
Abstract
The list of successful gene therapy trials using adeno-associated virus (AAV)-based vectors continues to grow and includes a wide range of monogenic diseases. Replication incompetent AAV genomes typically remain episomal and expression dilutes as cells divide and die. Consequently, long-term transgene expression from AAV is best suited for quiescent cell types, such as retinal cells, myocytes, or neurons. For genetic diseases that involve cells with steady turnover, AAV-conferred correction may require routine readministration, where every dose carries the risk of developing an adaptive immune response that renders treatment ineffective. Here, we discuss innovative approaches to permanently modify the host genome using AAV-based platforms, thus potentially requiring only a single dose. Such approaches include using AAV delivery of DNA transposons, homologous recombination templates into safe harbors, and nucleases for targeting integration. In tissues with continual cell turnover, genetic modification of progenitor cell populations will help ensure persistent therapeutic outcomes. Combining the safety profile of AAV-based gene therapy vectors with the ability to integrate a therapeutic transgene creates novel solutions to the challenge of lifelong curative treatments for human genetic diseases.
Introduction
The appeal of adeno-associated virus
Adeno-Associated Virus (AAV)-based viral vectors are firmly established gene therapy tools. In nature, infection of wild-type AAV (wtAAV) is considered nonpathogenic and is inherently replication incompetent in the absence of a helper virus, such as adenovirus or herpesvirus. In addition, a multitude of clinical trials using recombinant AAV vectors suggest that immune responses are typically mild. 1,2 Production of recombinant AAV vector is conceptually straightforward. The shuttle plasmid is readily modifiable allowing for quick adaptation and implementation. There are many distinct, naturally occurring serotype capsids and countless modified capsids used for directing cell-specific tropism. However, researchers must be mindful that tissue tropism may be species specific. The encapsidated, single-stranded DNA vector is stable, allowing for purification and concentration using ultracentrifugation, as well as long-term storage at low temperature.
Results from clinical trials and approved therapeutics using AAV vectors provide remarkable examples of gene delivery efficacy for a variety of genetic diseases and tissue types. In 2012, the first clinically licensed in vivo delivered AAV-based vector, Glybera, was approved by the European Medicines Agency. This AAV1-based vector was used to successfully treat lipoprotein lipase deficiency. In 2017, the first vector to be approved by the U.S. Food and Drug Administration (FDA) was Luxturna. Luxturna is an AAV2 vector expressing the retinal pigment epithelium-specific 65 kDa protein (RPE65) that is injected into the retina as a treatment for Leber's congenital amaurosis. 3 More recently, an AAV9 carrying survival motor neuron 1 (SMN1), termed Zolgensma, was approved for children with spinal muscular atrophy type 1 (SMA1). This treatment is injected intravenously and results in remarkable protection of motor function and prolongs the life of the patients. 4 Phase 3 trials for Duchenne muscular dystrophy highlight the benefits of muscle injections of a novel vector termed AAV2.5. 5,6 Of note, retinal, neuronal, and muscle tissues all consist of cell populations that are mitotically quiescent. These are only three examples of the rapidly growing list of success stories using AAV-based vectors to treat genetic diseases.
The field of gene editing has exploded in recent years with the goal of in situ repair of defective genes. Editing tools such as TALENs, ZFPs, CRISPR/Cas, and base editing systems have expanded the field of gene therapy beyond gene addition (reviewed in Refs. 7 –9 ). In many cases, AAV-based vectors have become the vehicle of choice for cellular delivery of editing tools. For multiple genetic diseases, AAV may set the bar for both successful gene addition and gene editing strategies.
Limitations of AAV
Although AAV contains many promising therapeutic features, it is unlikely that a single administration will be sufficient for all monogenic disease models. For example, AAV gene delivery to the liver of neonatal mice exhibited rapid diminishment of episomal expression due to high levels of cellular proliferation during development. 10 Similarly, AAV delivered to the airways of rabbits resulted in differential persistence across cell types, with expression waning in epithelia over a 60-day period while the surrounding smooth muscle retained expression. 11 Another study examining persistence delivered AAV8 expressing green fluorescent protein (GFP) via intravenous injection to mice. They showed high copy numbers of viral genomes 3 days postdelivery in the liver, where expression decayed to less than 1 copy/cell by 2 months. In contrast, genome persistence was much more stable in both cardiac and skeletal muscle tissues over the same time-course. 12 Nakai et al. demonstrated the role of episomal expression in the liver of adult mice given human factor IX (hFIX) via AAV8. 13 At 12 weeks postinjection, cell proliferation was induced via partial hepatectomy, and hFIX levels were measured for the subsequent 6 weeks. Mice with partial hepatectomy showed a significant loss in hFIX levels without returning to prior expression levels, indicating loss of episomal expression. 13 These data suggest that chromosomal integration of the transgene within a progenitor cell population may be essential for persistent expression from a single-dose delivery in nonquiescent tissues.
The packaging capacity of AAV can be a limiting factor for gene expression cassettes exceeding ∼4.7 kb. In the context of using AAV to deliver Cas9 for gene editing, Staphylococcus aureus Cas9 (Sa Cas9, ∼1 kb) is often opted for over the more traditional Streptococcus pyogenes Cas9 (Sp Cas9, ∼4 kb). Sa Cas9 is more easily packaged in AAV than Sp Cas9, especially if guide RNAs (gRNAs) are included. However, Sa Cas9 is less efficient than Sp Cas9 and requires a longer NNGRRT PAM sequence than the NGG PAM motif of Sp Cas9. 14 There are multiple strategies to circumvent the packaging limitation of AAV-based vector for delivery of Cas9 expression vectors (reviewed in Ref. 15 ). For example, trans-splicing takes advantage of the propensity of AAV genomes to concatemerize and incorporates splice donors and acceptors into codelivered vectors. 16 –18 More recently, protein trans-splicing elements termed split inteins have been incorporated into AAV vectors. Split inteins are peptide elements that perform a protein trans-splicing function to join N-terminal and C-terminal ends of partial protein peptide sequences. 19
Naturally Occurring Chromosomal Integrations of AAV Genomes
AAV has a single-stranded DNA genome flanked by repetitive sequences termed inverted terminal repeats (ITRs). All wild-type strains of AAV contain two open reading frames (ORFs) termed Rep and Cap. From the Rep ORF, four nonstructural proteins are expressed. Rep78 and Rep68 are involved in viral replication, while Rep58 and Rep40 are involved in genome packaging and processing. The Cap ORF encodes three structural proteins VP1, VP2, and VP3 that assemble to form the viral capsid and direct viral tropism.
In cell culture, wtAAV can integrate into a specific locus in human chromosome 19 known as AAVS1. 20 –22 Of the integrated virus, less than 1% of viral genomes will integrate within the AAVS1 site; however, up to 10% of integrations have been mapped to a 33 kb region surrounding the AAVS1 locus in cultured cells. 23 Of note, the vast majority of virus remains episomal. DNA sequences within the AAVS1 locus are necessary and sufficient for AAV integration that occurs as a result of homology to ITR sequences and a Rep78/68-dependent enzymatic reaction. 23,24 AAVS1 contains a tandem GCTC repeat element that can serve as a binding site for the Rep 68/78 proteins to mediate complex formation between AAVS1 and the AAV ITR. 25
Unlike wtAAV, recombinant AAV lacks any endogenous viral genes. Rep and Cap are deleted and replaced with a transgene expression cassette flanked by the AAV ITRs (typically from AAV2). Without Rep proteins, recombinant AAV no longer preferentially integrates into the AAVS1 locus. 26,27 However, recombinant AAV will integrate at low efficiency at alternative genomic sites other than AAVS1. The majority of the vector genomes remain episomal, with <1% integrating into chromosomal DNA. 28
Strategies to Permanently Modify the Host Genome Using AAV
The low-level frequency of recombinant AAV integration is likely insufficient for therapeutic benefit following multiple rounds of cell division. Intentionally increasing the occurrence of AAV integrations is a potential strategy that could lead to lifelong persistence of transgene expression. Here, we discuss varying strategies to achieve genome modification using AAV.
AAV-delivered DNA transposons
Recombinant “cut-and-paste” DNA transposons are a two-part system composed of terminal repeat (TR) elements flanking a gene of interest and a transposase that catalyzes transposon integration into the host genome. 29,30 Research with the DNA transposon Sleeping Beauty pioneered the use of a recombinant transposon system to achieve genomic integration of a transgene. 31 Other commonly used transposon systems include Tol2 and piggyBac . 32,33 In the presence of the requisite transposase enzyme, a DNA transposon will efficiently integrate into the genomes of human cells 34 –36 and mediate long-term expression in vivo. 37 –40
In a recombinant system, the transposon and transposase are typically codelivered by plasmid transfection; however, delivering naked DNA to somatic cells in vivo is inefficient compared with viral vector-mediated delivery. AAV can be used to deliver the transposon element and transposase in separate vectors. The resultant vector is often termed a hybrid transposon/AAV (Fig. 1). Zhang et al. first reported the incorporation of DNA transposon components into AAV vectors. Using Sleeping Beauty/AAV, they demonstrated that the AAV genome can successfully serve as a transposition template in human cells. 41 We reported that piggyBac/AAV conferred persistent transgene expression in immunocompetent mouse airways in vivo 42 and corrected the cystic fibrosis respiratory phenotypes in CFTR null pigs in vivo. 43 Without transposase, the hybrid transposon/AAV vector functions no differently than a standard AAV vector. In vitro cell passaging studies and sequence mapping confirmed persistent expression in the presence of transposase, while in the absence of transposase, expression waned. In vivo studies using piggyBac/AAV in mice show persistent reporter gene expression in the lung 42 that continued following a polidocanol treatment 3 months postdelivery. Polidocanol treatment accelerates lung cell turnover by ablating surface epithelial cells; thus, continued expression suggests that integration occurred in an airway progenitor cell population. These studies suggest that a single dose of an integrating AAV vector can lead to persistent expression for the life of a mouse.
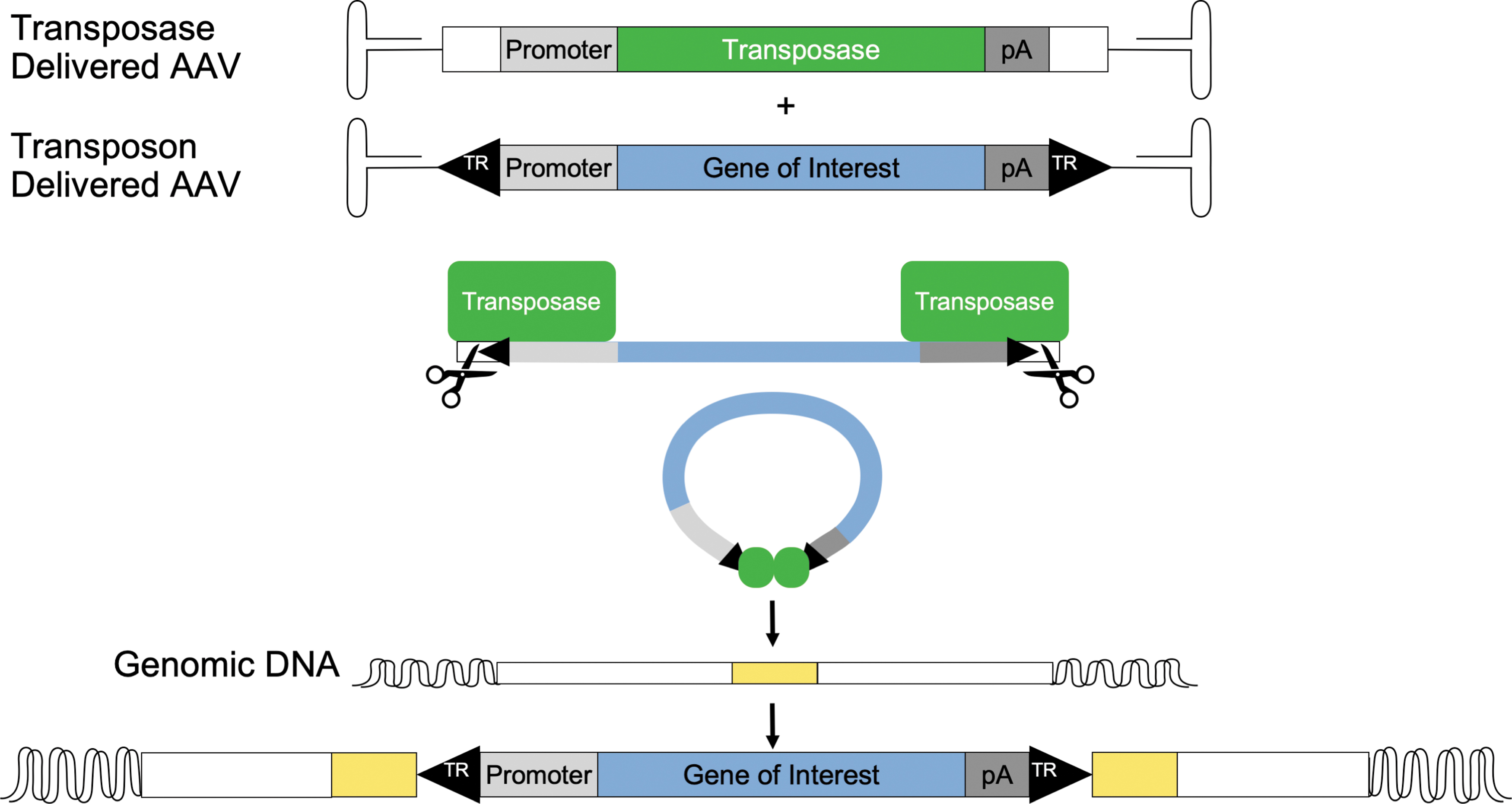
Transposon delivery via AAV vectors. An expression cassette is flanked by transposon TRs (black triangles) and is recognized by the transposase enzyme (green) supplied in trans. Transposase will facilitate the excision of the transgene cassette from within the AAV vector and integrate it into a short requisite DNA motif sequence of the genome (yellow). AAV, adeno-associated virus; TR, terminal repeat.
Typically, the transposon and transposase are delivered by separate vectors. If both components are delivered by the same vector, codelivery in all transduced cells would be guaranteed. This would lead to a predicted boost in efficiency. Yet, there are at least three reasons to deliver the transposon and transposase from separate vectors: (1) Production: if the transposase is expressed in producer cells, the transposon may be transposed from the vector genome before it can be packaged. This would result in empty capsids. This limitation can be overcome using a tissue-specific promoter that is not functional in the producer cell line. (2) Safety: if the transposon and transposase are on the same vector, there is the unlikely possibility that the transposase may be inadvertently integrated into the target cell genome. This undesired situation could result in persistent transposase expression that would theoretically repeatedly mobilize the transposon. (3) The aforementioned size constraints of AAV preclude single vector delivery.
Transposase is required for the initial transposition into the host genome, but transient transposase expression may be preferred to avoid the possibility of transposon “hopping.” A self-inactivating (SIN) piggyBac transposase strategy has been described, in which the promoter and transposase are separated by the piggyBac TR. 44 Although viral vectors are an efficient way to deliver the transposase, other delivery options may be considered. For example, transposase delivery by mRNA is a promising way to improve transposition efficiency and the quality of integration. 45,46
Promoterless gene targeting
A study aiming to restore normal factor IX (FIX) levels for the treatment of hemophilia B used AAV8 to deliver FIX flanked by homology arms targeted to 5′ of the albumin stop codon. Rather than a promoter sequence, FIX was preceded with a P2A read through sequence, minimizing disruption of the endogenous albumin (Fig. 2). In this method, the vector undergoes homologous recombination (HR) at the albumin locus that results in robust liver expression of the FIX transgene. Plasma FIX levels were maintained between 5% and 20% of normal levels among both adult and neonatal mice. Examination of the coagulation efficiency by activated partial thromboplastin time revealed restoration to wild-type levels. 47 As the liver does not have a true “stem” cell, cell turnover is followed by proliferation of hepatocytes rather than from a progenitor cell compartment. 48 The time of AAV administration during development and the health of the liver play a role in persistent transgene expression. 49,50 The lack of loss of expression within the developing neonatal liver further demonstrates the efficacy of AAV-integration on persistent expression.
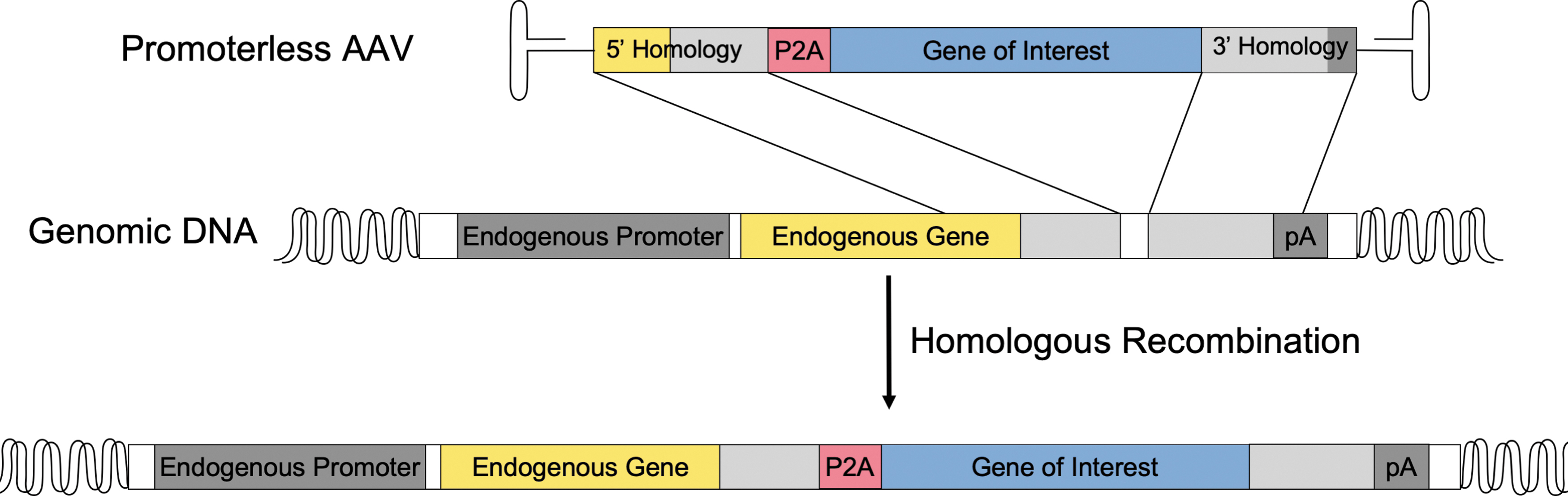
Promoterless gene targeting. An AAV vector carrying a gene of interest is designed without an exogenous promoter, rather a P2A ribosomal read through sequence shown in red. Flanking the 5′ and 3′ regions of the gene of interest are homology sequences targeted at a highly expressed gene in the genome. The homology arms facilitate homologous recombination within the genomic DNA at the targeted site of integration. Following integration, both the endogenous gene and the gene of interest are driven off the endogenous promoter and transcribed as one mRNA producing two proteins following translation.
This technology was further developed to increase efficiency of integration. 51 Cells were transduced with both the promoterless AAV carrying a transgene and another AAV carrying CRISPR/Cas9 and gRNA against the albumin gene, near the region targeted by the homology arms. The rationale was that CRISPR-induced cleavage will facilitate a more efficient integration. In cell culture, integration efficiency increased 26-fold over HR alone. Neonatal Crigler–Najjar mice, deficient in an enzyme required for adequate metabolism of bilirubin, were simultaneously treated with promoterless AAV carrying the therapeutic UGT1A1 gene, and an AAV carrying CRISPR/Cas9 targeting the albumin locus. Mice were treated with both vectors, promoterless vector only, or no treatment, and followed for 10 months after delivery. Untreated mice died before day 19, while mice given promoterless UGT1A1, with or without CRISPR, survived. Only mice given both promoterless UGT1A1 and CRISPR showed bilirubin at wild-type levels for the duration of the study. 51
Although this approach was adopted to direct high-level liver expression using the albumin promoter, the strategy is potentially adaptable to other tissues by using other genes. For example, BPIFA1 (aka SPLUNC1) is highly expressed in the large airways of the lung. Integrating a promoterless AAV into the BPIFA1 gene would, in theory, drive the expression of a protein of interest in the surface epithelium of the trachea and main stem bronchi.
Rep-dependent targeting of AAVS1
Another design for AAV transgene integration has been developed from the endogenous integration profile of wtAAV. Using Rep-mediated integration, some in vitro studies have shown that coinfection of wtAAV (AAV2) with a recombinant AAV2 vector carrying a gene of interest promotes integration of the transgene into the AAVS1 region in vitro (Fig. 3). However, there were also nonspecific integrations induced by this model as well as integration of the wtAAV used to deliver Rep. 52
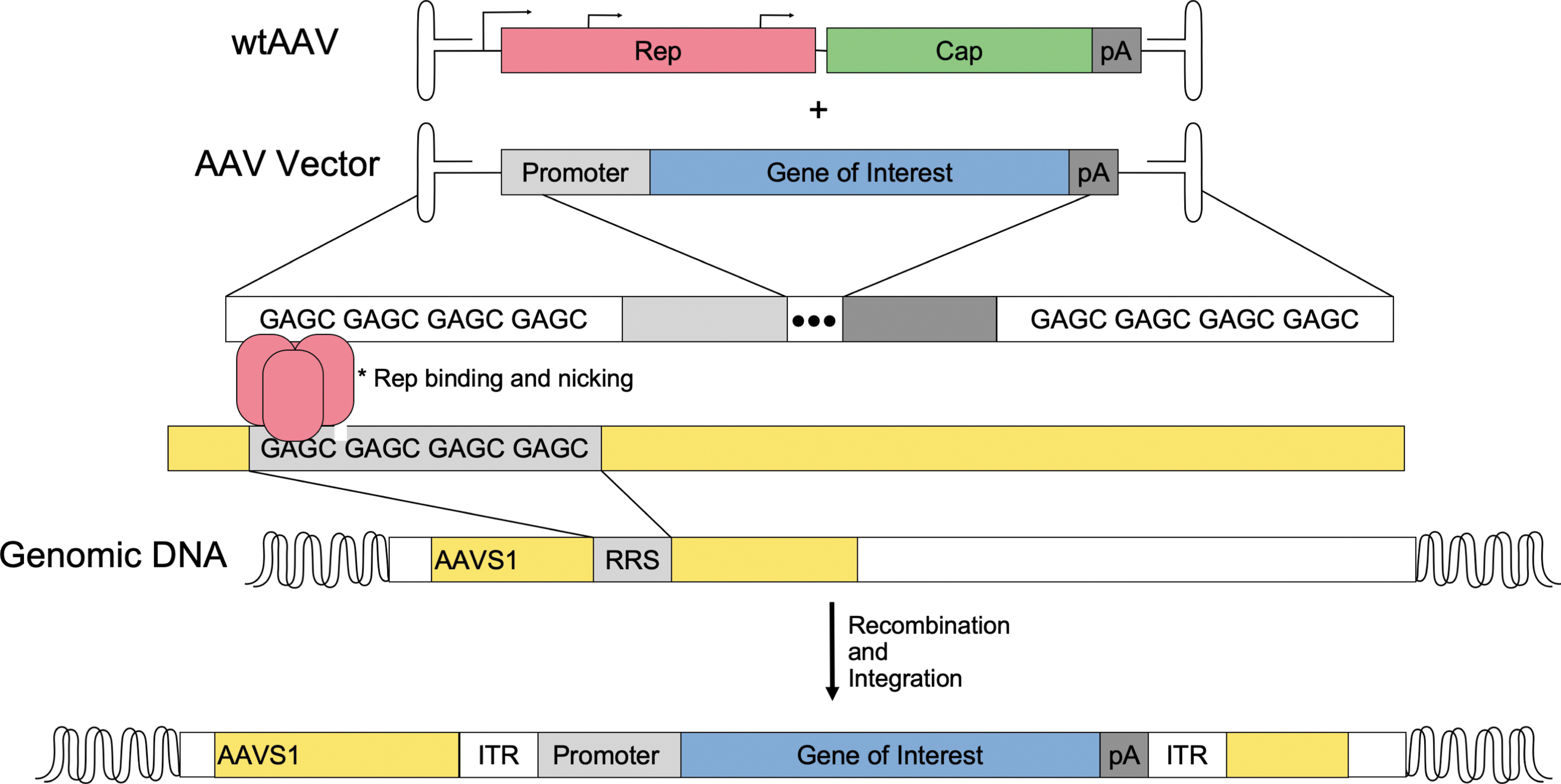
Rep-dependent targeting of AAVS1. wtAAV and AAV vectors carrying a transgene of interest are coinfected within a cell. Once transduced, wtAAV produces the Rep protein depicted in red. Rep binds the rep recognition sequence of both the ITR of the AAV genome and AAVS1. Rep-dependent nicking of the target strand induces homologous recombination of the AAV vector ITRs and AAVS1, resulting in integration of the gene of interest within AAVS1. ITR, inverted terminal repeat; wtAAV, wild-type AAV.
Similarly, fusion proteins of the transposases Sleeping Beauty, piggyBac, and Tol2 to the DNA binding domain of Rep, were developed in an effort to target integration toward the Rep-recognition sequence (RRS) within AAVS1. This study reports a modest increase in biased integration patterns for Rep/Sleeping Beauty and Rep/Tol2 fusion protein within the RRS of AAVS1 in human cells in vitro. The piggyBac variants, however, showed very weak redirection of integration by fusion to Rep. 53 These experiments provide a proof-of-concept for targeted integration of a transgene into the AAVS1 site.
Nuclease targeted gene addition of AAV genomes
As previously mentioned, AAV vector genomes can integrate within double-strand breakage sites induced by CRISPR/Cas9 editing. SpCas9 and gRNAs were codelivered by separate AAV1 vectors in a study mapping AAV integrations in mouse brain, cochlea, and muscle. Sequencing results identified that fragments of AAV ITRs had integrated in SpCas9-induced indels at the gRNA target both in vitro and in vivo. 54,55 AAV integration frequencies ranged from 10% to 39% of all indel formations. This work also used a novel sequencing strategy by synthesizing a very short AAV (465 bp) termed AAV-λ465 to allow for the more efficient sequencing methods compared with ∼3–5 kb. Genome-wide off-target AAV integration frequencies were unaltered by individual expression or coexpression of Cas9 and gRNA, confirming that AAV integration requires nuclease-mediated double-stranded breaks. 54 These experiments not only highlight implications for CRISPR editing in the context of AAV delivery, but also show promise as a molecular tool for targeting integration of transgenes within genomic safe harbors.
Mapping Integrations
Risk of insertional mutagenesis
Integrating vectors undoubtedly conjure concerns about insertional mutagenesis and cancer. The theoretical risks of viral vector-mediated insertional mutagenesis became a reality following a clinical trial using a murine leukemia virus (MLV)-based γ-retroviral vector. The trial was initiated in France and the United Kingdom in 1999 for X-linked severe combined immunodeficiency (SCID). Twenty patients 56 –58 had their hematopoietic stem cells collected, transduced with a γ-retroviral vector ex vivo, and then returned back to the patients. The vector expressed the common cytokine receptor γ chain (γc) driven off an long terminal repeat (LTR) promoter. Of note, this disease model was an attractive early target for gene therapy because corrected cells had a selective survival advantage. 59 –61 Although the initial results were very promising, 56 –58 over the next few years, 6 of the 20 patients developed clonal T cell lymphoproliferations, resulting in a leukemia-like disease. 62 –65 One patient died, but the others successfully responded to cancer treatments. Of patients who experienced clonal T cell lymphoproliferation, insertions mapped to proto-oncogenes (LMO2, BMI1, CCND2). 62,63,66
Similar serious adverse events were observed in clinical trials using γ-retroviral vectors for two other immunodeficiencies, X-linked chronic granulomatous disease (X-CGD) and Wiskott–Aldrich syndrome. Insertional mutagenesis in three people treated for X-CGD led to myelodysplasia 67,68 and seven patients treated for WAS developed acute leukemias. 69 Although oncogene-activating insertional mutagenesis is a serious unintentional consequence of treatment, a great number of patients from these trials continue to benefit from the treatment 20 years later. 70 The pathophysiology of an immunodeficiency and the selective advantage conferred to corrected cells likely facilitated clonal expansion of cells with integrations near oncogenes.
The adverse events in the X-linked SCID clinical trials led to a vigorous pursuit of an improved integrating vector design. For example, improved retroviral vector safety features now include SIN features that lack enhancer/promoter sequences. 71,72 Since additional safety modifications were introduced, there have been zero reported cancers in over 40 treated patients, 73 –77 suggesting that the risk of insertional mutagenesis using γ-retroviral vectors has been reduced. Indeed, Strimvelis, a γ-retroviral vector for the ex vivo treatment of adenosine-deaminase deficiency, was approved by the European Medicines Agency in 2016.
Viral vector chromosomal integrations have been mapped in multiple cell lines, species, and tissues (reviewed in Refs. 78,79 ). With good agreement, γ-retroviral integration is enriched near enhancer and promoter regions of actively transcribed genes. 80,81 Lentiviral vectors are also enriched in transcribed genes but are predominantly found in introns. 82 Clearly, integration site preference is virus-specific. 80,83 –86 Whenever DNA is introduced into a cell, there is (1) a finite probability for insertional mutagenesis, (2) a smaller probability for activation of oncogenes, and (3) a smaller probability of clonal proliferation leading to cancer. Vectors that intentionally insert their DNA cargo into the host cell chromosome will have an increased probability for an adverse event. However, to date, no lentiviral or AAV-based vector has resulted in oncogenesis in a clinical trial. Montini et al. demonstrated that lentiviral integrations, even at high vector titer loads, did not accelerate tumorigenesis in tumor-prone mice. In contrast, γ-retroviral vector transduction triggered a dose-dependent acceleration of tumor onset. 87 The evidence to date suggests that despite careful attention, clonal expansion of corrected cells has not been observed with modern integrating vectors. However, novel integrating vectors are breaking new ground in terms of integration mechanisms. Integration mapping and delivery to tumor-prone mice are approaches to remain vigilant to the risks.
Compared with retroviral or lentiviral vectors, much less effort has been afforded to evaluating the risk of insertional mutagenesis from AAV vectors. Recombinant AAV integrations can cause hepatocellular carcinoma in mice. These integrations were mapped to the Rian locus and associated with age, dose, promoter/enhancer selection, and capsid choice. 88 In total, AAV2 posed the greatest risk of insertional mutagenesis. 89 In a large study of 695 mice evaluating serotypes 1, 2, 5, 7, 8, and 9, only one liver tumor was observed resulting in an incidence of 0.14%. 90 To date, there have been no reports of AAV-induced tumor formation in humans; however, insertional mutagenesis is a potential risk when DNA is delivered to cells.
Recombinant AAV integrations
Considerable effort has been dedicated to mapping recombinant AAV vector integration patterns. An early large-scale analysis of AAV vector integration was performed with a vector capable of recovery in the form of a bacterial plasmid after restriction enzyme digestion of isolated cellular genomic DNA from transduced human primary fibroblasts. The authors obtained 977 unique integration junctions between ITR and host chromosomal DNA. Hotspots of AAV integration included CpG islands and ribosomal DNA repeats, with the latter accounting for up to 8% of total integration sites. 91 Subsequent studies mapped integration sites in mouse livers in vivo. 92,93 The mouse studies confirmed that integrations occur in vivo and preferentially target transcriptionally active genes. Inagaki et al. mapped ∼1,000 integration sites from the liver, heart, and skeletal muscle of mice. 94 These and other studies (reviewed in Ref. 95 ) suggest that nonhomologous integration of AAV vectors is nonspecific but not truly random.
Preferential integration in certain chromosomal regions suggests a mechanism dependent on host factors. A key integration step is the availability of free chromosomal ends that can ligate to the AAV genome. Initial reports suggested that AAV vectors can cause chromosomal double-strand breaks; however, later studies revealed that the formation of double-strand breaks was the rate-limiting step for integration. 96 Thus, AAV integration occurs in chromosomal regions prone to genomic instability, such as CpG islands, ribosomal DNA repeats, palindromes, and transcription start sites. 97 –99 AAV integration prefers sites of double-strand breakage. 91,96,100 In summary, the evidence suggests that AAV vectors do not cause chromosomal breaks but integrate at pre-existing chromosomal damage, providing free DNA ends for the host cell machinery to initiate nonhomologous end-joining repair.
Mapping AAV-delivered DNA transposons
Using linear amplification-mediated PCR (LAM-PCR), Sleeping Beauty/AAV transposition events (1,840 integration events in total) were mapped in mammalian cells resulting in a near random integration profile. 101 Similarly, we mapped piggyBac/AAV transposase-mediated integrations in HeLa cells in vitro or mice in vivo. 42 Recovered integrations from piggyBac/AAV-transduced HeLa cells were compared with reads recovered from HeLa cells transfected with standard piggyBac transposon and transposase expression plasmids. Only verified genomic integrations that included the junction between the TR and chromosomal DNA were included in the analyses. A subtle difference in the integration profile was observed following a comparison of piggyBac/AAV integrations, plasmid piggyBac integrations, and computationally selected matched random controls. PiggyBac/AAV showed a very modest increase in integration near gene-dense regions or CpG islands and a modest decrease in integrations near transcriptional start sites. 43 To date, chromosomal mapping analyses of AAV-delivered transposons suggest a nonspecific pattern of integration, similar to traditional transposition systems. 43
Targeting Stem Cells
The ability of a viral vector to integrate into the host cell DNA alone is unlikely to provide a lifelong therapeutic benefit in nonquiescent tissues. A stem cell that can self-renew, divide, and differentiate would need to be transduced. Naturally occurring AAV variants are often inefficient at transducing many cell types, particularly stem cells. 102 –104 In truth, all viral vectors have room for improvement, including their transduction efficiency, cellular tropism, and species tropism.
Directed evolution can be applied to natural viruses or viral proteins to fine-tune their functions for specific cell or tissue applications. Directed evolution of AAV capsids is a common strategy for generating novel capsids for targeting cell or tissue types (reviewed in Ref. 105 ). Indeed, multiple studies demonstrate that directed evolution yields novel AAV variants that enhance gene delivery and gene targeting in stem cells. Asuri et al. used this approach to create a novel AAV mutant to more efficiently transduce human pluripotent stem cells. 104 This variant termed AAV1.9 harbors a single R459G mutation in the AAV2 capsid. Similarly, Jang et al. created a novel AAV serotype, AAV r3.45, through directed evolution, that is proficient at infecting both murine and human neuronal stem cells (NSCs), while maintaining the cell progenitor markers indicating retention of cell immaturity. 102 This variant, derived from AAV2, carried a [LATQVGQKTA] peptide insertion within the heparin binding domain along with a V719M mutation. This allowed for a 14–50-fold increase in transduction efficiency of NSCs compared with AAV2 or AAV5. 102
Another study highlighting the benefits of vector serotype optimization on stem cell tropism found that two mutations (Y731F, Y445F) of the seven exposed tyrosine residues of AAV6 provided a significant increase in transduction efficiency within human CD34+ hematopoietic stem cells. 106 When these novel serotypes transduced human hematopoietic stem cells and were subsequently transplanted to a nonobese diabetic/SCID xenograft mouse model, vector persistence was found to be 11.2–12.7% from the tyrosine mutant transduced cells, whereas wild-type vector-treated cells ranged from 0.3% for AAV2 to 6.5% for AAV6. 106
Transduction of progenitor cells with the capacity to repopulate the surface airway epithelium has important implications for cystic fibrosis gene therapy. Compelling evidence from both in vitro and in vivo studies indicate that basal cells are multipotent proximal airway progenitor cells that repopulate pulmonary epithelia (reviewed in Refs. 107 –109 ). However, tracheal, bronchial, and alveolar epithelia are likely maintained by regionally distinct progenitor cell lineages. Different AAV serotypes may transduce specific airway cells with varying efficacy, and thus, screening for serotype tropism or directed evolution are potential strategies to maximize transduction efficiency. We asked if there were AAV serotypes with tropism for airway basal cells. Primary cultures of bronchial basal cells were freshly isolated from human lung donor tissue. The population of primary basal cells was confirmed to be ∼98% pure based on expression of the basal cell marker cytokeratin-5. 110 By expressing the reporter GFP, we compared seven different naturally occurring AAV serotypes and three AAVs previously generated by directed evolution for the ability to transduce primary basal cells (Fig. 4). Of the naturally occurring serotypes, AAV1, AAV2, and AAV6 resulted in the greatest number of GFP+ cells. However, AAV6.2, a product of directed evolution, 111 clearly resulted in the best levels of transduction. Interestingly, AAV2.5t showed poor transduction efficiency in primary human airway basal cells, even though this serotype has excellent transduction properties in well-differentiated primary human airway cells. 112

The indicated GFP-expressing AAVs were used to transduce human primary basal cells from human donor lungs (multiplicity of infection = 10,000). Cells were pretreated with doxorubicin and N-acetyl-L-leucyl-L-leucyl-L-norleucinal proteasome inhibitors for 2 h before transduction. Three days postdelivery, GFP expression was documented using an inverted fluorescent microscope and a 20 × objective. GFP, green fluorescent protein.
Conclusions
Undeniably, gene therapy holds enormous therapeutic potential for a wide variety of genetic diseases. A gene delivery vehicle should (1) efficiently, (2) safely, and (3) persistently express a therapeutic transgene in an appropriate cell type.
Efficiency
An abundance of preclinical and clinical studies describe methods for AAV production and purification, improvements in expression cassettes, generation of more efficient serotypes, and improved in vivo delivery techniques. AAV vector products have successfully progressed from early-stage development, through clinical trials, and emerged as FDA-approved therapeutics. Of course, gene delivery efficiency needs to be defined for every tissue and transgene cargo, but there is a clear precedence for successful gene delivery to a wide array of targets.
Safety
There are multiple factors to consider when designing integrating vectors, including (1) insertion site preference or prevalence of off-target effects; (2) neighborhood effects on gene expression; (3) conferred selective survival advantage of corrected cells; and (4) adaptive or innate immune responses to the vector capsid or transgene products. The three gene therapy clinical trials resulting in oncogenesis from insertional mutagenesis involved immunodeficiencies and first-generation MLV-based vectors harboring LTRs with strong enhancer/promoter sequences. Thus, the vector and disease setting will influence the risks for insertional mutagenesis and subsequent clonal expansion. Lentiviral vectors are transitioning from primarily an ex vivo delivery option to direct in vivo gene therapies depending on the disease model. As such, lentiviral vectors may pave the way for a next generation of integrating AAV vectors. With any novel vector configuration, there is a need to discern integration patterns and risk of cellular transformation; however, integrating vectors should not be dismissed out of hand.
Persistence in an appropriate cell type
If persistence is defined as a lifelong expression from a single dose, then genomic integration into a stem cell population may be a requirement for nonquiescent tissues. Efficient gene correction of pluripotent stem cells has the important benefits of self-renewal and differentiation into the appropriate lineages for therapeutic correction. Successful AAV gene therapy treatments have been achieved in cells with a low turnover. However, even in terminally differentiated cells with long half-lives, ultimate waning of expression is likely inevitable. In cells with short half-lives, the waning of expression will be accelerated. As discussed, there are multiple strategies to achieve genomic integration using AAV-based vectors.
The proven utility and safety profile of AAV-based gene therapy vectors hold great therapeutic promise for monogenic diseases. With the development of integrating AAV vectors, genomic therapy has added potential for a lasting therapy. Genome modifiers such as transposable elements and editing tools will continue to push the envelope of the efficacy of AAV gene therapy. In conclusion, integrating a therapeutic transgene to the host genome using an AAV vector enriches the gene delivery toolbox and provides novel solutions to the challenge of a lifelong curative strategy for genetic disease.
Footnotes
Acknowledgments
We thank Miguel Ortiz, Ambur Vu, Laura Marquez Loza, Brajesh Singh, and Rosarie Tudas for their critical review of the article. The AAV vector was produced at the University of Iowa Viral Vector Core.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by the National Institutes of Health [NIH P01 HL-51670, NIH R01 HL-133089]; the Center for Gene Therapy of Cystic Fibrosis [NIH P30 DK-054759]; and the Cystic Fibrosis Foundation [SINN19XX0, COONEY18F0].