
Abstract
Progress in antiretroviral therapy has considerably reduced mortality and notably improved the quality of life of individuals infected with HIV since the pandemic began some 40 years ago. However, drug resistance, treatment-associated toxicity, adherence to medication, and the need for lifelong therapy have remained major challenges. While the development of an HIV vaccine has remained elusive, considerable progress in developing innovative cell and gene therapies to treat HIV infection has been made. This includes immune cell therapies, such as chimeric antigen receptor T cells to target HIV infected cells, as well as gene therapies and genome editing strategies to render the patient's immune system resistant to HIV. Nonetheless, all of these attempts to achieve a functional cure in HIV patients have failed thus far. This review introduces the clinical as well as the technical challenges of treating HIV infection, and summarizes the most promising cell and gene therapy concepts that have aspired to bring about functional cure for people living with HIV. It further discusses socioeconomic aspects as well as future directions for developing cell and gene therapies with a potential to be an effective one-time treatment with minimal toxicity.
Introduction
The development of antiretroviral therapy (ART) has started in the late 1990s and gradually transformed infection with HIV from a fatal condition into a manageable chronic disorder. However, the inability of ART to eradicate latent HIV reservoirs entails that drug treatment must be continuous and lifelong. Accordingly, major efforts in the advancement of novel HIV treatments aim at reducing dependence on lifelong multidrug therapy by designing medicines that induce a functional cure. Along with approaches based on vaccination or broadly neutralizing HIV-specific antibodies, cell and gene therapies have been at the forefront of new strategies that strive for viral clearance. This review summarizes the most promising cell and gene therapy concepts that target the virus lifecycle to bring about functional cure for people living with HIV.
The HIV
Since first patients with AIDS were reported in 1981, HIV infection has developed into one of the greatest pandemics in modern times with devastating socioeconomic and demographic consequences. Currently, a total of 75 million people has become infected with HIV worldwide and 32 million have died of AIDS. In 2019, 1.7 million individuals became newly infected and some 38 million live with HIV, of whom not all have access to ART (
HIV type 1 (HIV-1), and the much rarer HIV-2, are retroviruses belonging to the family of lentiviruses. They infect CD4 T lymphocytes, regulatory T cells, monocytes, macrophages, and dendritic cells expressing the glycoprotein CD4, the main receptor of HIV-1 and HIV-2. After binding to CD4, the interaction with a coreceptor is needed to mediate membrane fusion of the viral membrane with the host cell membrane (Fig. 1A). The clinically relevant coreceptors are C-C chemokine motif receptor type 5 (CCR5) and C-X-C chemokine receptor type 4 (CXCR4). 1 Coreceptor usage determines whether the virus is considered a macrophage (M)-tropic strain (CCR5-dependent, also called R5-tropic) or a T lymphocyte (T)-tropic strain (CXCR4-dependent, also called X4-tropic). 2 Some strains are dual tropic and can use either chemokine receptor to enter the cells. M-tropic viruses are the predominant strain responsible for HIV transmission and found in early stages of the infection, whereas X4-tropic strains are associated with rapid depletion of CD4 T lymphocytes, causing accelerated disease progression and found in late stages of the infection. 3
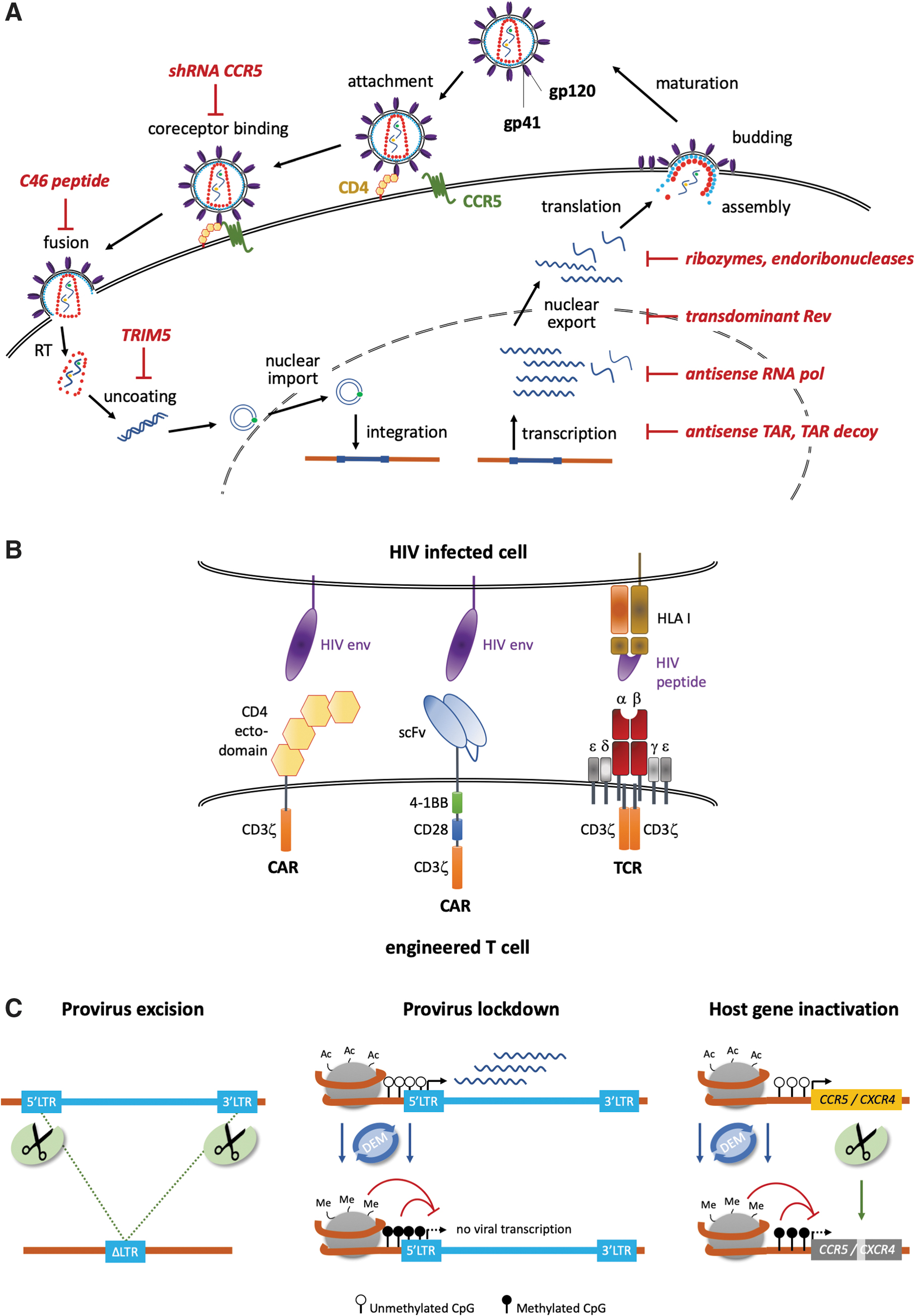
Strategies. The gene therapy approaches to treat HIV can be subdivided in defensive, offensive, and targeted strategies.
It is well established that a naturally occurring 32 nucleotide long deletion (Δ32) in the CCR5 locus leads to resistance to R5-tropic HIV-1 strains. 1,4 People with a homozygous Δ32 mutation live a normal life and are clinically unremarkable. 5 The Δ32 mutation is rare throughout the world and only found more frequently in North-Eastern Europe where allelic frequencies reach 10%. 6
Following membrane fusion, the viral capsid disintegrates and the viral RNA is released into the cytoplasm of the target cell, where the viral reverse transcriptase (RT) transcribes the viral RNA into proviral DNA. After transportation of the proviral HIV DNA to the nucleus, the viral integrase mediates integration of the HIV DNA into the host cell genome. In productively infected cells, the cell's normal transcription machinery produces both viral messengers RNA (mRNA) and genomic RNA, using the integrated HIV provirus as a template. The viral mRNA is translated into long polyprotein chains, which are cleaved by the viral protease into functional proteins, resulting in the assembly of infectious viral particles that bud from the host cell membrane.
Acute HIV-1 infection is characterized by high levels of viral replication, a sharp drop in peripheral blood CD4 T lymphocytes, and the establishment of a reservoir of latently infected CD4 T cells. Symptomatic individuals with acute HIV-1 infection may present with mononucleosis-like disease, but symptoms may be absent in up to 60% of cases. 7 Following acute infection, patients enter an often asymptomatic period of chronic HIV infection with relative stabile viremia and progressive decline of CD4 T lymphocyte counts. 8 After an average time of 8–10 years after HIV infection, patients exhibit profound immunodeficiency with CD4 T lymphocyte counts below 200 cells/μL, putting them at risk for life-threatening opportunistic infections.
ART can effectively control virus replication. If accessible, life expectancy of people living with HIV and responding to ART is similar to the general population, mainly as a result of substantial reduction of AIDS-related and non-AIDS-related mortality. 9 Entry inhibitors, nucleoside or nucleotide reverse transcriptase inhibitors (NRTI), non-nucleosidic reverse transcriptase inhibitors (NNRTI), protease inhibitors (PI), and integrase inhibitors (INSTI), are components of ART that target different steps in the HIV life cycle. First-line ART classically consists of three drugs (typically two NRTI + one INSTI, PI, or NNRTI) and is generally well tolerated. Initiating ART reduces plasma viremia below detection limit of routine assays, leading to increasing counts of CD4 T cells, which is associated with the restoration of the immune system. On the other hand, ART has important limitations: it is a livelong therapy, requiring 90% adherence to avoid failure and emergence of drug-resistant virus. 10 Moreover, ART has side effects, including bone or renal toxicity, dyslipidemia, insulin resistance, and accelerated cardiovascular disease. It is important to note, too, that ART fails to eradicate HIV, and cessation of ART is followed by viral rebound within weeks in the majority of patients. 11
Approaching Cure
The latent HIV-1 reservoir is perceived as the main obstacle for a curative approach in HIV-1 therapy. 12 The reservoir mainly consists of CD4 T lymphocytes, dendritic cells, and macrophages that are latently infected with transcriptionally inactive, but replication-competent HIV-1 proviruses. 13 The contribution of microglia and astrocytes to the latent HIV-1 reservoir is controversial. 14 The reservoir cells typically reside in tissues where ART has poor penetration, such as the central nervous system 15 –17 and the gut-associated lymphoid tissue (GALT). 18 Maintenance of the cellular HIV reservoir has been primarily attributed to the homeostatic clonal expansion of the latently infected T cell pool. It was shown that HIV provirus integration was preferentially found in genes associated with cancer or cell cycle regulation. On the other hand, it is important to mention that HIV does not preferentially integrate in such loci, but rather that the integration in these loci confers a selective proliferative advantage, so contributing to the expansion and persistence of those cells. 19 –21 While the molecular mechanisms leading to HIV latency are still not completely understood, but typically involve the formation of repressive chromatin surrounding the proviral integration site, 22 the latently infected cell reservoir was shown to be very stable with a half-life of 44 months on suppressive ART, making eradication of the reservoir with regular ART unlikely. 23
Early attempts to reduce the size of the latent reservoir were based on intensifying ART through the addition of integrase or entry inhibitors, but did not result in meaningful effects. 24 On the other hand, initiating ART very early during acute HIV-1 infection has been associated with a reduced reservoir size. In the so-called viro-immunological sustained control after treatment interruption (VISCONTI) cohort, 25 14 people living with HIV started a prolonged ART in the 1st month after infection. They had reduced HIV blood reservoirs and controlled HIV-1 replication for several years after cessation of ART. 25 Nonetheless, isolated reduction of reservoir sizes, even if radical, appears to be insufficient to achieve cure in most of HIV-1-infected individuals. A cohort study in Thailand included 8 people living with HIV, who started ART during earliest phase of HIV-1 infection. Although the test subjects had remarkably low HIV-1 reservoir sizes, all participants experienced viral rebound after analytical treatment interruption (ATI). 26 A further example is given by a case report of two men with HIV-1 infection on suppressive ART, who underwent allogeneic hematopoietic stem cell transplantation (HSCT) to treat hematologic malignancy. 27 Following allogeneic HSCT, HIV-1 DNA was undetectable in blood and gut tissue specimens, but both patients experienced a late viral rebound 84 and 225 days after ATI. A similar case report was reported in 201728: a 55-year-old man suffering from B-lineage acute lymphoblastic leukemia. Following allogeneic HSCT, HIV remained undetectable and the patient underwent ATI. He experienced viral rebound 288 days later, suggesting that allogeneic HSCT can reduce the viral reservoir size and HIV replication for a limited amount of time.
The so-called “shock & kill” strategies aim at reactivating HIV in the latent reservoir to ultimately eliminate all infected cells. 29 Even though promising, an increasing body of evidence suggests that this strategy has major limitations in both the “shock” and the “kill” aspects. First, HIV reactivation is a largely stochastic phenomenon, making it unlikely that all integrated proviruses are transcriptionally activated simultaneously. Second, the likelihood of “killing” the entire reservoir is slim, considering the size of the reservoir and the failure to remove latently infected cells in clinical trials. 30 Another study in which HDAC inhibitors were used to broadly activate host gene expression resulted in re-activation of HIV proviral transcription, but the HIV positive cells persisted. 31 In summary, conventional strategies with the goal to reduce the size of the latent HIV reservoir have been unsuccessful thus far.
Cure is Possible
The multiple strategies to cure HIV infection can be roughly subdivided in two groups: a “sterilizing cure” refers to the complete eradication of HIV-1 from the patient's body, while a “functional cure” aims at long-term control of HIV replication with preservation of CD4 T lymphocyte counts in the absence of ART. To date, two individuals, the so-called “Berlin patient” and “London patient,” have been reported to be cured from HIV after allogenic HSCT. In both cases, the patients received a stem cell graft of donors with a homozygous CCR5Δ32 mutation. 32,33 A third putatively cured patient, the “Düsseldorf patient,” was presented at conferences. 34 All patients suffered from a hematologic malignancy constituting the indication for allogenic HSCT. After ART was discontinued, patients were closely monitored for HIV persistence, but no sign of viral RNA or proviral DNA was detected to date. 33 –35 These cases illustrate that it is possible to transfer HIV resistance from a donor to a patient and to achieve sterilizing cure. However, the essential factors contributing to successful HIV cure remain elusive. In allogenic HSCT, the reconstitution of an intact immune system is preceded by a conditioning regimen suppressing the malignant disease, depleting the autologous hematopoietic stem cells (HSCs) and lymphocytes, as well as a graft-versus-host prophylaxis preventing an overreaction of the grafted immune system. The Berlin patient received two allogenic HSCT within 13 months; conditioning regimens consisted of polychemotherapy (fludarabine, cytarabine, and amsacrine) succeeded by cyclophosphamide and total body irradiation (TBI) before first graft, and gemtuzumab ozogamicin, fludarabine, and TBI before second graft. For graft-versus-host prophylaxis anti-thymocyte globulin, cyclosporine A and mycophenolate were used. The London patient received polychemotherapy (lomustine, cyclophosphamide, cytarabine, and etoposide) as conditioning regimen and the anti-CD52 antibody alemtuzumab combined with cyclosporine A and methotrexate for T cell depletion and graft-versus-host prophylaxis. Upon reactivation of Epstein–Barr virus after HSCT, he received the anti-CD20 antibody rituximab. Both patients experienced graft-versus-host disease, which may mediate depletion of the “autologous HIV reservoir.” It is hence likely that the intense conditioning regimens facilitated both the eradication of HIV reservoirs 36 and the reconstitution of an HIV-resistant CCR5Δ32 immune system, so enabling the cure of these patients. The study by Kordelas et al. highlights the importance of screening such patients for the absence of X4-tropic virus. 37 Following allogeneic HSCT with a Δ32 graft, a patient experienced rebound of X4-tropic HIV during ATI. Beside these reported cases, there have been more attempts with allogeneic CCR5Δ32 HSCT. Due to transplantation-related issues or secondary infections, these patients deceased before ATI could be performed. 38 In any case, the limited availability of human leukocyte antigen-matched CCR5Δ32 donors and the side effects of allogeneic HSCT preclude a broader use of this approach. Even if in the future, chemotherapy-free and radiation-free conditioning regimens 39 –41 might be available to bypass some of the severe adverse effects, these novel approaches need to be combined with an effective depletion of the HIV reservoir and genetically engineered autologous HSCs to render the immune system resistant to HIV.
HIV Gene Therapy Trials: A Retrospective Analysis
Various academic laboratories and biotechnology companies have developed innovative approaches to fight HIV infection based on gene transfer or genome editing. The first step in an autologous gene therapy scenario consists of isolating CD4 T cells or CD34 HSCs from the HIV-positive individual. Subsequently, these cells are rendered HIV resistant through a genetic intervention, either by retroviral/lentiviral gene transfer or targeted genome editing. Alternatively, the cells are equipped with additional features to eliminate HIV-infected cells.
Since 1994, 36 clinical trials have been registered on
Gene therapy clinical trials targeting HIV
CAR, chimeric antigen receptor; CCR5, C-C chemokine motif receptor type 5; mRNA, messenger RNA; n.a., not available; NCT, national clinical trial; shRNA, short hairpin RNA; ssRNA, single-stranded RNA; TAR, trans-activation response element; TCR, T cell receptor; TRIM5α, tripartite motif-containing protein 5 alpha; ZFNs, zinc finger nucleases.
Adoptive transfer of chimeric antigen receptor T cells or T cells with affinity-enhanced T cell receptors
The first gene therapy trial, initiated in 1994, was based on the adoptive transfer of T cells that were equipped with a kind of chimeric antigen receptor (CAR) targeting HIV-infected cells (Fig. 1B). The CAR was composed of the extracellular and transmembrane domain of CD4 linked to the intracellular ζ-signaling chain of CD3. 42,43 In both studies neither an impact on the viral reservoirs nor significant changes in plasma HIV RNA or proviral DNA levels were observed. Even so, trafficking to gut mucosa and stable persistence of the gene-modified T cells in blood were reported. Persistence of the CD4ζ-CAR T cells was observed for up to 11 years after infusion, with an estimated half-life of those cells of >16 years. 44 Multiple reasons and technical issues might have limited the efficacy of this early CAR T cell approach, like suboptimal CAR design, low transduction efficiency of cells with the used gene transfer vector, and poor ex vivo manipulation of cells. Today, these technical issues have been solved and more efficacious CAR T cells can be manufactured. A third-generation CAR that contained a single-chain variable fragment recognizing the CD4-binding pocket of numerous HIV-1 isolates 45,46 was used in the most recent trial (NCT03240328). No clinical results were published thus far, but the efficacy of these novel CAR T cells was demonstrated in a preclinical setting ex vivo. 47 Another trial is evaluating a combinatorial approach that combines CD4 CAR T cells with CCR5 gene disruption (NCT03617198). A different study (NCT00991224) has planned to evaluate the effects of transferring CD8 T cells modified with a Gag-targeting T cell receptor (TCR). Two different types of TCRs have been included: the natural form and an affinity-enhanced version targeting the Gag p17 epitope. Due to reported severe adverse events in an unrelated trial, in which two patients deceased due to off-target effects using an affinity-enhanced TCR, 48 this trial was closed before any patient was recruited. Recently, a novel approach based on concomitant expression of two different CD4-CARs, which is a combination of the CD4 ectodomain with either the 4-1BB/CD3ζ or CD28/CD3ζ endodomain, showed promising results in preventing CD4+ T cell loss in a humanized mouse model. 49 While these new generations of CAR T cells were able to lyse HIV-1-infected CD4+ T lymphocytes, it is still not clear how to activate these deep reservoirs in patients using currently available methods.
Fusion inhibitors
An interesting approach is the use of peptide inhibitors to prevent viral entry. In 2001, it was reported that the expression of membrane-anchored peptides derived from HIV-1 gp41 inhibited the fusion of HIV-1 with the host cell membrane 50 (Fig. 1A). Several trials have engaged this tactic and used constitutive expression of the fusion inhibitor peptide C46 either alone or in combination with a small interfering RNA that downregulates CCR5 expression. In a first trial, 10 patients were transplanted with CD4 T lymphocytes expressing the C46 peptide. Contrary to the in vitro results, there was no selective advantage of cells harboring the C46 peptide in vivo. The low levels of gene marking (<0.01% of total leukocytes) and suboptimal manufacturing conditions might have contributed to the missing impact on HIV load. 51 Other studies entailed the combined infusion of CD4 and CD34 cells, both modified to express C46 and a short hairpin RNA (shRNA) targeting CCR5 expression. While the results have not been published yet, the preclinical studies demonstrated potency in nonhuman primate and murine models. Transplantation led to long-term, multilineage engraftment of the modified CD34 cells, and positive selection of marked cells was observed after challenge with HIV. 52 –54 A detailed clinical protocol was published 55 for the most recent study that is still recruiting patients and that will include ATI under specific circumstances. However, a drawback of these approaches is the need to maintain a sufficiently high transgene expression level, which could trigger an immune response and so negatively impact on the antiviral activity. In fact, it is known that most HIV-infected patients have antibodies against gp41 epitopes. 51,56 While these antibodies are non-neutralizing at the beginning of the infection, 57 they can convert into broadly neutralizing antibodies later on. 58,59 It was shown that this problem can be overcome by the rational design of synthetic peptides that retain their function of inhibiting HIV entry, but show reduced immunogenicity. 60,61
Trans-dominant proteins
Some early trials involved the use of trans-dominant proteins. The viral regulatory proteins Rev and Tat can be reengineered to act in a trans-dominant negative manner 62 (Fig. 1A). Inhibition of HIV-1 replication is achieved by blocking the export of viral RNA from the nucleus of infected cells. In trial NCT00005785, one patient underwent allogenic transplantation with CD34 cells that expressed a trans-dominant negative Rev version. The patient remained in complete remission until she died 3 years later. Because ATI was not included, no data on the efficacy of this approach are available. 63,64 Other trials aim at infusing engineered cells that express trans-dominant negative Rev in combination with either a TAR antisense-RNA or an antisense-RNA targeting the pol transcript. None of these studies, however, reported long-term effects. 65 –69
Ribozymes
Ribozyme approaches were among the first to enter the clinic. Ribozymes are catalytically active RNA molecules designed to cleave the complementary sequences of the target RNA. These molecules have been used widely to target different regions of HIV-170 (Fig. 1A). In a first proof of concept, CD4 T cells were modified to express a tat targeting ribozyme, which was previously shown to suppress HIV infection in vitro. 71,72 Gene marking was low (≤0.1%), but modified cells could be detected up to 6 years after infusion. No mutation in the tat gene was detected in the proviral DNA of patients' cells, suggesting that no escape mutant has developed. 73 Later, infusion of tat ribozyme expressing CD34 cells has proven to be well tolerated, and persistence of those cells was detected up to 2.5 years postinfusion. Analysis of ribozyme target sequence showed that no escape mutant was selected up to week 104. 72 In a phase II study, modified CD34 was infused and two ATIs were performed to enable positive selection of the modified cells (NCT00074997). Although the primary efficacy endpoint was not reached, a significant lower HIV-1 viral load was detected in the treatment cohort. 74
Endoribonucleases
The Escherichia coli-derived MazF endoribonuclease (also known as ChpAK, chromosomal homolog of pem killer) cleaves single-stranded RNA at 5′-ACA sequences, making it an attractive tool to target HIV-1 RNA (Fig. 1A). Although MazF was shown to cleave mammalian mRNA, too, in vitro studies demonstrated that MazF restricts viral replication without cellular toxicity. Preclinical studies demonstrated both its efficacy in impairing HIV replication in vitro, 75 as well as the persistence of MazF-expressing CD4 T lymphocytes in the presence of simian/HIV (SHIV) in a non-human primate model. 76 Preliminary results of a phase I study (NCT01787994) suggested that autologous modified CD4 T cells are safe and able to persist up to 6 months postinfusion with stable CD4 counts.
Antisense strategies
Short and long antisense RNA anneal with complementary viral mRNA and form nonfunctional duplexes that block HIV replication (Fig. 1A). Adoptive T cell therapy was performed in two studies utilizing engineered CD4 T cells that express a 937 nucleotide long antisense RNA complementary to the env gene. In vitro, this antisense strategy exercised selection pressure on the virus and the resulting escape mutants were replication defective. 77 The clinical evaluation of the lentiviral modified CD4 cells expressing this antisense product proved to be safe with no sign of insertional mutagenesis. 78 The modified CD4 T cells engrafted in the GALT and persisted for up to 5 years. Unlike the preclinical data, virus rebound was observed in all patients who underwent ATI due to escape mutations. 79,80
Combinatorial strategies
A trial initiated in 2016 (NCT02797470) combines three approaches in autologous CD34 cells: expression of a shRNA downregulating CCR5 expression to target viral entry, tripartite motif-containing protein 5 alpha (TRIM5α) to prevent viral uncoating, and a TAR decoy targeting viral gene expression. 81 Preclinical studies in xenografted mice showed multilineage hematopoiesis and a selective advantage of the derived human CD4 T cells after challenge with an R5-tropic HIV-1 strain. 82 The trial is still recruiting patients and results have not been published yet.
In another study, a lentiviral vector expressing three antiviral agents, an shRNA targeting tat/rev, an RNA decoy targeting TAR, and a ribozyme targeting CCR5 expression, was used to transduce CD34 cells. Four patients received modified CD34 to treat an AIDS-related lymphoma, and gene marking was detected in multiple lineages up to 24 months postinfusion. 83 Grafts were well tolerated and subjects are in remission, but better transduction rates may be needed to reach a therapeutic effect.
Genome Editing: The Not-So-New Kid in Town
In the context of HIV, all major classes of programmable nucleases have been employed: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-Cas nucleases. 84 While ZFNs and TALENs are purely protein-based enzymes, CRISPR-Cas complexes are RNA-guided nucleases. A short RNA sequence (guide RNA or gRNA), which is complementary to the target sequence, mediates binding of the Cas nuclease to the target site. 85
An evident approach in genome editing was to excise the integrated HIV provirus from the host genome (Fig. 1C). High excision frequencies, ranging from 40% to 80%, were achieved ex vivo. 86 –88 However, a single designer nuclease is unlikely to recognize the provirus sequences of all HIV strains. This could be resolved by characterizing the pool of proviral genomes present in a patient, followed by the design of a set of gRNAs to target the majority of them. 89 Nonetheless, a major limitation of employing genome editing to remove integrated HIV provirus is the potential of creating escape mutants, resulting, for example, from error-prone repair of the nuclease-induced cut when excision fails. This was observed when using ZFNs or the CRISPR-Cas platform to target the RT gene. 90,91 A multiplexed approach improved the emergence of resistances. 92 In vivo excision of proviral DNA from HIV-infected humanized mice was demonstrated using AAV vectors expressing the CRISPR-Cas system, 93 even to an extent that enabled the clearance of latently infected cells when combined with sequential long-acting slow-effective release antiviral therapy. 94 It will be interesting to see how easily such an approach can be transferred to a human setting. While multiplexing allows for a broader target range, thorough analysis of off-target effects, especially translocations, will be indispensable.
Methods that avoid an intermediate DNA double-strand break represent therefore a valuable alternative. An interesting approach that differs from designer nuclease-based genome editing is engineering with recombinases. These prokaryotic enzymes generally recognize and bind to a short (30–40 bp) target site consisting of a spacer flanked by two palindromic sequences. Upon binding, the sequence between two recognition sites is excised. 95 Designer recombinases, like the Cre-based Brec1, were engineered in vitro to recognize long terminal repeat (LTR) sequences to drive the excision of the provirus from latently infected cells and have raised great interest, 96 and clinical translation is imminent.
More recently, the RNA-targeting CRISPR-Cas13a system has been presented as a tool to inactivate HIV RNA. 97 This system relies on the persistent expression of an active surveillance complex capable of recognizing and destroying the viral RNA. While this innovative approach opens new avenues in preventing the spread of RNA viruses in general, immunological reaction against the Cas13a protein and the reported off-target activity of CRISPR-Cas13a need to be addressed before clinical translation.
An alternative strategy follows the principle of HIV eradication in the Berlin patient. 98 Several studies have employed designer nucleases to knock out CCR5 (Fig. 1C). Early preclinical studies used ZFNs to disrupt the CCR5 locus either in CD4 T cells or CD34 cells. CCR5 knockout resulted in significant reductions of viral replication and virus load in xenografted mouse models 99,100 and nonhuman primates. 101 Importantly, the latter study demonstrated that CCR5-edited T cells derived from edited stem cell grafts traffic to and reduce the size of the viral reservoirs in secondary tissue sites. Editing of CD34 cells therefore represents an ideal treatment scenario as CCR5-edited stem cells will give rise to all lineages of the blood and immune system, including CD4-positive T cells and macrophages. On the other hand, severe side effects associated with myeloablative conditioning regimens, as outlined above, as well as the higher risk of genotoxicity due to nuclease related off-target effects must be considered in a balanced risk assessment. The first clinical results of infusing CCR5-edited CD4 T cells were published in 2014. 102 Transfusion was well tolerated and the modified cells homed to the gut mucosa and persisted long term. However, during ATI, the survival advantage of CCR5-edited T lymphocytes was minor, and virus rebound was delayed in a single subject who was heterozygous for the CCR5Δ32 mutation. Frequency of CCR5 disruption in the transplanted graft varied between 11% and 24%, which might be too low to sustain an antagonistic effect on viral replication. Another study (NCT02500849) has evaluated the use of ZFN to modify CD34 cells. Results have not been published yet, but targeting frequencies measured in manufacturing validation runs reached up to 70% gene disruption. A drawback of this study is the reported high off-target activities at the CCR2 gene and other loci (>20% of alleles), suggesting a relevant genotoxic potential. 103 A similar study was performed in an HIV-positive patient with acute lymphocytic leukemia, who was transplanted with CRISPR-Cas9-modified CD34 cells (NCT03164135). The CCR5 editing frequency in the graft was around 18% and was further diluted by the concomitant transplantation of unedited stem cells. Not surprisingly, virus rebound was observed during ATI. 104 In order of such an approach to be successful, CCR5 editing in CD34 cells must be both highly efficient and highly specific, which can be achieved by TALEN and CRISPR-Cas nucleases. 105,106 A mathematical model predicts that >50% biallelic knockout is necessary to provide the patient with a sufficient number of HIV-resistant immune cells. 107
Of note, synthetic HIV resistance through CCR5 disruption does not prevent the infection of those cells with CXCR4-tropic virus strains. For instance, X4-tropic strains were shown to rebound in a patient after allogenic transplantation of a Δ32 homozygous graft. 37 Although strategies that consider the simultaneous disruption of both loci are warranted, CXCR4 knockout in CD34 cells is intolerable because the receptor is, among other things, essential for homing to the bone marrow. On the other hand, the fact that CXCR4 function is dispensable in T cells has prompted strategies to disrupt CXCR4—alone or in combination with CCR5—in human CD4 T cells. 108 –110 These studies provided first in vitro evidence that broad resistance to multiple HIV types can be attained, but concomitant generation of multiple DNA cuts considerably increase the probability of translocations and thus the risks of malignancies.
Activity at unintentional sites, so-called off-target activity, represents a major challenge for the use of designer nucleases. The so-called off-target sites often differ from the desired target sequence in only a few bases. Consequently, high knockout efficiency must be combined with high specificity of the customized nuclease to avoid genotoxic side effects that could lead to transformation, especially in multipotent stem cells. In recent years, immense effort has been made to increase the specificity of programmable nucleases. 111,112 The mutations caused by cuts at off-target sites are particularly problematic if they are located, for example, in tumor suppressor genes as they might represent a first hit toward malignant transformation of the cell. In addition, open DNA ends between the target locus and off-target sites were shown to promote chromosomal rearrangements. 105,113 While differentiated T lymphocytes seem to tolerate chromosomal aberrations well, 114,115 induced translocations in CD34 HSCs can transform these multipotent stem cells and induce leukemia. 116,117
Alternatives to Genome Editing: Epigenome Editing
An alternative molecular treatment strategy is the inhibition of HIV entry or sustained HIV suppression in latently infected cells by means of epigenome editing (Fig. 1C). Such “lockdown” strategies entail to prevent the transcription of either the viral genome or essential cellular host factors through deposition of marks that promote chromatin condensation. Both engineered zinc fingers as well as a catalytically dead CRISPR-Cas variant (CRISPR-dCas) were fused to transcriptional repressor domains and used to target the viral LTRs. 118,119 While the mechanism by which HIV repression was achieved in these studies is still elusive, preventing the binding of factors that promote its transcription may have contributed to the observed results. Since the HIV genome contains multiple CpG islands (regions with a high frequency of CG-dinucleotide sequences), strategies aiming at sustainable methylation of those regions were hypothesized to prevent HIV reactivation as well. Indeed, fusion of a DNA methyltransferase to an engineered zinc finger that targeted the viral LTRs was shown to suppress viral replication in a cellular model of HIV infection. 120 Even though more efforts are necessary to investigate its curative potential in preclinical in vivo models, the results suggest that editing the epigenome in a targeted manner might represent a novel curative paradigm. Importantly, this approach can also be explored to achieve stable silencing of host factors. We and others have shown in primary human T cells that designer epigenome modifiers (DEMs) are capable of depositing repressive epigenetic marks in a targeted manner. 121,122 Moreover, transient DEM expression was sufficient to silence both HIV co-receptors, CCR5 and CXCR4, with high specificity. 122,123 Evaluation of their efficacy in preclinical models of HIV infection will be instrumental to establish the potential of this strategy to reach functional cure in future clinical application.
Concluding Remarks
In the last three decades, substantial progress has been made in developing cell and gene therapies to treat HIV. Nonetheless, no attempt has achieved functional or sterilizing cure in patients with HIV infection thus far. The challenges at hand are numerous: cell-based therapies must take into account the latent virus reservoirs, viral heterogeneity within a single host, virus escape mutants, technical hurdles related to effective gene transfer or genome editing, as well as the in vivo persistence of the genetically modified cells in sufficiently high numbers. The latter includes potential immunogenicity of expressed transgenes and the cytotoxicity associated with the genetic modification. The choice of the genetic target is demanding, too. Even aiming for highly conserved regions of HIV is not always crowned by success.
Given the remarkable advances in cell and gene therapy over the past years, the field is well positioned to address these challenges. Effective gene transfer protocols, improved manufacturing conditions, and smart choice of the starting material to ensure long-term survival of the modified cells after infusion, clever targeting that comprises tailored adjustment to cover 100% of an individual's viral repertoire, innovative genome editing interventions, as well as improved “shock & kill” strategies that combine optimized conditioning regimes with CAR T cell technology are all part of the current tool box that will contribute to increased therapeutic efficiencies of current and future approaches. Analogous to ART, strategies that combine multiple antiviral approaches should be envisaged to avoid escape mutants.
The socioeconomic incentives to push forward certainly exist: currently, the costs for the health care system to treat one HIV-infected patient with conventional therapy exceeds 20,000 € per year. Over the course of a patient's life, this adds up to a significant sum, in addition to the social burden, drug toxicity, compliance challenges, and the resulting development of drug resistance. Even the relatively high up-front costs of a cell therapy can be more economical than conventional ART in the long run, particularly if a one-time treatment approach will prove to be sufficient. Taken together, cell and gene therapies have come a long way, and their immense potential will open new avenues for developing a cure for HIV.
Footnotes
Acknowledgments
We thank all the members of our laboratories for their continuous support and critical discussions.
Author Disclosure
T.I.C. and
Funding Information
Research in the laboratories of T.I.C., C.M., and