Linked Immunosorbent Assay (ELISA) Kit and homogenized at 4°C (Fisher Bead Mill). The eGFP protein was quantified using the GFP ELISA Kit (cat. no. ab171581; Abcam). Total protein levels were quantified using the BCA Protein Assay Kit (Pierce). The levels of eGFP were normalized to total protein. Genome titers were determined by quantitative real time PCR (7500 Real-Time PCR System; Applied Biosystems) using TaqMan Universal Master Mix (Thermo Fisher) with primers specific for the polyadenylation signal. Vector levels were expressed as genomes per microgram protein.
Micron IV in-life imaging
At 4 weeks postinjection, animals were anesthetized using 120 mg/kg ketamine and 6.7 mg/kg xylazine delivered intraperitoneally. OU were dilated using ∼1–3 drops of 1% Mydriacyl (Alcon) administered topically ∼10 min before testing to allow for complete mydriasis. A small drop of goniotaire was placed on the cornea to avoid dehydration. Mice were placed on a platform and their eyes were aligned to a Micron IV fundus camera. Images were taken once the optic nerve head was visible and were focused on the retina. For capturing GFP fluorescence, we used the same gain settings across all samples. Anesthetics were reversed by injecting atipamezole (“Antisedan”) and mice were placed on warm pad until they recovered.
NHP retinal explants
All animal procedures were conducted in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, the Office of Laboratory Animal Welfare, and in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Fresh monkey eyes without known ocular diseases were obtained from Biomere (Worcester, MA), where they were enucleated 15 min after animals were sacrificed. The eyes were placed in Neurobasal medium and transported immediately on ice. Under aseptic conditions, all extraocular connective tissues were removed, and the eyes were disinfected with 70% ethanol followed by washing with PBS. Before starting the experiment, six-well transwell culture plates were preincubated with 2 mL of complete Neurobasal medium (cat. no. 21103049; Thermo Fisher Scientific, Waltham, MA), supplemented with 1% N-2 Supplement (cat. no. 17502048; Thermo Fisher Scientific), 2% B-27 Supplement (cat. no. 17504044; Thermo Fisher Scientific), 1% GlutaMAX Supplement (cat. no. 35050-061; Thermo Fisher Scientific), 0.2 μg/mL of recombinant human β-nerve growth factor (cat. no. 256-GF-100; R&D Systems, Minneapolis, MN), and 0.4 μg/mL recombinant human epidermal growth factor (cat. no. 236-EG-200; R&D Systems) beneath the transwell insert and 0.5 mL in the transwell insert in humidified cell culture incubators at 37°C and 5% carbon dioxide (CO2). An incision was made with an 18G needle around 5mm below the limbus. By using this incision as an entry point for the scissors, the anterior part of the eye, cornea, lens, and vitreous, were removed from each eye leaving posterior eyecups consisting of intact neural retina, choroid, and sclera. Next, three flaps were made to open the eye cup by making three cuts toward the optic nerve head. While the whole eye cup is submerged in the Neurobasal complete medium, an 8 mm biopsy punch was used to cut equatorial, full-thickness posterior segment explants. The retina was subsequently peeled off by gently applying a piece of dry sterile filter paper onto the ganglion cell layer, lifting off the neural retina, and placing the filter paper with attached retina onto the culture insert, photoreceptors facing down, and filter paper was gently removed with fine forceps. After 24 h, the medium was replaced with fresh complete Neurobasal medium and half of the AAV was injected directly beneath each retinal explant, creating a bleb similar to what is formed in vivo when performing therapeutic subretinal injections. An additional half of virus was added to the culture medium that was placed beneath the transwell insert.
Finally, the explants are incubated at 37°C and 5% CO2. AAV vectors at a dose of 1.8 × 1011 vgs was used for each explant. The medium was changed every other day and cultures were maintained for 6 days posttransduction. After 7 days of culture, retinal explants were rinsed in 1 × PBS and fixed for 3 h in 4% PFA. Explants were washed three times with PBS to remove the residual PFA and cryoprotected in graded sucrose 10–30%, after which they were frozen in optimum cutting temperature compound at −80°C. Thirteen micrometer-thick sections were cut using a cryostat (CryoStar NX70 cryostat; Thermo Fisher Scientific) and mounted with VECTASHIELD DAPI (Vector Laboratories, Peterborough, United Kingdom). The native eGFP expression was observed and images were captured using an inverted fluorescence microscope (Axio Observer Z1; Carl Zeiss, Inc., Oberkochen, Germany) using appropriate excitation and detection settings.
Statistical analyses
All data shown in the present article are reported as mean ± standard deviation. GraphPad Prism 7.0 software was used for statistical analyses. The statistical tests used were unpaired Student's t-test for two-group comparisons. p < 0.05 was considered significant. The statistical analyses performed for each dataset is indicated in the figure legends. For all figures, *p < 0.05, **p < 0.01, ***p = 0.001, ****p < 0.0001.
Results
Translational fidelity of AAV2-HBKO in the NHP retina
Previously, we reported on the transduction activity of a novel AAV2 capsid variant in the mouse retina. 20 The AAV2 variant had amino acids R585A and R588A mutated, which are required for AAV2 binding to its receptor, HSPG, to generate a variant known as AAV2-HBKO. In contrast to parental AAV2, the AAV2-HBKO variant displayed low transduction activity following intravitreal delivery to the mouse eye; however, following its subretinal delivery, AAV2-HBKO resulted in significantly greater photoreceptor transduction. Frequently, the AAV transduction profile in mouse is not always predictive of transduction potential in the NHP, thus, the performance of AAV2-HBKO in the NHP retina was evaluated to determine if this novel variant demonstrated a similar improvement in retinal transduction in a primate. The objective of the study was to compare eGFP expression in photoreceptors from an AAV5 and an AAV2-HBKO vector, when administered as a single dose through subretinal injection to male cynomolgus monkeys. Male cynomolgus monkeys (M. fascicularis) were assigned to two groups, and AAV5-eGFP or AAV2-HBKO-eGFP were administered at a dose of 1 × 1012 vg/eye as described in Table 1. The vector preparations were analyzed using a series of optimized assays to confirm quality (Supplementary Fig. S1).
Description of dosing regimen for subretinal delivery of either AAV5-eGFP or AAV2-HBKO-eGFP to nonhuman primates
Both eyes of each animal were dosed. Animals were dosed at a volume of 120 uL/eye.
Dose concentrations were based on the test article as supplied.
AAV, adeno-associated viral; eGFP, enhanced green fluorescent protein; HBKO, heparan sulfate binding knockout; OU, both eyes; vg, vector genome.
Six weeks postvector administration, the animals were euthanized and the eyes were processed for histological sectioning. The section that passed through the fovea was selected for analysis (Fig. 1A). The assessment of eGFP expression in photoreceptors, following administration of AAV5-eGFP or AAV2-HBKO-eGFP, was evaluated by FAF and IHC. Both vectors harbored the same expression cassette, which consisted of a human rhodopsin promoter-driving expression of eGFP. The animals were euthanized ∼6 weeks following vector administration and the eyes processed for paraffin embedding and histological sectioning. The slides were stained for the immunofluorescence detection of eGFP (Fig. 1B), and images were sequentially collected from one edge of the subretinal bleb to the other. Expression of the eGFP transgene appeared to be uniform across the subretinal bleb. In areas where there were fewer rod photoreceptors (adjacent to and within the fovea), the transgene expression was diminished, confirming the fidelity of the human rhodopsin promoter in restricting expression to the rod photoreceptors. Previously, AAV5 was shown to transduce cone photoreceptors when transgene expression was under the control of the rod and cone-specific human rhodopsin kinase promoter. 45 Quantification of the percentage of photoreceptor transduction under the bleb revealed that AAV5 and AAV2-HBKO demonstrate equal photoreceptor transduction activity in the NHP retina (an average of 61% of the photoreceptors were transduced by both AAV5 and AAV2-HBKO) (Fig. 1C).
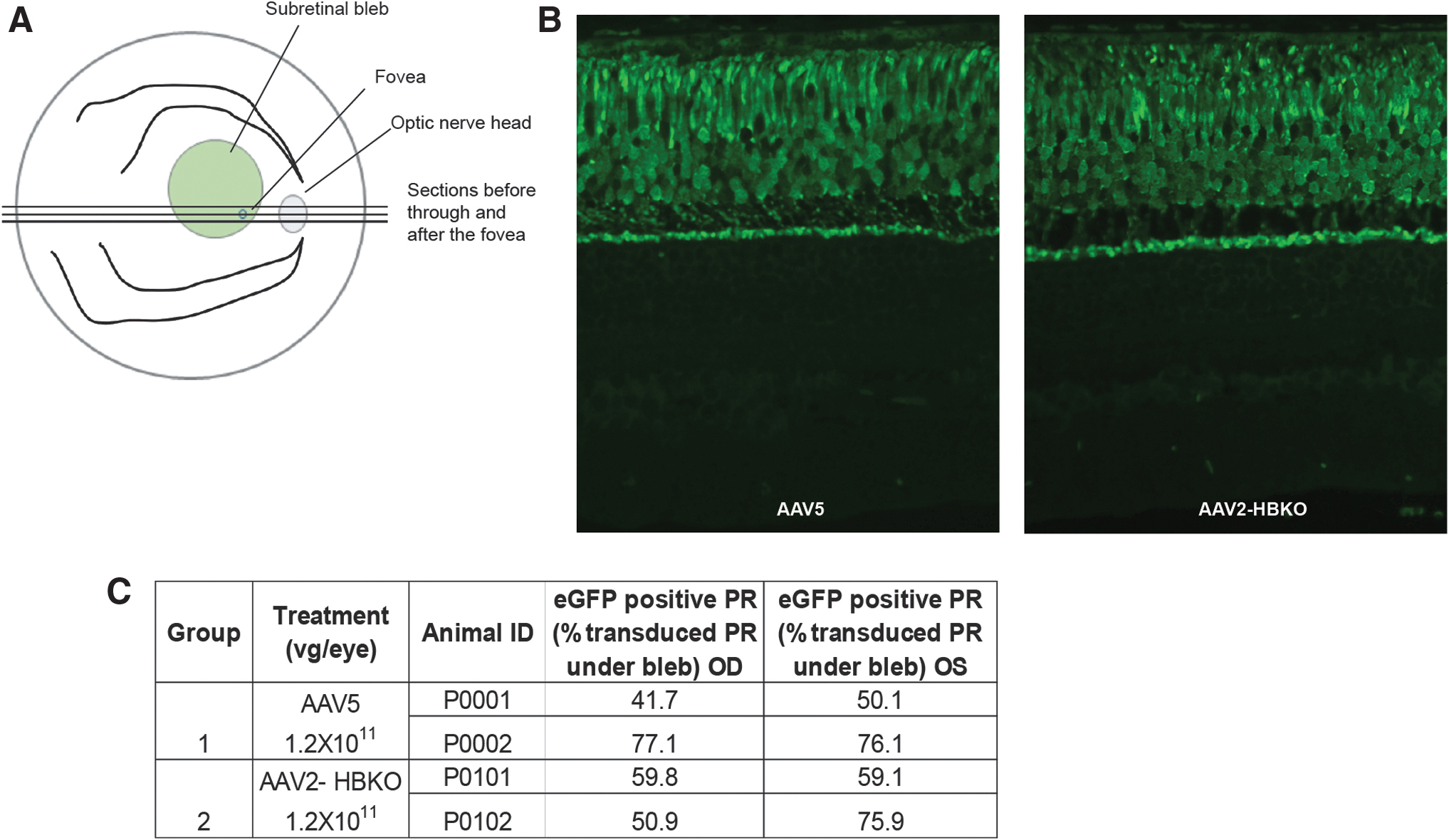
AAV2-HBKO- and AAV5-mediated eGFP expression in the NHP retina 6 weeks postsubretinal injection.
Additionally, the expression of eGFP was monitored using spectral domain optical coherence tomography with autofluorescence imaging capabilities; eGFP was observed 2 and 4 weeks following vector administration. The intensity of eGFP signal increased with time and was confined to the retina, within the margin of the subretinal bleb in the eyes treated with AAV5-eGFP (Fig. 2A), while in contrast, expression of eGFP from the eyes treated with AAV2-HBKO-eGFP extended well past the margin of the subretinal bleb (Fig. 2B). Separate slides were costained with an anti-eGFP antibody and anti-rhodopsin antibody for the immunohistochemical detection of eGFP as well as rhodopsin; this analysis was done to confirm that only rod photoreceptors were transduced. Figure 3 shows that histological survey of the subretinal bleb. Immunohistochemistry for rhodopsin (red) and eGFP (brown), in paraffin-embedded tissue, reveals that the transduction with the AAV5 vector does not spread from the margin of the subretinal bleb, the transition at the margin is abrupt (Fig. 3A). In contrast, AAV2-HBKO-eGFP transduction spreads beyond the bleb and tapers off in areas not lifted by the injection process (Fig. 3B; Supplementary Fig. S2). Colocalization of eGFP and rhodopsin indicate that the transduced cells are rod photoreceptors. The overall architecture of the retina is preserved, although there are changes to the RPE, including hypertrophy, displacement of pigment, and displacement of cells. These signals of intolerability are likely due to expression of the foreign eGFP reporter protein.
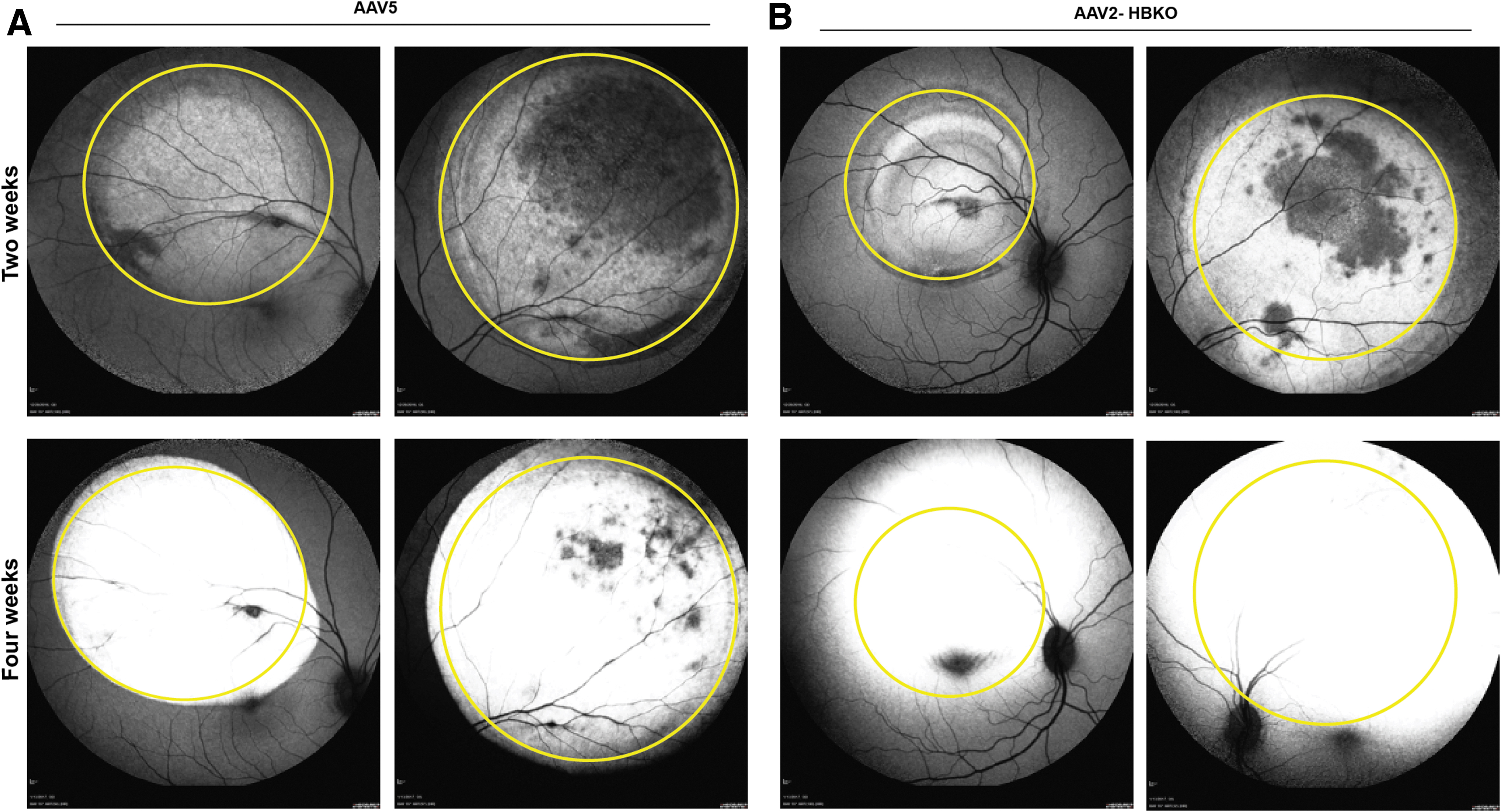
AAV-mediated eGFP expression in NHP eyes by FAF. FAF showing the eGFP fluorescence in the bleb area (yellow circle) treated with AAV5-eGFP
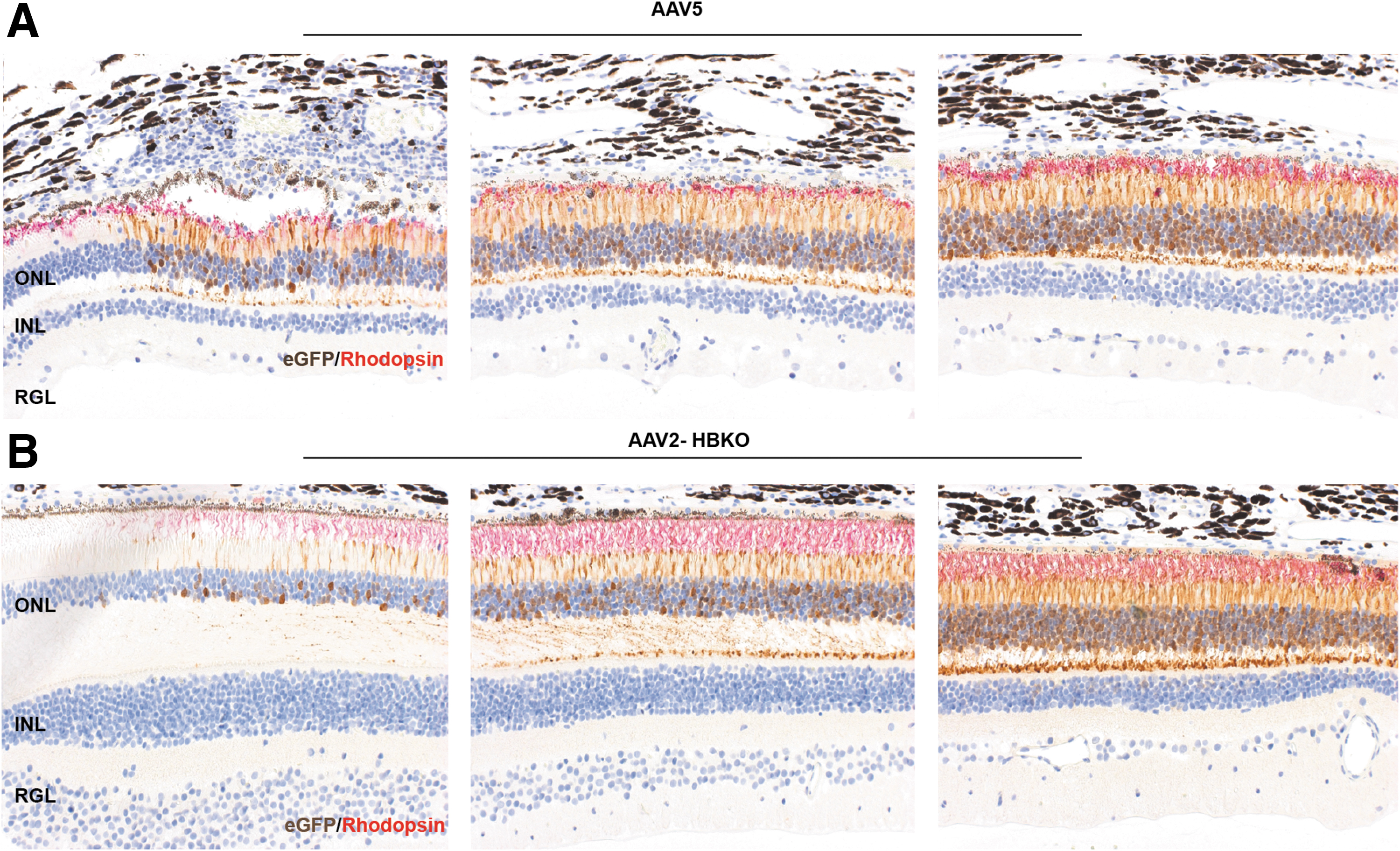
Subretinal injections in NHP show superior transduction ability of AAV2-HBKO over the AAV5 in retina. Relative transduction efficiencies of AAV5 and AAV2-HBKO in rod photoreceptors were compared following subretinal injection. Paraffin-embedded retinal sections were immunolabeled with anti-GFP antibody and anti-rhodopsin antibody for the detection of eGFP (brown) as well as rhodopsin (red). Immunohistochemistry analysis around the bleb region showed that the transduction of the AAV5 vector does not appear to spread from the margin of the subretinal bleb, the transition at the margin is abrupt
Evaluation of the transduction potential of AAV5 arginine variants in the retina
The AAV2-HBKO variant revealed the importance of arginines, and by extension surface charge, on transduction activity in the retina. The effect of altering surface arginines was further explored with another capsid, AAV5, a serotype that has a high affinity for photoreceptors when delivered subretinally. 43 AAV5 variants AAV5G474R, AAV5N564R, and AAV5N573R were generated and their tropism in the mouse retina, following intravitreal and subretinal delivery, was evaluated. The surface map of AAV2 was aligned to that of AAV5, 14 and amino acids in the AAV5 capsid were selected for mutation, to generate arginine-rich AAV5 variants. Of note, the AAV5 variants produced at yields that were comparable or twofold less than yields achieved with parental AAV5, but variants retained the same capsid protein ratio as parental AAV5 (Supplementary Fig. S3). First, subretinal delivery of the AAV5 arginine variants was evaluated in the wild-type mouse retina and their transduction activity compared with that of the parental AAV5 capsid. Figure 4A and C shows the performance of AAV5, AAV5G474R, AAV5N564R, and AAV5N573R following subretinal delivery of 1 × 109 vgs of each vector harboring the identical CBA-eGFP expression cassette. Notably, there was no apparent difference in the ability of the AAV5 arginine variants to transduce the retina following subretinal delivery, when compared with parental AAV5 as evidenced by in-life fundoscopic (Fig. 4A) and fluorescence microscopic analysis (Fig. 4C). Analysis of native eGFP fluorescence in transduced retina revealed that the AAV5G474R, AAV5N564R, and AAV5N573R variants transduced the ONL and RPE cells, to the same level as parental AAV5-eGFP (Fig. 4C). Additionally, the AAV5 arginine variants were further evaluated following intravitreal delivery to the mouse retina, at the same vector dose of 1 × 109 vgs/eye. Following intravitreal delivery, the AAV5 variants demonstrated minimal transduction of the retina compared with AAV2 revealed by Micron IV in-life fundoscopic analysis (Fig. 4B). The eGFP fluorescence analysis of the transduced retinae (Fig. 4D), confirmed that all AAV5 arginine variants, AAV5G474R, AAV5N564R, and AAV5N573R, had acquired a novel tropism for corneal endothelial cells, and in contrast, parental AAV5 showed no transduction activity with intravitreal delivery (Fig. 4D).
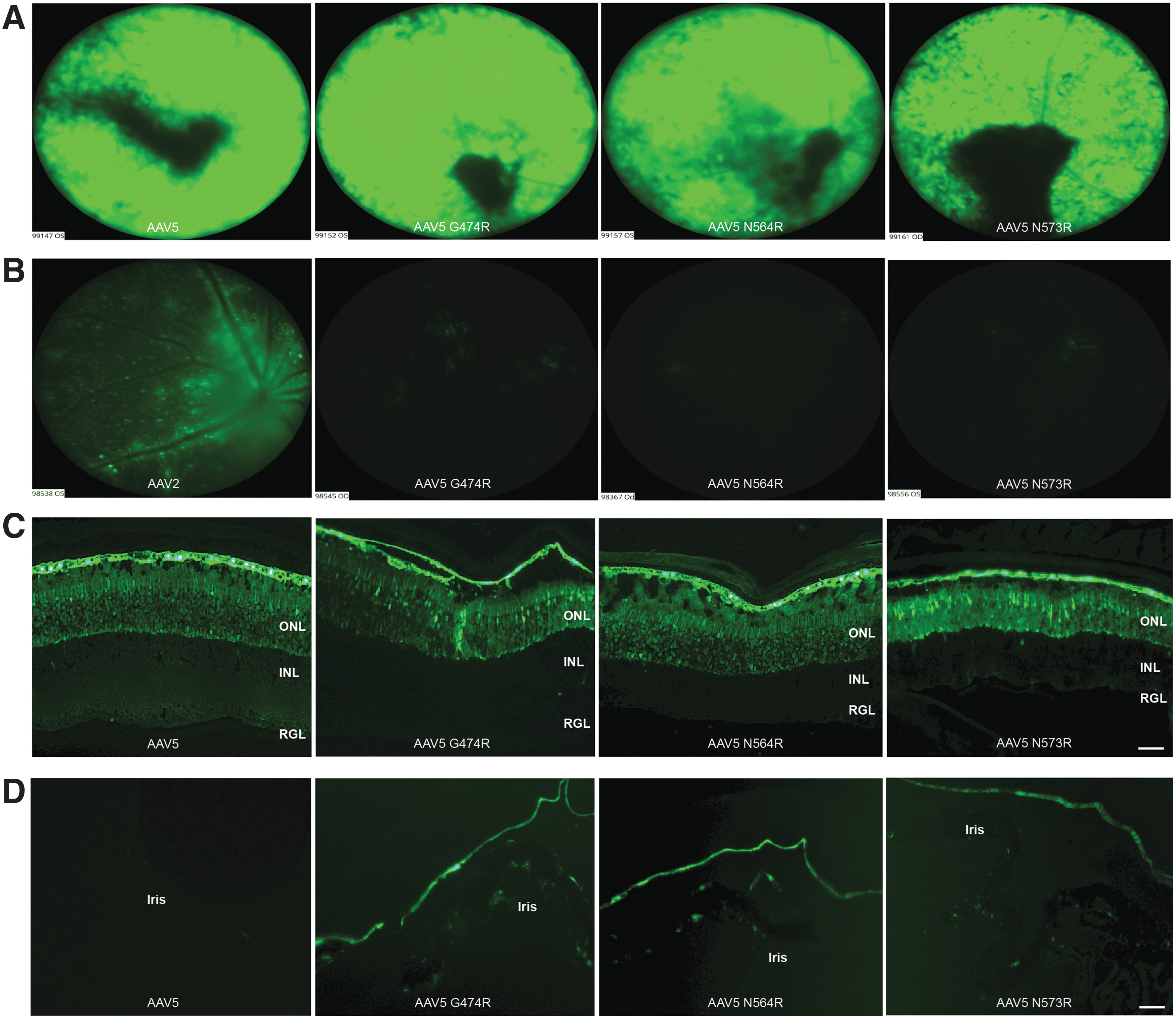
Comparison of retinal and corneal transduction efficiency between AAV5 and AAV5 arginine variants.
Probing the role of acetylation on AAV transduction
The AAV2-HBKO and AAV5 arginine variants were generated using a rational design approach, using knowledge of receptor binding and surface charge, respectively. Next, additional variants were generated using knowledge we gained from LC/MS analysis of the AAV capsid, which identified novel PTMs, including N-terminal acetylation on VP1 and VP3 capsid proteins. 29 To further explore this attribute, a series of AAV5 acetylation variants was generated to elucidate the role of this significant PTM on AAV5 biology in the retina. The mutations that were introduced into the AAV5 capsid sequence are described in Fig. 5A; this included changing the amino acid, after initiating methionine, from one that has a high frequency of acetylation (alanine or serine) to an amino acid with a low frequency of acetylation (glycine or proline). These changes were performed separately on the AAV5 VP1 and VP3 capsid proteins; in addition combined changes were made in both AAV capsid proteins. The AAV capsid protein VP2, showed no evidence of acetylation in previous studies, 29 thus the VP2 sequence remained unchanged from the parental sequence. AAV5 acetylation variants were analyzed by LC/MS to confirm acetylation status and the results are shown in Fig. 5A. Irrespective of the amino acid change, that is, glycine or proline, all AAV5 acetylation mutants were confirmed to have reduced acetylation. LC/MS analysis confirmed the correct molecular weights for each AAV5 capsid protein mutants. No acetylation was observed in the S–P mutants while 10% acetylation was observed in S–G mutants. Notably, the acetylation mutants showed equivalent packaging efficiencies, compared with wild-type AAV5, suggesting that the novel amino acid changes had not adversely affected AAV vector production, or capsid protein ratios (Fig. 5B).
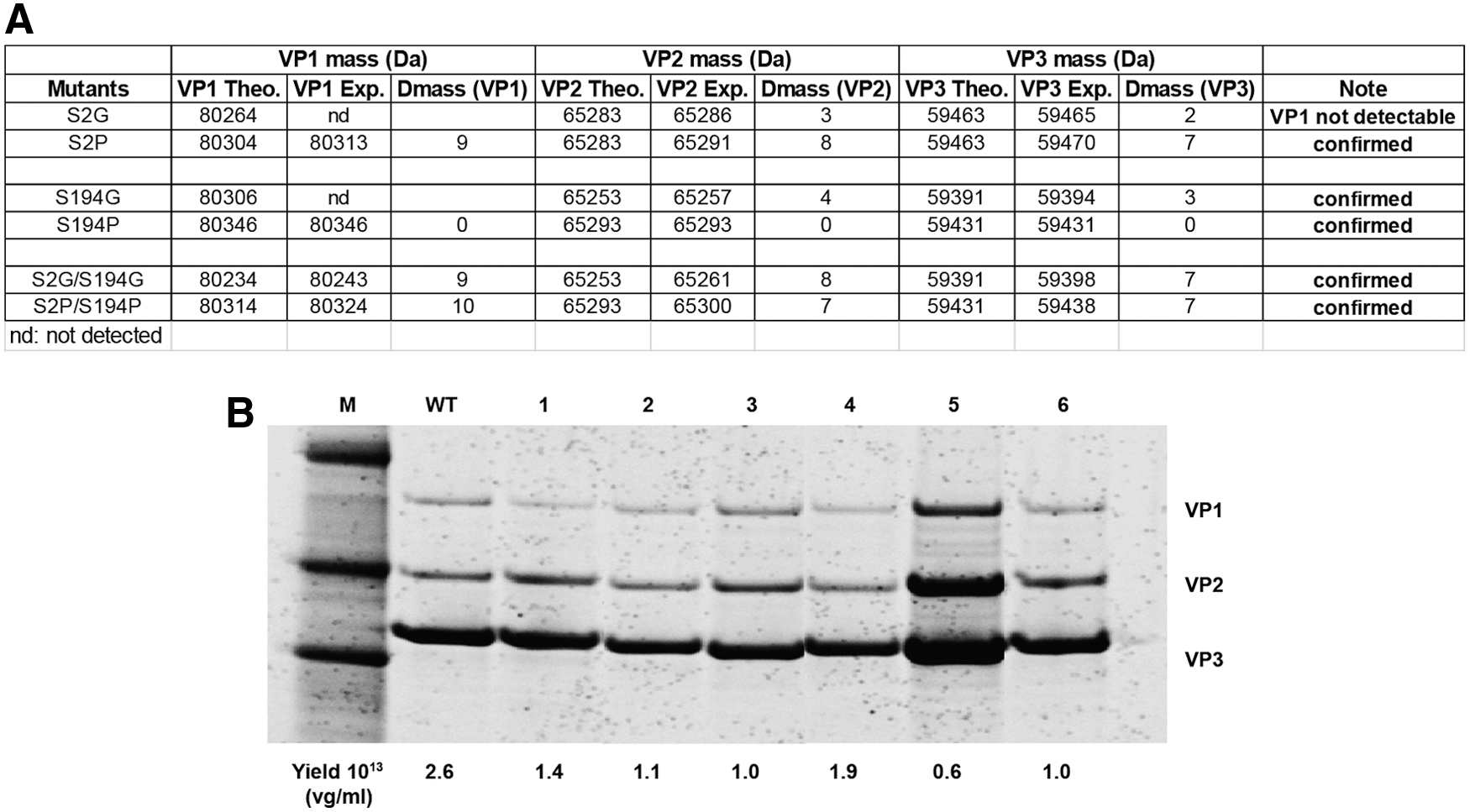
Characterization of AAV5 acetylation mutants by LC/MS and SDS-PAGE maintain WT levels of capsid protein expression and vector yields.
Next, the transduction efficiencies of the AAV5 acetylation variants were compared with wild-type AAV5, in vivo, following subretinal injection to the mouse. Wild-type mice were injected with 1 × 109 vgs AAV5-CBA-eGFP or one of each of the AAV5 acetylation mutants, harboring the same CBA-eGFP expression cassette. As previously shown (Fig. 4A), subretinal injection of AAV5-eGFP resulted in robust eGFP expression in the outer retina (Fig. 6A). In contrast, the acetylation mutants AAV5S2G, AAV5S2P, AAV5S194P, AAV5S2G/S194G, and AAV5S2P/S194P showed reduced levels of eGFP expression in the retina, as evidenced by eGFP fluorescence of injected retinas (Fig. 6A) and eGFP protein levels (ELISA) (Fig. 6B). Notably, the acetylation mutant AAV5S194G-eGFP, showed a dramatic increase in eGFP expression in photoreceptor cells compared with parental AAV5-eGFP (Fig. 6A). This did not translate to a statistically different increase in eGFP protein expression, as measured by ELISA (Fig. 6B), so the performance of the AAV5S194G variant at different dose levels was explored (Fig. 7). Analysis of the vg copy number in transduced retina, revealed little difference between groups for the acetylation variants compared with parental AAV5 eGFP vector in treated retinas (Fig. 6C), at a vector dose of 1 × 109 vgs.
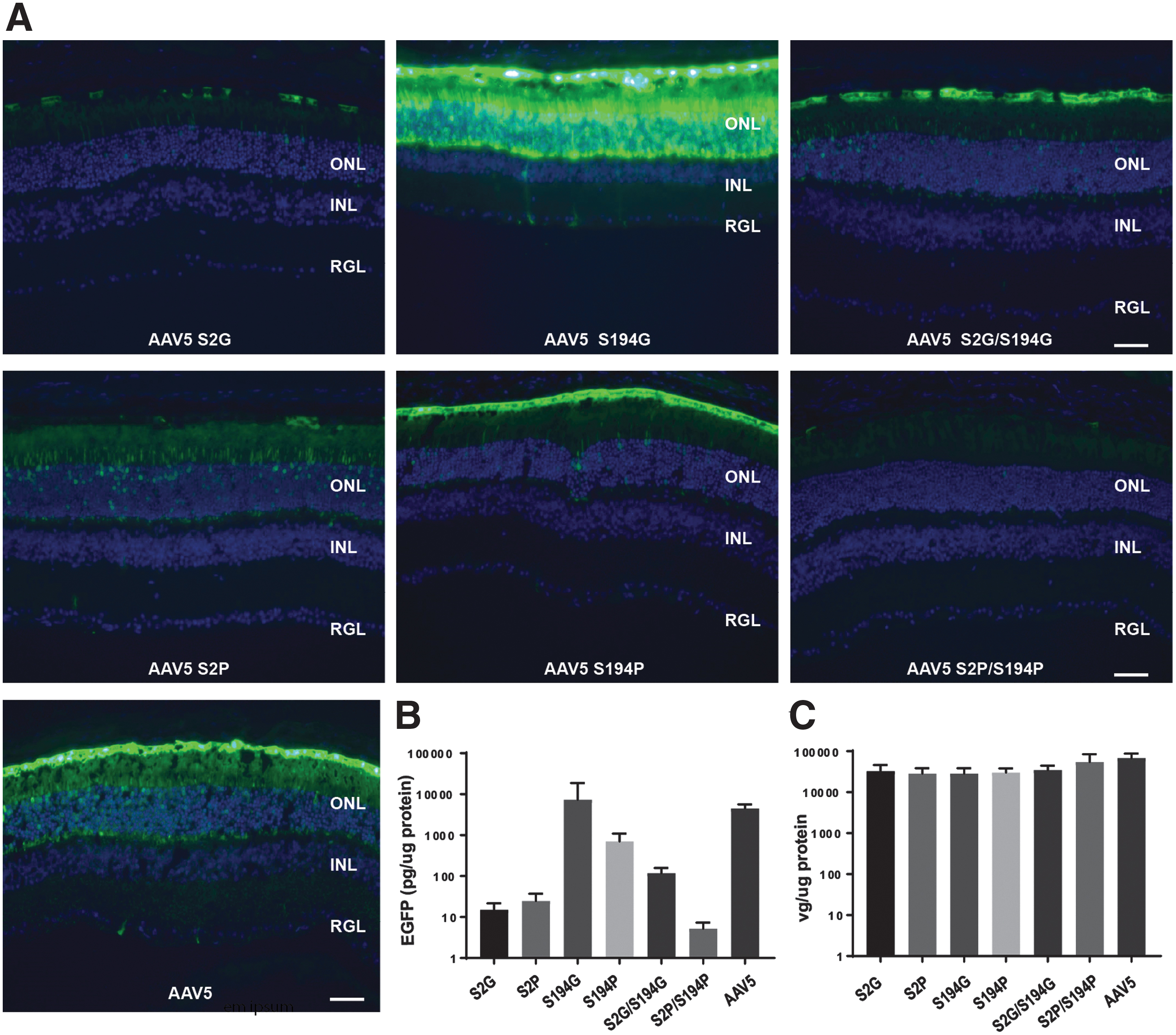
Effect of AAV5 acetylation on photoreceptor transduction.

Comparison of eGFP expression after subretinal delivery of AAV5 acetylation variants in a dose-dependent manner.
The transduction properties of the acetylation mutants, AAV5-S194G-eGFP and AAV5-S194P-eGFP, were further evaluated in a dose-response study and their performance compared with AAV5-eGFP. Increasing doses, 1 × 108–1 × 109 vgs of AAV5-S194G-eGFP, AAV5-S194P-eGFP, or AAV5-eGFP, were administered subretinally to a wild-type mouse. At a dose level of 1 × 109 vgs, the AAV5 vector showed robust transduction of photoreceptor cells (Fig. 7A); in contrast the acetylated mutant AAV5S194P, as seen in the previous experiment (Fig. 6), showed a dramatic reduction in expression compared with AAV5, whereas the AAV5-S194G variant, demonstrated a significant increase in photoreceptor cell transduction (Fig. 7A). At lower doses of vector, 5 × 108 and 1 × 108 vgs, there was a marked decrease in eGFP expression in the retinas that had received the AAV5-eGFP vector, whereas the retinas transduced with AAV5-S194G-eGFP had robust eGFP expression in photoreceptors; even at doses as low as 1 × 108 vgs. Notably, AAV5S194P-eGFP showed little transduction of photoreceptors at all doses evaluated (Fig. 7A). The eGFP protein levels were confirmed to be statistically higher, by ELISA, in retinas treated with the AAV5S194G-eGFP compared with AAV5-eGFP at, at a dose of either 5 × 108 or 1 × 108 vgs. The difference in expression between the AAV5 acetylation variants and the wild-type AAV5 did not reach statistical significance at a dose of 1 × 109 vgs, presumably because of saturation of eGFP expression at this higher vector dose (Fig. 7B), and as seen in the previous experiment, study (Fig. 6B). Analysis of vg copy number revealed a dose-response for both vectors, retinas transduced with the AAV5S194G-eGFP acetylation variant, at a dose of 5 × 108 or 1 × 108 vgs, had higher vg copies, compared with retinas treated with AAV5-eGFP at a similar dose (Fig. 7C).
Evaluation of AAV5 capsid variants in NHP retinal explants
We evaluated the use of an ex vivo NHP organotypic explant system to test the transduction efficiency and the tropism of our novel variants and to assess translational fidelity of AAV variants between species. To that end, retinal explants from NHPs were established and the transduction efficiency of the acetylation variants AAV5S194G & AAV5S194P was evaluated. Seven days after plating, the cultured retinas retained normal architecture, including intact rod and cone photoreceptor inner and outer segments, a full ONL, and outer to inner retinal connectivity (Fig. 8A). The transduction efficiency of the AAV5 and AAV5 acetylation variants in the retinal explants mimicked the transduction efficiency observed in the mouse retina (Fig. 6A), and for unmodified AAV5, mimicked the transduction activity also seen in the NHP retina (Fig. 3A). Confirming what was seen in the mouse retina (Fig. 7), the AAV5S194G variant showed superior transduction efficiency in the NHP ONL compared with the AAV5 parental vector (Fig. 8A, C). In contrast, compared with the parental AAV5, the AAV5S194P variant revealed decreased transduction efficiency in the NHP retinal explant, similar to its performance in the mouse retina (Fig. 7). The authenticity of the organotypic culture was confirmed by the performance of the AAV5-eGFP parental vector, which demonstrated robust transduction of the ONL (Fig. 8A, C), similar to the transduction performance seen with subretinal delivery of AAV5-eGFP vector to the NHP retina (Fig. 3A). Finally, evaluation of the acetylation mutants in the NHP retinal explant model revealed an additional benefit to the AAV5S194G variant over unmodified AAV5. This capsid variant selectively transduces photoreceptor cells, with little transduction observed in other cell types (Fig. 8A, C). The eGFP protein levels in the NHP retinal explant cultures were quantified by ELISA (Fig. 8B). There was no difference in eGFP expression between the AAV5 and AAV5S194G capsid. The eGFP ELISA quantifies the total GFP protein in all cell types in the retinal explant culture and as a result does not highlight the differences in tropism between these two capsids observed at the cellular level in the fluorescence images (Fig. 8C). There was no detectable eGFP in the NHP retinal explants infected with the AAV5S194P vector (Fig. 8B).
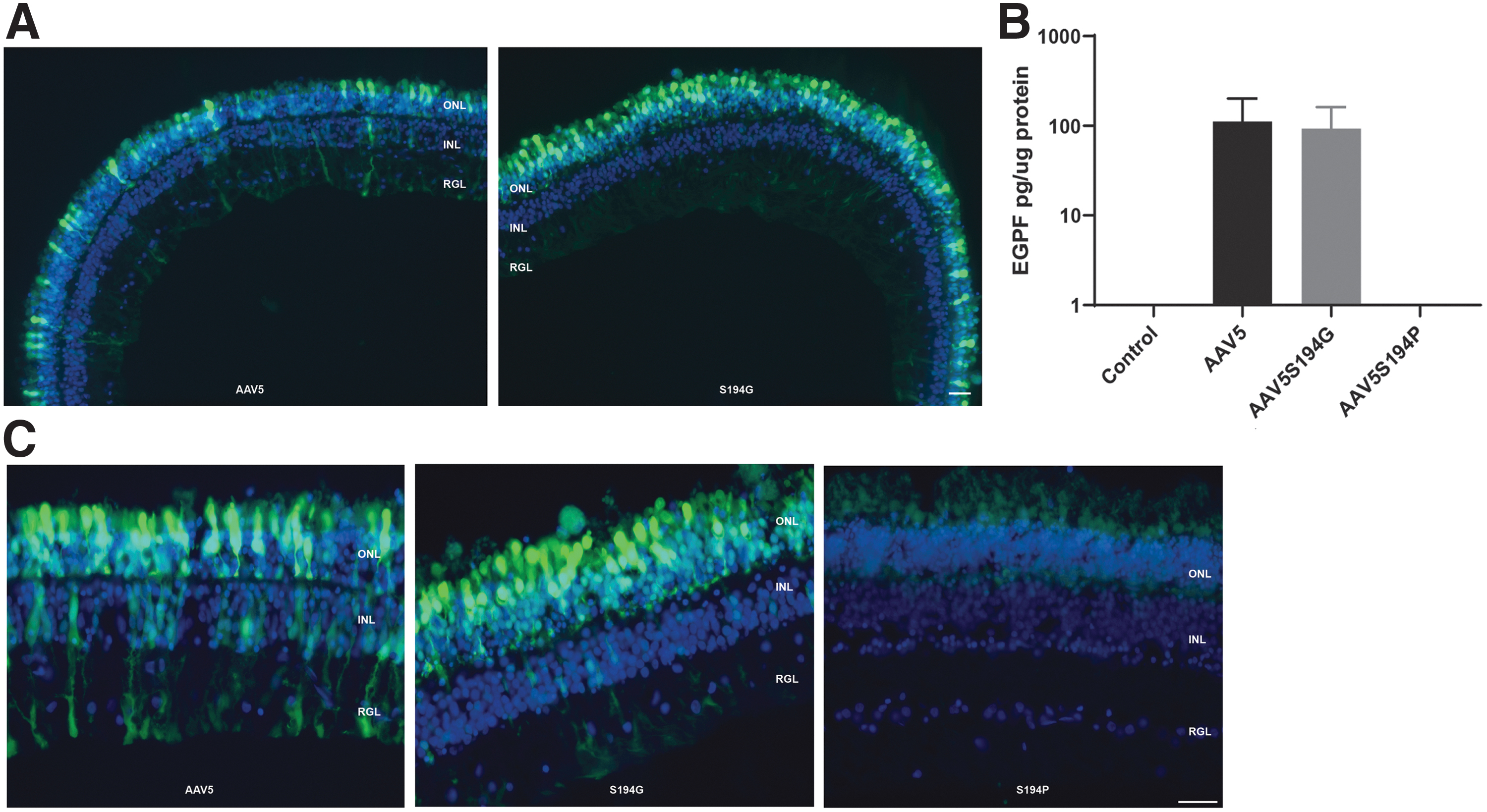
Comparison of native eGFP fluorescence following ex vivo administration of AAV5 and AAV5 acetylation variants in NHP retinal tissue.
Evaluating the role of deamidation on AAV2 transduction in the retina
Analysis of quality attributes of AAV vectors generated using the PCL production platform, including PTMs on the AAV capsid proteins, revealed several interesting observations. Specifically, the PCL process, in the context of AAV2 vectors, resulted in AAV vector preparations that consistently had a protein running below VP1, as compared with a similar AAV2 vector produced through the triple transfection production platform (Fig. 9B). LC/MS analysis revealed the protein to be a truncated form of VP1 protein (tVP1), lacking the first 34 amino acids, with acetylated A35 confirmed to be the N-terminal amino acid (article in preparation). Deamidation, in which side chain amide groups, typically asparagine, are converted to aspartic acid, is a common PTM, which often signals a protein for degradation. We hypothesized that tVP1 was a result of deamidation at a neighboring asparagine, N57, resulting in the proteolytic cleavage of VP1 at A35 to generate tVP1 (Fig. 9A). LC/MS analysis confirmed the deamidation status of N57, in the context of the AAV2 PCL-derived vector, to be higher at 18.4%, compared with 6.7%, for a comparable AAV2 vector generated by triple transfection (Fig. 9D). Notably, there was no measured difference in deamidation at other potential deamidation sites in the AAV2 capsid sequence, including N511G or N717G. Interestingly, the infectivity of the AAV2 PCL vector trended lower in an analytical in vitro assay (Fig. 9D), thus deamidation mutants were generated to further explore the role of deamidation on AAV2 infectivity in the retina. To that end, the N57 deamidation site was mutated to aspartic acid, N57D, to generate a capsid that would have complete deamidation at that residue. Additionally, variant G58D was generated to control for the effects of introducing an aspartic acid in this region of the AAV2 capsid sequence. AAV2N57D-eGFP and AAV2G58D-eGFP variants were generated and produced using the triple transfection production method. The AAV2 variants had similar packaging efficiencies and capsid protein profiles (Fig. 9C, E), as the parental AAV2 capsid. LC/MS analysis of the deamidation variants confirmed that the AAV2G58D-eGFP was 1.1% deamidated (to become aspartic acid), wild-type AAV2-eGFP was 5.7% deamidated, whereas the AAV2N57D-eGFP was 100% deamidated (Fig. 9E).

Altering deamidation levels within the PLA2 domain of AAV2 VP1 does not impact capsid protein expression or vector yields but impacts potency.
The deamidation variants were next evaluated in vivo to assess the role of this PTM on AAV2 transduction activity. Intravitreal delivery of AAV2-eGFP vector to wild-type mice resulted in significant transduction of retinal ganglion cells (Fig. 10C). A similar result was observed with the AAV2G58D-eGFP variant, in contrast, intravitreal delivery of AAV2N57D-eGFP resulted in poor retinal transduction as measured by eGFP fluorescence (Fig. 10C) or ELISA (Fig. 10A). The vg copies of retinal tissue transduced with the AAV2N57D-eGFP variant trended lower than the levels measured with unmodified AAV2-eGFP or AAV2G58D-eGFP, suggesting a possible correlation to the reduced transgene expression measured with the AAV2N57D-eGFP variant (Fig. 10B).

Effect of AAV2 capsid deamidation on retinal transduction.
Discussion
We have shown, using a rational design approach and knowledge of AAV receptor binding, surface charge, and capsid protein PTMs, that AAV variants with novel tropism and transduction for retina and cornea can be generated (Table 2). Previously, we reported on the improved transduction performance of our AAV2-HBKO variant in the context of the mouse retina and central nervous system (CNS). 20 The AAV2-HBKO variant has key surface arginines mutated in regions of the capsid that facilitate the binding of AAV2 to its cognate receptor, HSPG. This heparan-binding knockout variant demonstrated novel transduction patterns in the mouse CNS and retina, 20 and in follow-up studies, the novel CNS transduction properties were confirmed to translate to the NHP brain. 46 However, the translatability of the transduction performance of the AAV2-HBKO variant from mouse to NHP retina was not confirmed. In this study, we show that the AAV2-HBKO variant exhibited high levels of transduction and transgene expression to the outer retina, when the identical vector was delivered subretinally to either mice or NHP. Previously, subretinal delivery of AAV2 vectors to the NHP retina demonstrated limited photoreceptor cell transduction, with most of the transgene expression restricted to the RPE; this was demonstrated in the context of a ubiquitous CBA promoter-driving expression of a human RPE65 transgene. 47 Thus, the ablation of HS binding of AAV2-based vectors, as shown here, is an effective strategy for enhancing biodistribution to primate photoreceptors. Further restriction of transgene expression to rod photoreceptors was achieved with the use of a rhodopsin promoter. Rod photoreceptor transduction was observed across the area of the subretinal bleb, with transduction dramatically decreasing in the cone-rich area of the fovea. In the NHP retinal study, the transduction profile of the AAV2-HBKO variant was compared with that of an AAV5 vector harboring the identical eGFP expression cassette, and although the extent of rod photoreceptor transduction was equivalent for both vectors, notably in the NHP eye, the AAV2-HBKO vector demonstrated the ability to spread past the border of the subretinal bleb. This was confirmed by both in vivo imaging and postmortem histology.
Summary of adeno-associated viral capsid variants generated for these studies
IVIT, intravitreal delivery; VP, viral protein; SR, subretinal.
It can be hypothesized that the improved transduction and spread observed with the AAV2-HBKO variant, is partially because this variant is not sequestered by HSPG and can traffic beyond the borders of the bleb. One caveat to the interpretation of the transduction activity of the AAV2-HBKO in the NHP retina, is that the n value (4) is limited, and a more comprehensive dose-ranging study with a larger n value is needed to confirm these preliminary observations. Altogether, these results establish that the ablation of HS binding of AAV2-based vectors may be an effective strategy for enhancing biodistribution to photoreceptors and promoting vector spread in the primate retina, a valuable attribute when treating inherited retinal diseases that require a more global correction of the retina.
The AAV2-HBKO variant revealed the importance of arginines, and by extension capsid surface charge, on transduction activity in the retina. The AAV2-HBKO vector has two positively charged arginine residues (R585A and R588A) substituted for two hydrophobic alanine residues on the surface of the AAV2 capsid. These substitutions significantly altered the tropism of AAV2 in the retina. In contrast to parental AAV2, AAV2-HBKO lacks tropism for inner retina following an intravitreal administration, 20 whereas it acquires a novel tropism for photoreceptors following subretinal delivery.
One hypothesis to explain the different tropism AAV2-HBKO demonstrated with intravitreal delivery is that, AAV2 surface arginines are important for navigating the proteoglycan-rich region of the inner limiting membrane, a barrier separating the vitreous from the neural retina. 35 We questioned if adding arginines could impact the tropism and transduction activity of a different capsid, AAV5, which has relatively few surface arginines compared with AAV2. It is known that while subretinally delivered AAV5 demonstrates superior photoreceptor transduction (current study and Ref. 45 ), its transduction activity is limited following intravitreal surgery. 48 Thus, the effect of altering surface charge on the AAV5 capsid was explored to determine if improved transduction activity in the retina could be achieved, both in the context of intravitreal and subretinal delivery. Intravitreal delivery of AAV5 arginine variants, AAV5G474R, AAV5N564R, and AAV5N573R, to the mouse resulted in limited retinal transduction, comparable to the pattern observed following intravitreal delivery of the AAV5 parental capsid. However, in contrast to the AAV5 parental capsid, all AAV5 arginine variants acquired a novel tropism for and improved transduction activity in corneal endothelial cells. This is the first report of an AAV vector that can transduce corneal endothelial cells following a noninvasive procedure such as intravitreal surgery. Serotypes, such as AAVAnc80L655 and AAV8G9, 49 first-generation serotypes, AAV1–AAV9, 50 and AAV2 tyrosine variants, 6 have been reported to transduce corneal endothelial cells in rodents, but only following intracameral delivery, a surgical procedure that requires puncturing the cornea, with the concomitant risk of damage to the anterior chamber of the eye. The enhanced AAV5 vectors described in this study will need to be further tested to determine if the gains in transduction from the vitreous, as seen in mouse, translate to similar improvements in the primate eye. If translatability is confirmed, these novel AAV5 arginine variants show promise for the treatment of inherited corneal dystrophies or systemic diseases affecting the cornea. 50
The AAV2-HBKO and AAV5 arginine variants were generated by a rational design approach using knowledge of receptor binding and surface charge, respectively. Next, additional variants were generated using knowledge gained from LC/MS analysis of the AAV capsid, where PTMs, including acetylation on AAV5VP1 (Ser2) and VP3 (Ser194) capsid proteins were revealed. 29 To understand the general function of N-terminal acetylation on AAV capsid proteins, we examined the role of N-terminal acetylation on AAV5 transduction in the retina. Proteins in which the second amino acid is either a proline or a glycine are the most unfavorable proteins for N-terminal acetylation. 51 Thus, a change in serine at position 2 in the AAV5 VP1 protein, to glycine or proline (variants AAV5S2G and AAV5S2P, respectively), resulted in reduced acetylation on VP1, as verified by LC/MS, and a concomitant reduction in mouse retinal transduction. This reduced transduction was evident when the N-terminal acetylation of VP1 was decreased in isolation, or in combination with reduced N-terminal acetylation in the AAV5 VP3 protein, that is, variants AAV5S2G/S194G and AAV5S2P/S194P. Notably, when VP1 N-terminal acetylation was preserved and VP3 N-terminal acetylation reduced, by changing serine 194 to a glycine (variant AAV5S194G), there was a significant improvement in retinal transduction compared with parental AAV5; the improved transduction was evident in both the mouse retina and NHP retinal explants. These data underscore the importance of VP1 N-terminal acetylation in AAV5 transduction and suggests that this PTM maybe a requirement for AAV endosomal release, a process that requires a pH conformational change in the VP1 N-terminus, before trafficking to the nucleus. 27
N-terminal acetylation has been shown to influence the localization of other proteins. Arl3p, an “Arf-like” (Arl) GTPase, which is crucial for the organization of membrane traffic, requires its Nα-acetyl group for correct targeting to the Golgi membrane. 52 In contrast, N-terminal acetylation on VP3 is redundant, as evidenced by the improved transduction activity of the VP3 N-terminal acetylation variant AAV5S194G. However, when VP3 N-terminal acetylation was reduced by substituting S194 with the bulkier proline, the resulting variant, AAV5S194P, demonstrated limited transduction in the mouse retina and NHP retinal explant model. The reason for the difference in transduction activity between AAV5S194G and AAV5S194P variants, maybe best explained by the difference in size of the substituted residues, proline versus glycine, which in turn influences the local flexibility of the peptide backbone. Glycine is a hydrophilic amino acid that provides significant flexibility, in contrast proline is nonpolar and likely provides more rigidity to the capsid; this rigidity may impede the process of AAV5 capsid uncoating, having a negative impact on AAV5 transduction in the retina. Altogether, these data underscore the importance of maintaining the N-terminal acetylation on VP1 for AAV5 transduction activity, additionally, N-terminal acetylation on the AAV5 VP3 protein is not necessary for transduction, but rather the flexibility of the amino acid at the second position in the VP3 N-terminus influences transduction activity by controlling capsid rigidity and likely capsid uncoating, a critical step in the AAV transduction process. This discovery highlights an area that might be further explored to improve transduction activity of other AAV serotypes.
Deamidation, in which side chain amide groups, typically asparagine, are converted to aspartic acid, is a common PTM that can signal a protein for proteolysis. 53 A common attribute with AAV2 vectors generated using the PCL process, 41,43 when compared with the same vector made by transient transfection, is the presence of a tVP1, and analytically, a trend for decreased in vitro potency. We hypothesized that the truncated VP1 species may have resulted from proteolysis signaled by deamidation on 57NG58 (Fig. 9A). LC/MS analysis confirmed increased 57NG58 deamidation for PCL-derived vectors, and the effects of genetically forcing deamidation on AAV transduction activity, was explored. Our AAV2N57D variant showed total loss of in vivo transduction activity, in both mouse and NHP retina. Additionally, changing N57 to either a lysine (N57K) or a glutamine (N57Q), ablated in vitro transduction (Supplementary Fig. S4). Surprisingly, mutating the neighboring glycine to an aspartate (variant G58D), had no effect on AAV2 transduction activity, suggesting that the G58 residue is tolerant of change, in the context of AAV2 function in the mouse eye. Altogether, these data underscore the importance of the N57 residue in AAV2 transduction activity, and suggest that changes in N57, either by deamidation or directed mutagenesis, result in functional loss.
VP1/VP2 protein N-terminal sequences harbor infection-relevant functional domains, such as a PLA2 catalytic subunit. 25,26,28 Importantly, the N57 residue lies in the calcium-binding loop of the PLA2 domain, suggesting a possible role for N57 in binding this essential cofactor of enzyme activity and viral endosomal escape, an obligatory step in AAV2 infection. 26 Alternatively, maintaining N57 at the VP1 N-termini is critical for the correct unfolding and surface exposure of the PLA2 domain. 10,27 Data to support the hypothesis that the defect in the AAV2N57D mutant is likely endosomal escape or uncoating are supported by the fact that there are measurable vgs in the cell, but no detectable transgene expression (Fig. 9). In summary, our data suggest that strategies to rationally design AAV2 vectors for enhanced transduction in the retina should avoid any changes in the N57 VP1 N-terminal residue, whereas changes in neighboring residues, that is, G58, are well tolerated, and suggest that a +1 site mutagenesis strategy may be used to stabilize 57NG58 asparagine from inadvertent deamidation. Interestingly, during our studies, Giles et al., 54 reported on NG deamidation—induced functional loss for AAV8, and evaluated strategies to stabilize amides to improve vector performance. The authors also highlighted the importance of controlling for deamidation during AAV manufacturing and recommended quantitating the level of deamidation as a critical quality attribute for clinical AAV vector lot release. 54
The current study describes the optimization of an NHP retinal organotypic explant system that facilitated rapid assessment of the transduction activity of our novel variants in a retina from a species other than mice. The lack of translation from mouse to NHP brain has been reported for the AAVPHP.B variant, 55 so with this precedent, we wanted to confirm if the improved transduction activity of AAV5S194G in mouse retina was translated to NHP photoreceptors. The fidelity of the NHP transplant model was confirmed using AAV5, and the superior transduction activity of the AAV5S194G variant in the explant model was demonstrated. Notably, evaluation of the AAV5S194G variant in the NHP retinal model revealed an additional benefit to this capsid; AAV5S194G transduction is predominantly in primate photoreceptors, in contrast parental AAV5 transduces photoreceptors, INL and GCL. One caveat to the NHP explant model is that it does not provide data relating to potential immunological findings for the novel AAV capsids, however, the model provides insights to the potential translatability of an AAV capsid for the treatment of human retinal disease.
In conclusion, this study underscores the utility of a rational design approach to engineer capsids for targeting and improving the efficacy of AAV vectors for ocular gene therapy. Prior knowledge of AAV receptor binding, surface charge, and PTMs was applied to the design of optimized capsids that provide improved retinal spread, a novel corneal cell tropism and increased photoreceptor transduction, respectively. Importantly, the attributes provided by these novel capsids will add to the efficacy, specificity, and safety of their potential use in gene therapy for a wide variety of human ocular diseases.
Footnotes
Acknowledgments
The authors would like to acknowledge Bindu Nambiar, Shelley Nass, Maryellen Mattingly and Denise Woodcock from the Gene Therapy Research Vector Core and Rachel Diangelo and Simon Godwin from the Gene Therapy Development and Manufacturing group.
Author Disclosure
All authors are employees of Sanofi.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.