
Abstract
We report here the development of oncolytic adenoviruses (Ads) that have reduced toxicity, enhanced tumor tropism, produce strong antitumor response, and can overcome resistance to immune checkpoint inhibitor therapy in breast cancer. We have shown that LyP-1 receptor (p32) is highly expressed on the surface of breast cancer cells and tumors from cancer patients, and that increased stromal expression of transforming growth factor β-1 (TGFβ-1) is associated with triple-negative breast cancer. Therefore, we constructed oncolytic Ads, AdLyp.sT and mHAdLyp.sT, in which the p32-binding LyP-1 peptide was genetically inserted into the adenoviral fiber protein. Both AdLyp.sT and mHAdLyp.sT express sTGFβRIIFc, a TGFβ decoy that can inhibit TGFβ pathways. mHAdLyp.sT is an Ad5/48 chimeric hexon virus in which hypervariable regions (HVRs 1–7) of Ad5 are replaced with the corresponding Ad48 HVRs. AdLyp.sT and mHAdLyp.sT exhibited better binding, replication, and produced higher sTGFβRIIFc protein levels in breast cancer cell lines compared with Ad.sT or mHAd.sT control viruses without LyP-1 peptide modification. Systemic delivery of mHAdLyp.sT in mice resulted in reduced hepatic/systemic toxicity compared with Ad.sT and AdLyp.sT. Intravenous delivery of AdLyp.sT and mHAdLyp.sT elicited a strong antitumor response in a human MDA-MB-231 bone metastasis model in mice, as indicated by bioluminescence imaging, radiographic tumor burden, serum TRACP 5b and calcium, and body weight analyses. Furthermore, intratumoral delivery of AdLyp.sT in 4T1 model in immunocompetent mice inhibited tumor growth and metastases, and augmented anti-PD-1 and anti-CTLA-4 therapy. Based on these studies, we believe that AdLyp.sT and mHAdLyp.sT can be developed as potential targeted immunotherapy agents for the treatment of breast cancer.
Introduction
Despite the latest breakthroughs in medical research, metastatic breast cancer remains an incurable disease and the frequent cause of cancer-related deaths worldwide. 1,2 In advanced breast cancers, the rapid acquisition of therapy resistance seems to limit the effectiveness of newly developed targeted therapy and immunotherapy. Therefore, it is important to develop novel therapies that can target tumor cells and signaling molecules specific for metastasis, and circumvent various resistance mechanisms. 3,4
Recently, there has been a growing interest in understanding the role of transforming growth factor β (TGFβ) signaling in promoting tumor growth and metastases and inducing therapy resistance. Under physiological conditions TGFβ is a potent growth inhibitor. However, aberrant TGFβ expression has been reported in advanced malignancies, including breast cancer. 5,6 It has been shown that aberrant TGFβ signaling can alter the tumor microenvironment (TME), promote angiogenesis and epithelial–mesenchymal transition (EMT) while suppressing antitumor immunity, and contribute significantly to metastasis and tumor immune escape. 7,8 Thus, several inhibitors of TGFβ pathway are currently being developed and tested in animal studies and clinical trials for many cancer types. 9 –14
Our laboratory has previously developed an oncolytic adenovirus Ad.sT, expressing soluble TGFβ receptor II fused with human IgG Fc fragment (sTGFβRIIFc).
15,16
sTGFβRIIFc acts as the decoy protein preventing TGFβ binding to tumor cells.
15,16
In immunodeficient mouse models, systemic delivery of Ad.sT inhibited skeletal metastases of human breast and prostate cancer cells.
16,17
In the breast and renal immunocompetent mouse models, direct inoculation of a similar sTGFβRIIFc expressing oncolytic virus
To treat metastatic cancers, the preferred route to deliver adenoviral vectors would be via systemic administration. Two key limitations in the use of Ad5-based adenoviruses (Ads) for systemic administration are the limited tumor tropism and liver/systemic toxicities. To enhance tumor tropism, in this study, a 9-amino acid-long tumor homing-cell penetrating LyP-1 peptide was inserted into the HI loop of Ad5 adenoviral fiber to generate AdLyp.sT. This novel AdLyp.sT vector will target LyP-1 receptor, which is expressed on the breast cancer cell surface, tumor macrophages, and tumor lymphatics, but not readily detectable on normal tissues. 19 –21
To bypass the hepatic uptake and produce minimum hepatic and systemic toxicity, we have previously engineered a liver detargeted oncolytic adenovirus (
The goals of this study were to examine the following: (i) the prevalence of LyP-1 receptor expression in breast cancer cells and in the tumors from breast cancer patients, and if aberrant TGFβ expression is associated with advanced breast cancer; (ii) whether modification of oncolytic viruses with LyP-1 peptide1 will enhance virus binding and replication, and sTGFβRIIFc production in breast cancer cells; (iii) if upon systemic delivery, mHAdLyp.sT will have reduced hepatic uptake and produce minimum hepatic and systemic toxicity; (iv) if systemic administration of AdLyp.sT and mHAdLyp.sT in human breast tumor models in immunodeficient mouse will have improved inhibition of the skeletal metastases and the tumor-induced bone destruction; and (v) if in immunocompetent model, intratumoral inoculation of AdLyp.sT will produce antitumor growth and antimetastasis effects, and augment anti-PD-1/anti-CTLA-4 antibody therapy.
We report here that breast cancer cells and human breast tumors ubiquitously express LyP-1 receptors and high levels of stromal TGFβ-1 proteins in triple-negative breast cancer (TNBC) patients. Inclusion of LyP-1 peptide in AdLyp.sT and mHAdLyp.sT improves the virus binding and replication, and transgene expression. Systemic administration of mHAdLyp.sT in mice showed reduced hepatic/systemic cytotoxicity, while both AdLyp.sT and mHAdLyp.sT produced effective antitumor and antibone metastasis activities in a human TNBC mouse model. Finally, our studies indicated that direct inoculation of AdLyp.sT into murine syngeneic tumors inhibited tumor growth and lung metastasis, and enhanced efficacy of anti-PD-1/anti-CTLA-4 therapy in a mouse TNBC model. Overall, our studies suggest that AdLyp.sT and mHAdLyp.sT can be potentially developed for metastatic breast cancer therapy and enhancing ICI therapy.
Materials and Methods
Case selection and tissue slide preparation
Fifty-two paired (tumor and matched normal tissue samples adjacent to tumors) formalin-fixed, paraffin-embedded (FFPE), breast tissue blocks with initial diagnosis from 26 breast cancer patients were derived by expert pathologists. These surgical specimens were collected from 2004 to 2011 in the NorthShore University HealthSystem (Illinois). The FFPE blocks were cut into 5-μm-thick sections and the sectioned tissue slides were prepared according to an established and verified standard protocol. Relevant clinical and pathological information of these patients was retrieved from the NorthShore Epic electronic health records (EHRs) by authorized personnel. To better understand the relationship between molecular targets of interest (LyP-1 receptor and TGFβ) with the human epidermal growth factor receptor 2 (HER2) and hormone receptor statuses of patients, we used the sample pool consisting of similar numbers of molecular subtypes. The protocol was approved by the NorthShore Institutional Review Board, and written informed consent was previously obtained from each patient for breast cancer research through the NorthShore Biospecimen Repository.
Cell lines and adenoviruses
Human mammary tumor cell lines, MCF-7 (ATCC, Manassas, VA), MDA-MB-231 (ATCC), and MDA-MB-231-luc2, 16 and the mouse mammary tumor cell line, 4T1 (ATCC), were maintained as described earlier. 28 All media components were purchased from Thermo Fisher Scientific (Waltham, MA).
To create oncolytic adenoviruses, viral or target genes were modified and cloned in a shuttle vector and subjected to homologous recombination with adenoviral genomic DNA derived from adenoviral mutant dl01/07 using published methods. 15 In particular, the LyP-1 receptor binding sequence encoding nine amino acids (LyP-1 peptide: CGNKRTRGC) was introduced into the HI loop of fiber to create p309-FibHI-Lyp-1. AdLyp.sT was obtained by homologous recombination between p309-FibHI-Lyp-1 and pTG-sTGFβRIIFc. The latter contains the sTGFβRIIFc gene to inhibit TGFβ signaling.
To create mHAdLyp.sT, p309 FibHI-Lyp-1-TGFβRIIFc was first generated to have both sequences of LyP-1 peptide and the TGFβ inhibitory ligand: sTGFβRIIFc. It was subjected to homologous recombination with a previously constructed Ad5/48 chimeric hexon containing vector, pTG hex5/48, 26 to generate mHAdLyp.sT. The other key components—mutant E1A (01/07), ADP (adenoviral death protein), and ITR (inverted terminal repeats), are sequentially positioned as described previously. 15,26 All adenoviral vectors were amplified in HEK293 cells (ATCC) and purified as described earlier.
Immunofluorescence, Western blot, and immunohistochemistry staining
For immunofluorescence staining, cells were seeded and, when reaching ∼50% confluences on Fisherbrand™ cover glasses in six-well plates, were subjected to staining. Cells were fixed with 3.7% paraformaldehyde and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS). One percent goat serum was used for blocking. Rabbit anti-GC1q R (p23, the receptor for LyP-1) antibody from Abcam (Cat#: ab101267, 1:200 dilution; Cambridge, United Kingdom) and goat anti-rabbit IgG H&L (Alexa Fluor® 488; Abcam) were used for detection. Cells were mounted with ProLong® Gold Antifade Reagent with DAPI (Invitrogen, Carlsbad, CA) and viewed using a Leica microscope equipped with epifluorescence illumination and appropriate fluorescence filters for DAPI and Alexa Fluor 488. Fluorescence images in each wavelength were collected using a CoolSNAP CCD camera and were relayed to the IPLab Spectrum image analysis software. Images were subjected to identical grayscale normalization and contrast enhancement and were then subjected to pseudocolor overlay to produce the color images.
Cells grown in 100 mm dishes were collected for Western blot using a protocol described previously, 27 using rabbit anti-GC1q R (p23) antibody (ab101267; Abcam) or anti-actin antibody (A2066; Sigma-Aldrich, St. Louis, MO) as the primary antibody.
For immunocytochemistry analysis, paraffin sections were deparaffinized and rehydrated by a standard protocol. 29,30 Proteolytic-induced epitope retrieval followed by saponin/hydrogen peroxide solution was utilized to promote epitope availability and block endogenous peroxidase activity. The tris phosphate buffer (TBS) blocking solution contained 5% goat serum, 5% BSA, 0.1% gelatin, 0.5% Triton X-100, and 0.05% Tween 20. The primary antibodies: rabbit anti-GC1q R (p23) antibody (ab101267, 1:200 dilution; Abcam) and mouse anti-TGFβ-1 antibody [TB21] (ab27969, 1:200 dilution; Abcam) in 2% BSA TBS were used for detection, followed by TBS containing 0.1% Tween 20 TBST washes and incubation with corresponding biotinylated secondary antibodies from the VECTASTAIN® Elite® ABC HRP Kit (Burlingame, CA). Vector DAB Peroxidase (HRP) Substrate Kit and hematoxylin Gill III (Newcomer, Middleton, WI) were utilized for chromogenic reaction and counterstaining. The slides were dehydrated, mounted, and observed by a Nikon Eclipse TE200 Inverted Microscope. The Nikon DS-Fi1 camera and NIS-Elements BR 3.10 software were used for documenting micrographs. Higher magnification was used when necessary for identification.
Adenoviral binding, replication, and sTGFβRIIFc expression in MDA-MB-231 and 4T1 cells
In vitro virus binding and replication assays were performed according to our established protocols as described earlier. 17 For virus binding assay, 1 × 105 cells per well were seeded in 24-well plates. The next day, cells were washed two times with ice-cold PBS and exposed to 2.5 × 104 viral particles (VPs) per cell corresponding viruses in ice-cold serum-free cell culture media for 1 h at 4°C. At the end of 1 h, the plates were switched to 37°C for 5 min to allow viruses to be internalized. Cells were then immediately washed twice with room-temperature PBS, followed by the incubation with complete cell culture media at 37°C for 24 h.
Cell fixing and sTGFβRIIFc staining were performed by the procedures previously described. 27,28 Goat anti-human IgG, Fcγ fragment-specific antibody from Jackson ImmunoResearch Laboratories (Cat#: 109005098; West Grove, PA) was used to detect sTGFβRIIFc. Positive cells were counted with 100 × magnification in the 2.54 mm2 field area in our digital image system for microscopes previously described. 28 Regular viral infection and protein expression assays were also performed according to our established protocol as described earlier. 17,27 In short, cell culture media were collected, and sTGFβRIIFc levels in the media were determined by enzyme-linked immunosorbent assay (ELISA) using previously described methods. 15,17,27
Animal studies
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the NorthShore University HealthSystem.
Toxicity studies and innate immune responses
Six-week-old female NU/NU nude mice (Charles River Laboratories, Wilmington, MA) were injected with 1 × 1011 VPs per mouse of corresponding adenoviruses via tail vein. Each group contained four mice. Mice were euthanized 3 days after the injection. The livers were photographed. The liver samples were collected and processed for hematoxylin and eosin (H&E) staining, immunohistochemistry (IHC) staining by the anti-human IgG, Fcγ fragment-specific antibody (Cat#: 109005098; Jackson ImmunoResearch Laboratories), and viral genome copy measurement by quantitative PCR by the methods described in our previous publications. 26 Mice spleens were also processed for H&E staining and IHC staining of sTGFβRIIFc.
Mouse blood samples were collected (0.5 mL) and serum samples were used to determine the sTGFβRIIFc levels by ELISA as described earlier. 16 The commercially available kits were utilized to determine serum levels of alanine transaminase (ALT) (EALT-100; BioAssay Systems, Hayward, CA), aspartate transaminase (AST) (EASTR-100; BioAssay Systems), and lactate dehydrogenase (LDH) (ab197000; Abcam, Cambridge, MA). Serum tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1β levels were determined by ELISA as previously described. 27
Bone metastasis model and bioluminescence imaging
To establish bone metastasis, MDA-MB-231-luc2 cells (2.0 × 105/mouse) were injected into the left heart ventricle of 4-week-old female nude mice on day 0, as described earlier. 17,31 On day 7, mice were subjected to bioluminescence imaging (BLI) in dorsal and ventral positions using Xenogen IVIS and the laboratory's standard protocol previously described. 16 Signal intensities were quantified as the total flux (photons/seconds) within regions of interests (ROIs) in both left and right hind limbs using Living Image software 3.0 (Caliper Life Sciences, Hopkinton, MA) as described. 16,17,31 We obtained 70 mice that had photon flux of ROIs within the range of 1.0 × 105–1.0 × 106 photons per second. Mice were divided into seven groups, with statistically indistinguishable BLI signals among each group (an average of about 2.8 × 105–6.2 × 105 photons/second per group).
For treatment groups, various viral vectors were administered via tail vein on days 8 and 12 as described in the Results section. In précis, we included four groups that received a low therapeutic dose (LTD) of Ad.sT, AdLyp.sT, mHAd.sT, or mHAdLyp.sT (a total of 5 × 1010 VPs/mouse), and two groups treated with a high therapeutic dose (HTD) of
X-ray radiography analysis, body weight measurements, and quantifications of calcium and TRACP 5b in serum
Mice were subjected to X-ray radiography in the prone position using Faxitron (Faxitron X-Ray Corporation, Wheeling, IL) on day 15 and 22. Radiographic tumor burden from bone lesions was quantified in the femur and tibia of both hind limbs using ImageJ software (NIH, Bethesda, MD) as described earlier. 17,31 All mice, together with control mice that did not receive MDA-MB-231-luc2 cells (normal, n = 10) or any treatments (buffer, n = 10), were also subjected to body weight measurement once a week. Quantification of calcium and TRACP 5b in mouse serum was performed using our published methods. 17,31
4T1 xenograft model and therapeutic analysis with/without ICIs
To establish the mouse mammary tumor syngeneic model, we injected 2 × 106 4T1 cells per mouse subcutaneously (day 0) into the dorsal right flank of 30 female BALB/c mice (4–6 weeks old). Mice were monitored every day. On day 6, tumor dimensions were measured (in mm) using a caliper. Tumor volumes were calculated by the following formula: tumor volume = (width 2 × length)/2. Then, tumor-bearing mice were divided into six groups, without statistical differences between each group (n = 5). On day 7, 9, and 11 post-tumor cell implantation, tumor-bearing mice were administered 100 μL of PBS buffer, Ad.sT, or AdLyp.sT (5.0 × 1010 VPs per mouse/each injection) intratumorally (10 mice for each virus). On days 8, 10, 13, and 15, anti-PD-1 and anti-CTLA-4 antibodies were administered together intraperitoneally (0.2 mg per mouse per antibody) in the groups indicated in the Results section.
The tumor volumes were monitored on day 10, 14, 17, 21, 24, and 27. The body weights were measured on day 6, 14, 17, 21, 24, 27, 31, and day 35. Mice were euthanized on day 35. The whole lungs were excised and photographed. Lung surface tumor lesions were counted for each group. Parts of the lungs were processed for H&E staining according to our published protocol. 18 Pulmonary metastatic burden was quantified by using ImageJ software (NIH) for each lung section.
Statistical analyses
All data were analyzed using GraphPad Prism software version 5 (GraphPad software, San Diego, CA). For prognostic and clinicopathological significance analysis, the chi-squared test and Fisher's exact test were used to assess for independence among variables. The chi-squared (χ 2 ) test applied an approximation assuming multiple samples, while the Fisher's exact test ran an exact procedure for two variables. Data are presented in the tables. For all other statistical analyses, data are plotted as the mean ± standard error of the mean. Longitudinal data were analyzed using a two-way repeated-measure ANOVA followed by Bonferroni post hoc pairwise tests for the data obtained over the time course. For multiple group analyses, one-way ANOVA followed by Bonferroni post hoc tests adjusting for multiplicity was performed. Student's t-tests were performed to compare two sets of data. A Fisher's exact test was used for the bone metastasis incidence data. Differences were considered significant at two-sided p < 0.05.
Results
LyP-1 receptor (p32) expression on the surface of TNBC cell lines and on tumor samples of breast cancer patients
We have examined the prevalence of two relevant molecular biomarkers, LyP-1 receptor (p32) and TGFβ-1, in breast cancer cell lines and in the human breast tumors. Immunofluorescence staining showed that the two human breast cancer cell lines, MDA-MB-231 and MCF-7, exhibit strong LyP-1 receptor staining on cell surface and internal membrane networks (Fig. 1A). LyP-1 receptors were localized more on plasma membrane and endoplasmic reticulum-like membrane networks in MDA-MB-231 cells (Fig. 1A, upper panel). In MCF-7 cells, LyP-1 receptors were enriched in Golgi-like juxtanuclear compartments (Fig. 1A, middle panel). 4T1, a highly tumorigenic and invasive mouse mammary tumor cell line, showed a staining pattern similar to that of MDA-MB-231 (Fig. 1A, lower panel). To be noted, both human MDA-MB-231 and mouse 4T1 are TNBC cell lines and are more aggressive than MCF-7, an estrogen/progesterone receptor-positive and HER2-negative cell line.

LyP-1 receptor (p32) and/or TGFβ-1 expression in breast cancer cell lines and patients.
The expression of LyP-1 receptors in these three breast cancer cell lines was further verified by Western blot analysis (Fig. 1B). LyP-1 receptors in mouse 4T1 cells exhibit a different banding pattern from those seen in human breast cancer cells, suggesting that human and murine Lyp-1 receptors, although they can react with our LyP-1 receptor antibody, show species specificity. We further analyzed LyP-1 receptor expression in human tumors derived from breast cancer patients. The FFPE samples obtained from 26 patients before they received any therapy were subjected to IHC staining of LyP-1 receptors. Tumor cells in all these samples showed medium (Fig. 1C, lower left panel) to strong (Fig. 1C, upper left panel) staining for LyP-1 receptor. The statistical analysis indicated that LyP-1 receptor expression levels are not associated with any clinical and/or pathological parameters in these patients.
Tumor stromal expression of TGFβ-1 is associated with TNBC and is a poor prognostic marker of overall survival in breast cancer patients
Because there is increased evidence in the literature suggesting that aberrant transforming growth factor β (TGFβ) signaling is closely associated with tumor growth and metastases in advanced cancers, including breast cancer, we analyzed for TGFβ-1 expression in these patient samples. Distinct TGFβ-1 expression patterns were observed among them. Some samples contained increased TGFβ-1 expressing cells in tumor stroma (Fig. 1C, upper right panel), while others had fewer TGFβ-1 expressing cells (Fig. 1C, lower right panel). Relevant clinical and pathological information of these patients was retrieved from the NorthShore Epic EHRs by authorized pathologists. Our sample pool consists of five patients who are negative for HER2&ER&PR (TNBC), five patients who are HER2 positive only, seven HER2 (−)/ER&PR (+) patients, and nine patients with all positive markers [HER&ER&PR (+)].
Our statistical analysis showed that a strong TGFβ-1 expression in tumor stroma was significantly associated with these molecular statuses (χ 2 = 10.85, p = 0.0126; Table 1). Furthermore, Fisher's exact test suggested that the difference in TGFβ-1 expression between TNBC and ER/PR/HER2 (+) tumors was most significant (p = 0.005). The tumor stromal expression of TGFβ-1 was also significantly associated with overall survival (OS) (p = 0.0084, Fisher; Table 1). Two other major clinical and pathological parameters significantly associated with progression-free survival and overall survival (OS) in this group were molecular subtypes and metastasis status (Supplementary Table S1).
Associations between transforming growth factor β-1 expressions in tumor-associated stromal cells by immunohistochemistry and clinical and pathological parameters
HER2, human epidermal growth factor receptor 2; TGFβ-1, transforming growth factor β-1; TNBC, triple-negative breast cancer.
Construction of AdLyp.sT and mHAdLyp.sT, viral replication, virus binding, and vector-mediated sTGFβRIIFc protein expression in TNBC cell lines
Our laboratory has previously developed a platform of oncolytic adenoviruses expressing sTGFβRIIFc. Ad.sT is the original cytomegalovirus (CMV) promoter-regulated Ad5-based virus expressing sTGFβRIIFc (named as
To enhance tumor tropism, we have now created AdLyp.sT and mHAdLyp.sT by introducing LyP-1 peptide, a 9-amino acid-long tumor homing-cell peptide, into the HI loop of the Ad.sT and
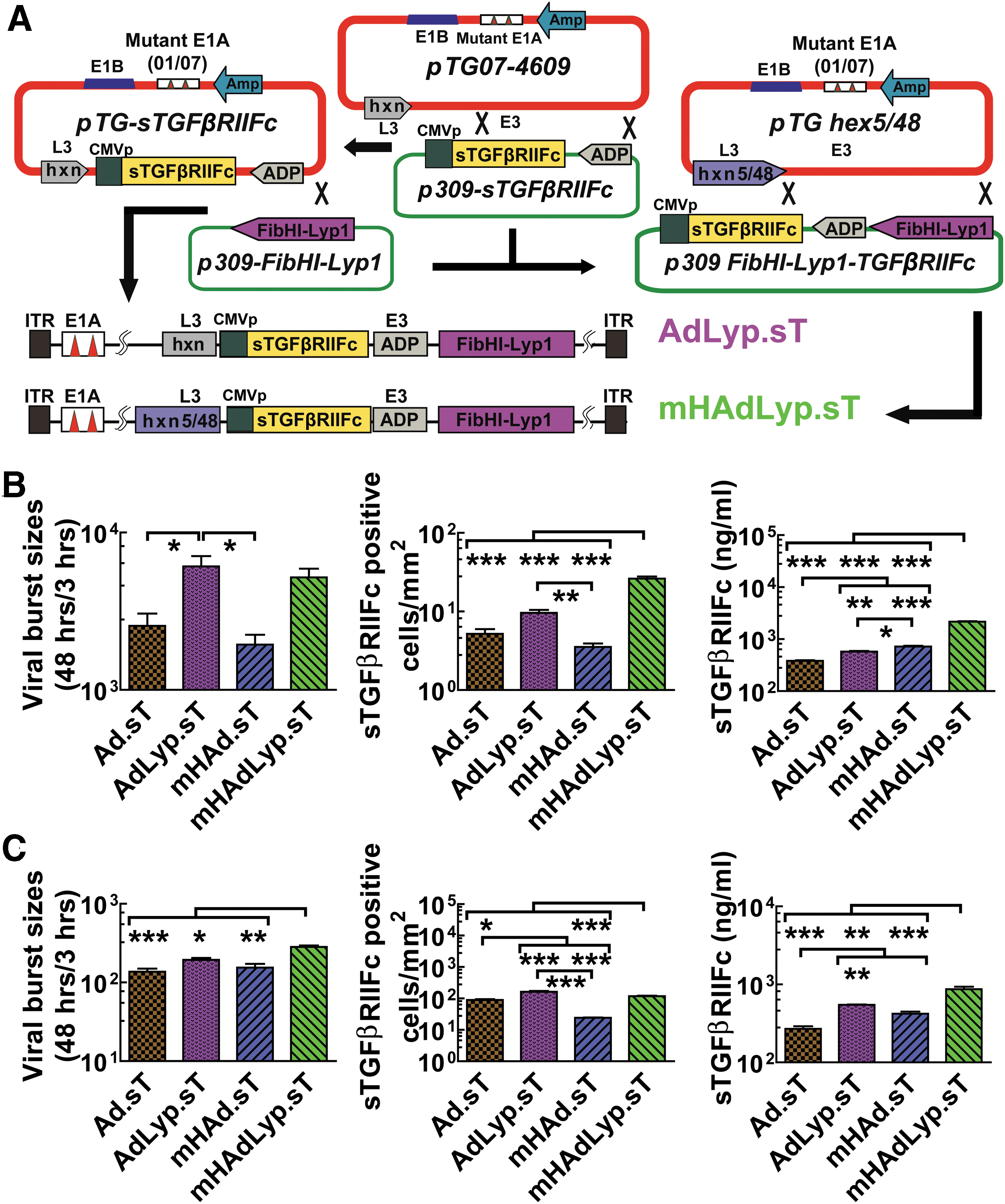
Construction of LyP-1-modified adenoviruses expressing sTGFβRIIFc (AdLyp.sT and mHAdLyp.sT) and their replication, binding, and sTGFβRIIFc expression in MDA-MB-231 and 4T1 cells.
AdLyp.sT and mHAdLyp.sT were examined for their replication potential and sTGFβRIIFc expression in breast cancer cells. We used human MDA-MB-231 and mouse 4T1 cells in this study because they are two very well-studied TNBC cell lines and we have documented their cell surface LyP-1 receptor expression. Two previously published adenoviruses, Ad.sT and mHAd.sT, were used as controls. To demonstrate their replication potentials, the viral burst sizes were calculated as the ratio of viral hexon protein production at 48 h relative to the levels seen at 3 h, as previously described. 27 In human MDA-MB-231 cells, the viral titers of AdLyp.sT and mHAdLyp.sT were significantly more than double that of Ad.sT and mHAd.sT, respectively (Fig. 2B, left panel, p < 0.05). This result suggested that the LyP-1 peptide facilitates viral spread and production in the breast tumor cells. Moreover, mHAdLyp.sT exhibited a higher viral replication potential in mouse 4T1 cells (Fig. 2C, left panel, one-way ANOVA test showed that the viral titer of mHAdLyp.sT was significantly higher than that of all other viruses).
Next, the viral binding and entry into the tumor cells were quantified by the viral binding assay. In both cell lines, mHAdLyp.sT produced a significantly higher numbers of sTGFβRIIFc-positive cells than Ad.sT or
Systemic administration of mHAdLyp.sT in nude mice resulted in reduced uptake in the liver and spleen, hepatotoxicity and systemic toxicity, and attenuated innate immune response
To compare the toxicity profiles of AdLyp.sT and mHAdLyp.sT, we administered these and Ad.sT and
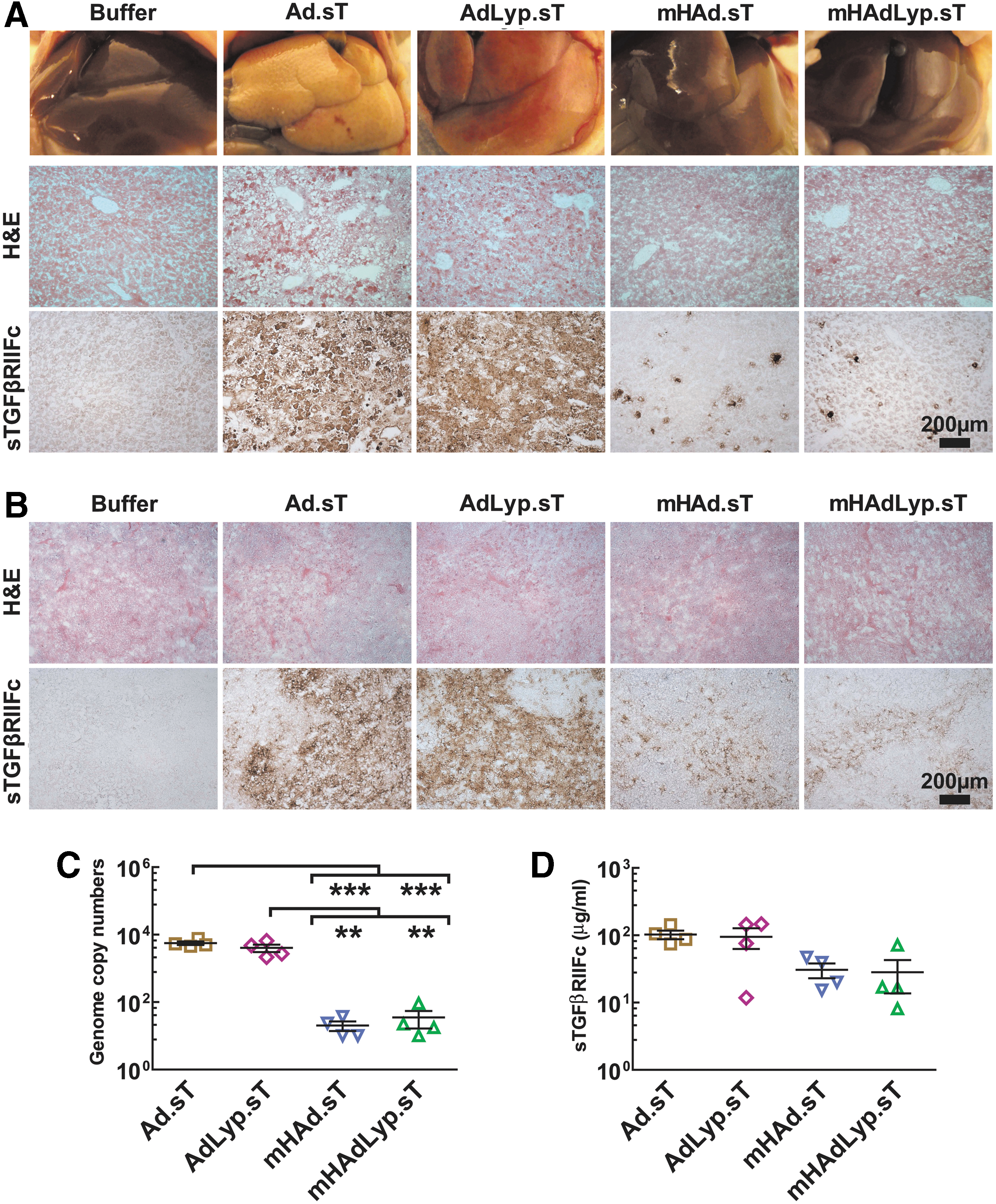
Systemic administration of
We also detected more sTGFβRIIFc-positive cells in
We further quantified adenoviral uptake in the liver by measuring viral genomic DNA. Viral DNA copy numbers in the
We next examined if the systemic administration of
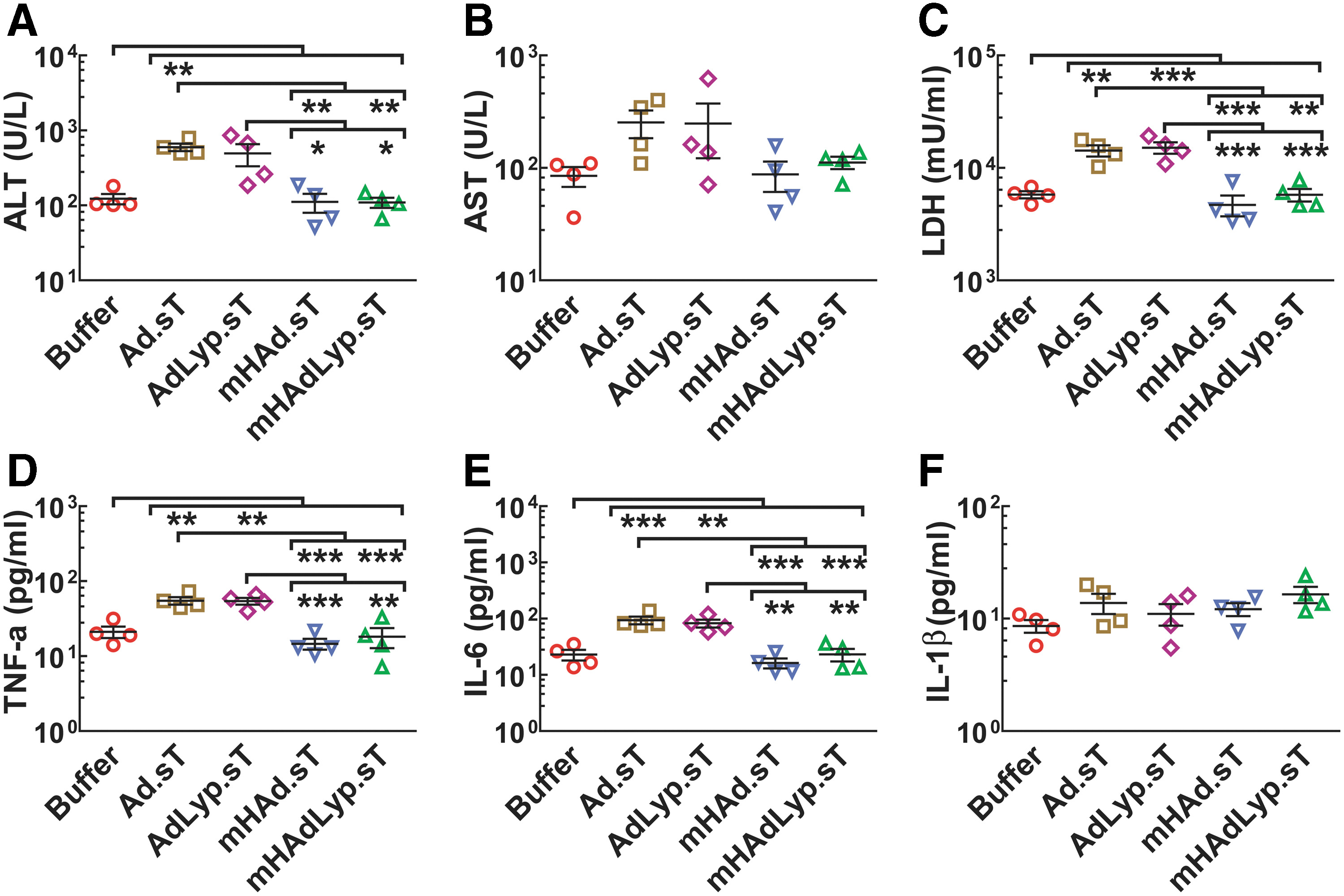
Systemic administration of
Next, we examined the serum LDH, TNF-α, IL-6, and IL-1β levels in 72-h samples. There was a significant increase of LDH levels in the
Similar results were obtained regarding the serum levels of TNF-α and IL-6, two proinflammatory cytokines that are closely associated with innate inflammation, suggesting attenuated inflammatory responses induced by
Systemic administration of LyP-1-modified AdLyp.sT and mHAdLyp.sT viruses inhibits bone metastases in a human MDA-MB-231 experimental metastasis model
Next, we investigated if following the systemic delivery of Ad.sT, AdLyp.sT, mHAd.sT, or mHAdLyp.sT, breast cancer metastasis can be inhibited in a mouse experimental metastasis model. MDA-MB-231-luc2 cells were inoculated in the left heart ventricle of female nude mice to produce skeletal metastases. Viral vectors were administered intravenously on day 8 and 12. Four animal groups received an LTD (two doses, 2.5 × 1010 VPs/mouse each, a total of 5 × 1010 VPs/mouse) of each virus. Given the superior safety profile of Ad5/48 chimeric hexon viruses shown in Figs. 3 and 4, two groups were treated with HTD (two doses, 1.0 × 1011 VPs/mouse each, a total of 2.0 × 1011 VPs/mouse) of
In our laboratory, previously we had conduced dose dependence toxicity studies for Ad.sT and

Systemic administration of low dose of AdLyp.sT and high dose of
The BLI signal intensity of combined dorsal and ventral ROIs in each mouse was quantified. In the buffer-treated group, there was a time-dependent increase in BLI signals (Fig. 5B). All five treatment groups exhibited significant reductions of signal intensities in these ROIs on day 22 when compared with the buffer group by two-way ANOVA followed by Bonferroni post hoc tests adjusting for multiplicity (p < 0.001, n = 10). Moreover, three treatment groups (the LTD of mHAdLyp.sT, the HTD of mHAd.sT, and the HTD of mHAdLyp.sT) exhibited significant reductions compared with the LTD of
Since MDA-MB-231-luc2 cells can also metastasize to the liver, lungs, and brain, we analyzed whole-body BLI photon flux on day 22. Again, four treatment groups (the LTD of AdLyp.sT, the LTD of mHAdLyp.sT, the HTD of mHAd.sT, and the HTD of mHAdLyp.sT) exhibited similar statistical significance against the buffer-treated group (p < 0.001 in one-way ANOVA and p < 0.01 in t-test, Fig. 5D). These results further indicated that Lyp-1-modified viruses have enhanced inhibitory effect against breast cancer metastases in the mouse experimental model.
To further examine the effect of Lyp-1-modified viruses to inhibit bone metastases, we also preformed radiographic analyses on day 15 and 22. Figure 6A shows representative X-ray images on day 22 from each group. Osteolytic lesions on the skeletal tumors are marked by yellow arrows. Lesion sizes in the hind limb bones for each mouse were quantified by using ImageJ software. Compared with the buffer group, a significant inhibition of lesion size progression was observed for each of the treatment groups on day 22 (p < 0.001 in two-way ANOVA followed by Bonferroni post hoc tests adjusting for multiplicity, n = 10, Fig. 6B). On day 15, only the HTD of mHAdLyp.sT exhibited significant inhibition (p < 0.05). Also, both the HTD groups of mHAdLyp.sT and

Effects of adenoviral vectors on MD-MB-231 tumor burden: radiographic, blood, and body weight analysis.
When we used one-way ANOVA and paired t-test to further analyze day 22 data (Fig. 6C), we found that only the LTD of
We also monitored the animal body weights once a week, as an indicator of cancer cachexia. On day 22, the average body weight of the buffer group mice dropped 22% from that on day 15; the average body weight was 17.68 g on day 22 versus 22.46 g on day 15 (Fig. 6E). It was significantly lower than that of all treatment groups and normal group (naive mice without tumors) (the HTD of mHAdLyp.sT and mHAd.sT, and normal: p < 0.001; all other treatment groups: p < 0.01; Fig. 6E). Of note, there was no significant difference in the day 22 body weight between the normal group and the HTD of mHAdLyp.sT or
To further investigate the effect of the HTD of mHAdLyp.sT to inhibit tumor-induced osteolytic bone destruction, we measured two biomarkers in mouse serum. Serum tartrate-resistant acid phosphatase 5b (TRACP 5b) is considered to be a good indicator of the osteoclastic bone resorption and turnover. Two treatment groups, the LTD of AdLyp.sT and the HTD of mHAdLyp.sT, as well as the normal group, have significantly lower serum TRACP 5b levels compared with that of the buffer group (p < 0.05 in one-way ANOVA; p < 0.001 in t-test; Fig. 6F).
Since osteolytic bone destruction results in hypercalcemia, the serum calcium levels were also measured. When compared with the buffer group by t-test, the HTD of mHAdLyp.sT and
Intratumoral delivery of AdLyp.sT improves the antitumor effects of Ad.sT and anti-PD-1 and anti-CTLA-4 immunotherapy in a highly aggressive 4T1 syngeneic immunocompetent mouse model
We have recently shown that in the 4T1 syngeneic mouse model, intratumoral inoculation of an oncolytic adenovirus expressing sTGFβRIIFc (rAd.sT) can significantly improve the inhibition of tumor growth and metastases by anti-PD-1 and anti-CTLA-4 antibodies. In rAd.sT, the viral replication is regulated by the human telomerase reverse transcriptase promoter (TERTp). In the oncolytic adenoviruses investigated in this study, Ad.sT and AdLyp.sT, a more robust and general promoter, CMV, drives the sTGFβRIIFc gene expression. Thus, in this study, we investigated the effects of intratumoral delivery of Ad.sT and AdLyp.sT in inhibiting tumor growth and metastases alone and in combination with ICIs. It is known that 4T1 tumors are resistant to anti-PD-1 and anti-CTLA-4 therapy, 33 –36 and our unpublished data (not shown here) have also confirmed that observation.
Moreover, in a preliminary study using intratumoral delivery of Ad.sT, our combination therapy with a single ICI agent, anti-PD-1 or anti-CTLA-4, did not produce significant improvement in the antitumor response (data not shown). Unfortunately, we did not conduct immunotherapy using a combination of AdLyp.sT and single ICI. Although anti-PD-1 and anti-CTLA-4 produce significant inhibition of tumor growth and metastases, we do not observe complete eradication of the tumors or metastases by this combination (Fig. 7). Therefore, we were very interested to further improve anti-PD-1 and anti-CTLA-4 in TNBC models such as the 4T1 tumor model.
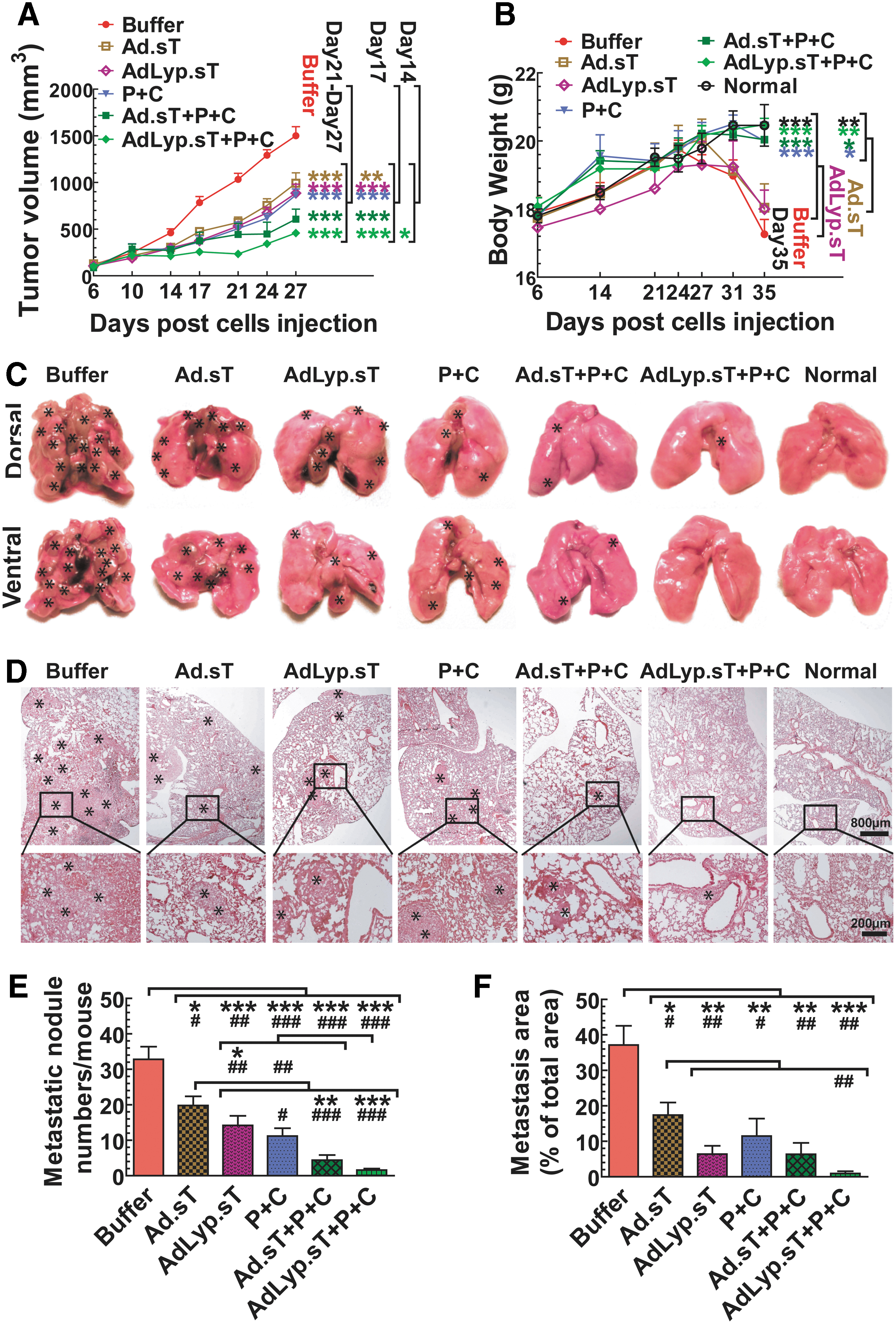
Intratumoral delivery of Ad.sT and AdLyp.sT enhanced the immune checkpoint inhibitor therapy on 4T1 tumor growth and spontaneous metastasis in the syngeneic mouse model.
4T1 tumors were established in BALB/c mice, and treated with Ad.sT or AdLyp.sT and anti-PD-1/anti-CTLA-4, either alone or in combinations, as described in the Materials and Methods section. Primary tumor growth was followed over time till day 27. In this model, from day 27 onward, severe ulcerations are produced in some of the primary tumors, and therefore, tumor volume readings beyond day 27 are less reliable. There was significant inhibition of tumor growth on day 21, 24, and 27 in mice treated with either Ad.sT or AdLyp.sT and/or anti-PD-1/anti-CTLA-4, compared with the buffer group (p < 0.001) (Fig. 7A). However, only the triple therapy combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 produced significant inhibition on day 14 (p < 0.05). These results suggested that the combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 was the most effective approach. On day 17, Ad.sT alone was effective (p < 0.01 compared with the buffer), however, all other treatment groups produced better inhibition of tumor growth (p < 0.001).
All mice were euthanized on day 35, by which time some of the mice had reached the level 2 of the body condition score. The body weights of
Next, we examined the effects of our therapy modalities on the spontaneous lung metastasis, as lung is the predominant metastatic site in this 4T1 mouse model. At the end of this study, mice were euthanized and all visible superficial lung metastatic foci from each mouse were photographed and counted by gross examination (Fig. 7C, E). There was significant inhibition of the numbers of macrometastases on the lung surface in all the treatment groups compared with the buffer group (p < 0.001, p < 0.01, or p < 0.05 by either one-way ANOVA or t-test; Fig. 7E). However, further analysis of data showed that only the combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 has p < 0.001 in both one-way ANOVA and t-test when compared with the buffer group or Ad.sT alone, the weakest treatment group. Thus, the combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 was also the most effective in inhibiting lung macrometastases.
We further conducted histological examination of lung metastases (Fig. 7D). The total lung micrometastatic burden in each group was determined as described in the Materials and Methods section (Fig. 7F). Again, while all the treatment groups significantly inhibited the lung micrometastatic burden compared with the buffer group (p < 0.001, p < 0.01, or p < 0.05 by either one-way ANOVA or t-test; Fig. 7F), the combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 was the most effective (p < 0.001 by one-way ANOVA). When compared with Ad.sT alone by t-test, the combination of AdLyp.sT and anti-PD-1/anti-CTLA-4 also produced significant inhibition (p < 0.01, Fig. 7F). These results indicated that in the 4T1 tumor model, intratumoral inoculation of AdLyp.sT can significantly improve anti-PD-1+anti-CTLA-4-mediated inhibition of tumor metastasis.
DISCUSSION
We are reporting here the construction of two novel oncolytic
A unique feature of mHAdLyp.sT is that HVRs 1–7 of Ad5 hexon were replaced by the corresponding Ad48 HVRs. Because of this genetic modification, systemic administration of mHAdLyp.sT bypasses the liver uptake and produces minimum hepatic/systemic toxicity. Our studies described here have shown that both AdLyp.sT and mHAdLyp.sT elicited strong antitumor and antimetastasis responses in the human breast cancer immunodeficient mouse model. These antitumor responses were in general superior to Ad.sT and mHAd.sT, the corresponding unmodified viruses that do not express the LyP-1 peptide. In addition, because mHAdLyp.sT can be administered systemically with the HTD, the best inhibitory effect was observed with this modality. In this study, we have also shown that intratumoral delivery of AdLyp.sT was effective in inhibiting the tumor growth and metastasis, and enhanced ICI therapy in an immunocompetent mouse tumor model.
TGFβ as a target for breast cancer therapy
In the recent years, there have been significant advances in understanding the key regulators of the TME and immune system. TGFβ is one of the key factors that have a direct influence on tumor evasion of immune response and in promoting tumor growth and metastasis. The outcome of the TGFβ signaling pathway is dependent on the cell and tissue context. In invasive tumors, TGFβ singling plays the oncogenic role and promotes tumor progression by several mechanisms. These include activating EMT and stimulating angiogenesis. 8,37 TGFβ can also recruit myeloid-derived suppressor cells, Tregs, and M1 macrophages, known to induce immunosuppression. 7 In addition, TGFβ can destroy the production and functions of immune cells such as NK cells, dendritic cells, and CD8+ CTLs that are critical in producing antitumor responses. 8,38 Thus, over the years, several TGFβ inhibitors, including receptor kinase inhibitors, antiligand antisense oligonucleotides, and monoclonal antibodies, have been explored to treat advanced tumors both in the preclinical setting and in the clinical trials in cancer patients. However, till to date, none of the anti-TGFβ inhibitors has been approved by the FDA for clinical usage.
To block the TGFβ pathway, we have previously constructed oncolytic adenoviruses that express sTGFβRIIFc. We have reported that intratumoral administration of one of these viruses, rAd.sT, can inhibit TGFβ-regulated genes such as CTGF, CXCR4, IL-11, PTHrP, N-cadherin, vimentin, VEGFA, and VEGFR in 4T1 tumors established in the syngeneic mouse. 18 These TGFβ targets are well-documented inducers/markers of tumor growth, EMT, angiogenesis, and metastases. Moreover, rAd.sT produced a favorable shift to the production and recruitment of antitumor immunomodulatory factors and immune cells in the mouse tumors, peripheral blood, and spleen, suggesting a systemic immune activation directed toward 4T1 tumors. 18 Thus, our approach of targeting aberrant TGFβ signaling is quite effective in disrupting the protumorigenic TME and overcoming immune tolerance.
The results presented in this study further document that our sTGFβRIIFc expressing oncolytic viruses elicit strong inhibitory effects in a human breast cancer metastasis model in immune deficient mice. Although we did not conduct bioactivity results of vector-induced sTGFβRIIFc expression in this study, previous studies from our laboratory as well as from others have documented the biological effects of sTGFβRIIFc. 18,28,39,40 Furthermore, our oncolytic viruses are also effective in inhibiting mouse mammary tumor growth and its spontaneous metastasis to the lungs in a syngeneic mouse tumor model.
Viral replication: an added advantage of using oncolytic adenoviruses for targeting TGFβ pathways
Although several approaches to target TGFβ are being examined in many laboratories, we believe that one advantage of using oncolytic adenoviruses expressing sTGFβRIIFc is that in addition to inhibiting TGFβ pathways, viral replication will directly cause tumor cell death. Whether the viruses are delivered intratumorally or via systemic route (as described in our studies here), viral uptake into the tumors will produce viral replication within the tumors, cause tumor cell death, release tumor antigens, and activate dendritic cells and tumor-specific cytotoxic immune cell responses. 41,42 Thus, our oncolytic viruses AdLyp.sT and mHAdLyp.sT can produce antitumor responses by targeting multiple steps—including targeting tumor cells, TME, and activating the immune system, both locally and systemically.
One point regarding the differences in the response to oncolytic adenoviruses by mouse and human cancer cells is worth discussing here. Because our oncolytic viruses do not exhibit significant replication in mouse cells (including 4T1 cells), while the effect on adenovirus binding and hence transgene expression can be enhanced by LyP-1 modification, no significant improvements in the viral replication were observed by LyP-1-modified viruses in 4T1 cells. However, oncolytic viruses do replicate in human cancer cells, and LyP-1-modified viruses bind better with human breast cancer cells; therefore, LyP-1-modified viruses produce both enhanced viral replication and transgene expression in human cancer cells.
Addressing key barriers in the systemic delivery of oncolytic adenoviruses
In this study, we have attempted to address the key barriers in the use of oncolytic adenoviruses—massive systemic toxicity induced by the systemic therapy of oncolytic adenoviruses, the prevalence of preexisting adenovirus neutralizing Abs, and the lack of their tumor specificity. 16,26,43,44 Regarding systemic toxicity, it is known that the commonly used Ad5-based viruses produce toxicity mainly from viral sequestration and destruction by the liver Kupffer cells. To address this issue, we have created the hexon chimeric Ad5/Ad48 virus (mHAdLyp.sT). Since Ad48 hexon HVRs 1–7 have much reduced factor X binding, the Ad5/Ad48 chimeric hexon virus will have attenuated hepatic uptake. 22 –27 In fact, we have shown here that mHAdLyp.sT indeed has the diminished capacity of liver sequestration and produces reduced hepatotoxicity and decreased systemic toxicity.
Although we have documented that in immune-deficient mice, the liver detargeted viruses have reduced uptake in the liver as measured by viral DNA copy numbers (Fig. 3C), in future, we will also conduct these distribution studies in multiple tissues in more relevant immunocompetent mouse models. Also, because functionally significant Ad5-specific neutralizing antibodies are directed primarily against the HVRs of Ad5 hexon in both human and mouse, the hexon chimeric Ad5/Ad48 virus can circumvent pre-existing antivector immunity. 24,25,45,46
To address the issue of tumor tropism, our new virus mHAdLyp.sT described in this study has LyP-1 peptide-enhanced breast tumor tropism. As shown, mHAdLyp.sT exhibits strong antitumor effects in the experimental bone metastasis model. Moreover, because of the improved safety profile, we could also administer a higher therapeutic dose of mHAdLyp.sT further improving its efficacy in targeting metastases. LyP-1-modified mHAdLyp.sT virus will target the LyP-1 receptor, which is expressed on the cancer cells, tumor macrophages, and tumor lymphatics, but not on the normal tissues. 19,20,47 Thus, in the potential future clinical trials in breast cancer, the systemic administration of LyP-1-modified Ad5/48 chimeric hexon-based mHAdLyp.sT is expected to have attenuated hepatic uptake causing reduced hepatic/systemic toxicity, but will have enhanced tropism for the breast tumors.
mHAdLyp.sT has two levels of tumor selective targeting/replication. First, the virus will target the tumor cells and the tumor lymphatics via LyP-1, a tumor-specific peptide enabling it to selectively target the breast tumors. Second, the viral backbone used to construct the oncolytic mHAdLyp.sT vector selectively replicates in the tumor cells, producing tumor lyses. Upon the arrival of mHAdLyp.sT in the tumors, large amounts of sTGFβRIIFc, the TGFβ decoy will be produced in the tumors. sTGFβRIIFc will be also secreted into the blood stream. sTGFβRIIFc will bind with TGFβ to inhibit aberrant TGFβ signaling, relieving TGFβ-induced immune suppression. Thus, mHAdLyp.sT will target tumor cells as well as remodel tumor stroma potentially enhancing T cell trafficking into the tumors. It should be also noted that in LyP-1-modified viruses, the LyP-1 peptide is generically linked with the H1 lops of fiber; we do not believe that the LyP-1 peptide can be dissociated from the fiber when it is being formed in the cytoplasm, as large amounts of LyP-1 viruses can be grown in the cell culture.
Another point worth noting is that following systemic delivery of oncolytic viruses, the level of transgene expression can be also possibly impacted by additional factors, including vector-induced cytotoxicity in the infected tissues. For example, while Ad5-based viruses have increased uptake in the liver and spleen, due to vector-induced toxicity, these vectors tend to destroy these tissues, thus potentially reducing sTGFβRIIFc production in the blood. Conversely, this will be true for liver detargeted viruses—because they have reduced uptake in the liver, could be more stable in those resident tissues, and continue to produce sTGFβRIIFc in the blood. Therefore, following adenoviral administrations, the amount of sTGFβRIIFc in the blood is somewhat difficult to interpret. Despite that, as shown in Fig. 3D, Ad5-based viruses do indeed produce about five times more protein in the blood (about 100 μg/mL), compared with Ad5/48-based viruses, which produce about 20 μg/mL in the blood. It is true that liver detargeted viruses have reduced uptake in the liver, which appear dramatically different than the Ad5-based viruses (Fig. 3A). However, it should be noted that liver detargeted viruses also produce reduced levels of sTGFβRIIFc in the blood (about fivefold), although it does not appear as dramatic (perhaps due to log scale used in Fig. 3D).
Targeting TGFβ to overcome resistance to anticheckpoint inhibitor therapy
In recent years, ICIs, including anti-PD-1, anti-PD-L1, and anti-CTLA-4, have shown promise as antitumor agents, and are approved for the treatment of several malignancies. 48 –51 Clinical trials in breast cancer patients have shown that about 20% of TNBC responded favorably to anti-PD-1 antibodies. 52 –55 However, many TNBC patients are resistant to anti-PD-1/PD-L1 and anti-CTLA-4 treatments. 52 –55 Therefore, there is a significant interest in exploring novel therapeutic approaches to improve anti-PD-1 and anti-CTLA-4 therapies for TNBC. Aberrant TGFβ signaling is one of the key factors in producing resistance to ICI therapy. 56
In this study, IHC analysis of the patient tumor samples showed that elevated stromal TGFβ-1 was associated with more aggressive TNBC phenotype and poor OS, indicating resistance to the current therapies. As discussed above, we believe that aberrant TGFβ signaling can produce a TME that is resistant to ICI therapy. So far, the only approved ICI therapy for TNBC is the combination of anti-PD-L1 antibody (atezolizumab) and nab-paclitaxel (Abraxane) for PD-L1-positive, unresectable, locally advanced, or metastatic TNBC patients. 57 However, TNBC patients with low levels of PD-L1 or even some patients with high PD-L1 are resistant to this combination therapy. 57 In that regard, our results that AdLyp.sT can augment anti-PD-1 and anti-CTLA-4 responses in a poorly immunogenic and ICI-resistant 4T1 tumor model 33 –36 are quite important.
In this initial study using our novel oncolytic viruses, we have focused primarily on describing their safety, toxicity, and therapeutic profile. In our next series of experiments, we will conduct additional studies to document intratumoral gene expression and to define the key factors in more relevant immunocompetent models as shown previously for the unmodified oncolytic virus rAd.sT. 18 We will also examine the effect of intravenous delivery of our oncolytic adenoviruses in improving the ICI therapy in immunocompetent mouse models. In the future, we also plan to elucidate the underlying mechanisms how AdLyp.sT and ICIs work collectively to convert immunologically cold tumors to into hot ones, thus activating the immune system to destroy cancer cells.
AdLyp.sT and mHAdLyp.sT have the potential of targeting several other malignancies
Although in this study we have focused on targeting TNBC, it is worth noting that many other cancers could also benefit from our LyP-1-modified oncolytic virus approach targeting TGFβ signaling. LyP-1 receptor has been shown to be overexpressed in many cancers, including breast, prostate, melanoma, glioblastomas, pancreas, and in general is considered an excellent target for cancer therapy. 19 –21,47 Likewise, aberrant TGFβ signaling has also been shown to promote tumor growth and metastases of many cancers, including prostate, kidney, and gastrointestinal cancers. 58,59 Therefore, we believe that our targeted oncolytic viral approach described here has the potential of an excellent alternative to these ongoing approaches 9 –14,60,61 to target multiple cancers and to improve ICI therapies.
In conclusion, based on our studies described here, we believe that both AdLyp.sT and mHAdLyp.sT can be further developed as promising treatment strategies to control metastatic breast cancer as well as to augment immunotherapy.
Footnotes
ACKNOWLEDGMENTS
We are thankful to the Kovler Family Foundation, Mr. and Mrs. Richard Hulina, Mr. Jimmie Alford and Ms Maree Bullock, Maxine and James Farrell, Carol Gollob Foundation, and an anonymous donor for their generous gifts. We are thankful to Drs. Bruce Brockstein, Michael Caplan, Janardan Khandekar, Charles Brendler, and Ted Mazzone for their continuous support.
AUTHOR DISCLOSURE
No competing financial interests exist for any of the authors.
FUNDING INFORMATION
The research was funded, in part, by grants from the Department of Defense Breast Cancer Research Program Award DAMAD17-03-0703, the National Cancer Institute grant R01CA127380, Cancer Gene Therapy funds from NorthShore, and the NorthShore Auxiliary Breast Cancer Research award to Prem Seth.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.