
Abstract
To test the effectiveness of repeat dosing, we sprayed two doses (1013 vg each) of AAV1Δ27-264-CFTR into airways of four rhesus monkeys at 0 and 30 days, followed by a single dose of 1013 vg of AAV1GFP on day 60. Monkeys were sacrificed on day 90. No adverse events occurred, indicating that AAV1 vectors are safe. An elevated anti-AAV1 neutralizing titer was established by the third dose. A positive ELISPOT to the adeno-associated virus (AAV) capsid but not to cystic fibrosis transmembrane conductance regulator (CFTR) occurred after the third dose in three monkeys. AAV1-CFTR and GFP vectors were detectable in all lung sections and in the heart, liver, and spleen. The CFTR protein was higher in treated monkeys than in an untreated monkey. GFP protein was detected in treated lungs. Lung surface and keratin 5-positive basal cells showed higher CFTR staining than in the uninfected monkey and were positive for GFP staining, indicating widespread gene transduction by AAV1CFTR and GFP. AAV1 safely and effectively transduces monkey airway and basal cells. Both the significant numbers of vector genomes and transduction from AAV1CFTR and GFP virus seen in the monkeys 3 months after the first instillation suggest that repeat dosing with AAV1-based vectors is achievable.
Introduction
Adeno-associated virus gene therapy
Adeno-associated virus (AAV) vector-based gene therapy continues to be developed for a number of genetic disorders, including cystic fibrosis (CF). 1 It has taken several years to overcome the well-documented challenges 2 –4 experienced in moving from a laboratory concept to approved therapy. The field achieved a milestone with the FDA approval of an AAV-based treatment for the RPE65 mutation associated with retinal dystrophy. This treatment is based on the AAV2 serotype and is a single-use therapy. 5,6 AAV gene therapy vectors are well known to persist for extended periods, 7 particularly in cells that do not have high rates of turnover. Thus, for some disorders such as retinal dystrophy, a single dose is therapeutic, but for other disorders such as CF, single treatments will not be effective in the long term.
Cystic fibrosis
CF 8,9 is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR), which functions as a chloride and bicarbonate conducting channel that is important for generating and maintaining mucosal fluids and maintaining the proper composition of sweat. 10 It plays a role in the movement of fluid across the cells that line the passageways of the lungs, pancreas, gastrointestinal tract, and sweat duct. 10 Mutations in the CFTR gene disrupt the function and trafficking of CFTR, 10 causing multisystemic disease featuring disrupted lung function, pancreatic insufficiency, intestinal blockage, and abnormal sweat Cl−. 11 The most common mutation in CF is a missing phenylalanine at position 508 (F508-del). Given the systemic nature of CF, a gene therapy for this disease would have to target several organs whose cells normally turn over, 12,13 and therefore, it requires repeated treatment.
Airway delivery of viral vectors for CF gene therapy
The prospect of gene therapy for CF was advanced soon after the discovery of the gene 14 –16 encoding CFTR. Since then, a number of viral as well as nonviral approaches have been tried (reviewed in Cooney et al. 17 ).
Among all these approaches, AAV-based vectors persist as a promising option because of their lack of toxicity and the number of serotypes that can be used to target various organs. 1 However, it has proven challenging to develop a lung-based strategy for AAV gene therapy. Several preclinical and clinical studies were originally performed using a first-generation AAV2 virus containing CFTR. This viral construct contained full-length CFTR and relied upon a weak promoter endogenous to AAV218; some of the studies yielded positive results, particularly in animals, but most of the clinical trials did not. 1
The first AAV clinical trials for CF conducted in humans involved single-dose administration, with AAV2-CFTR being instilled into one of the maxillary sinuses. 19 Restoration of CFTR function was detected, and no toxicity was observed. This initial positive result led to additional human studies, but unfortunately, in a follow-up study, the rate of sinusitis in the patients did not differ significantly between the placebo and treated groups, and no changes were detected in CFTR function. 20 In another study, AVV2-CFTR was administered to one side of the nose and to the superior segment of the lower lobe of the right lung. 21 Low levels of AAV2 CFTR were detected, but they did not produce any statistically significant differences in CFTR function between the treated and nontreated noses, as assessed by nasal potential difference. In another single-administration trial, AAV2-CFTR (1013 particles) was administered by nebulization to the lungs of CF subjects, and 0.6–0.1 vector copies per cell were detected at 14 and 30 days; however, by day 90, the number of copies per cell had declined to undetectable levels.
The feasibility and safety of repeat dosing were also evaluated in several studies, including repeat-dosing regimens tested in New Zealand white rabbits and rhesus monkeys. 22,23 In these studies, two doses of AAV2-CFTR, followed by a single dose of either AAV2-CFTR or GFP, were instilled into the lungs via a microsprayer. In the rabbits and monkeys, the presence of neutralizing antibodies in the serum, defined in terms of inhibition of wild-type AAV2 replication, increased after the first dose. By the third dose, all the animals had neutralizing antibody titers. However, although we detected GFP expression in both the rabbit and monkey studies, repeated dosing caused a significant drop in vector transduction, suggesting that the presence of neutralizing antibodies had interfered with transduction.
Two repeat-dosing clinical trials were conducted in CF patients, in whom the AAV2-CFTR vector was delivered twice via a nebulizer (1 × 1013 vector particles) at 30-day intervals. In the first study, 24 desired clinical outcomes were obtained, including an increase in forced expiratory volume (FEV1) and a reduction in interleukin-8 in the induced sputum samples. However, a later Phase IIB study of 102 subjects did not meet its primary endpoint for statistically significant improvement over placebo. Although these studies did achieve their positive safety endpoints, the lack of transduction made it clear that it was necessary to further improve on the first-generation AAV2-based vector.
Next-generation AAV vectors
One of the first steps to improve the efficiency of gene transfer was the inclusion of a chicken β-actin (CBA) promoter in the construct to boost CFTR expression. 25 However, given the combined size of the CBA promoter and the full-length CFTR, it was no longer possible for AAV to package the construct into viral particles. Thus, to package CFTR into AAV viral particles, CFTR had to be truncated so as to retain its function. 26 Thus, Sirninger and coworkers 26 developed a truncated CFTR (Δ264 CFTR) that was missing the first four transmembrane segments and showed that this construct was functional in Xenopus oocytes. 27 To improve expression, an additional 27 amino acids were then added to the N-terminus of CFTR to create Δ27-264 CFTR, 26 the truncated version of CFTR that was used in the present study. Δ264 CFTR and the new construct, Δ27-264 CFTR, do not conduct Cl− on their own, but they can restore the function of F508-del by binding to it and repairing its trafficking and function (reviewed in Cebotaru and Guggino 28 ) through a process known as transcomplementation.
Given the discovery of additional serotypes of AAV, 29 we decided to focus on AAV5, which showed a higher transduction efficiency than AAV2 in airways. 30 We have also studied AAV1, which transduces polarized primary human airway cells in culture with a preference toward transducing cells via the apical plasma membrane, in contrast to other AAVs, such as AAV6, that display a preference for transducing cells via the basolateral cell membrane. 31 Recognizing that AAV tropism may vary among animal species, we chose to study nonhuman primates, which are evolutionarily closer to humans. 30 In chimpanzees and rhesus macaques, we tested AAV1 and AAV5 in a simultaneous matchup, using a dual-luciferase reporter system with firefly and Renilla luciferase genes packaged into AAV1 and AAV5, respectively. Both serotypes were instilled simultaneously into the animals' lungs. Both AAV1 and AAV5 vector genomes were detected in all the lung samples more than 45 days after instillation, with the vector genome number for AAV1 being at least 10-fold higher than that for AAV5. Importantly for the present study, the neutralizing antibodies in serum were increased dramatically against both serotypes but were less abundant for AAV1 than for AAV5. No adverse events were noted, again indicating that AAV gene therapy is safe.
Given that our previous studies in chimpanzees and rhesus monkeys 30 were single-dose administration studies, the goal of the present study is to determine whether transduction is achievable after three doses using AAV1.
Materials and Methods
Viral particles
The vector consisted of an AAV gene delivery vector, AAV1-CBΔ27-264CFTR. The pseudotyped vector had an AAV serotype 1 capsid but contained the termini (inverted terminal repeats [ITRs]) of AAV serotype 2. The expression cassette consisted of the CBA transcription promoter, an intron, 30 CFTR with the sequence encoding amino acids 27 to 264 deleted (Δ27-264 CFTR), 33 and a synthetic polyadenylation sequence. A second vector contained AAV-CB-GFP (UF11). 34 The vectors were manufactured using a helper system 35 at the Vector Core Laboratory, Powell Gene Therapy Center (PGTC), University of Florida, Gainesville. AAV1-CBΔ27-264CFTR (Reference No. 6293) had a dot blot titer of 6.96 × 1012 vg/mL. The total volume of 16 mL in phosphate-buffered saline (PBS) was pooled from 38 cell stacks. AAV1-CBΔ27-GFP (Reference No. 6455-6458) had a dot blot titer of 1.21 × 1013 vg/mL. The total volume of 1 mL in PBS was pooled from four cell stacks.
Study design
Healthy juvenile rhesus macaques (four vector-treated, one control) were obtained from the Johns Hopkins University breeding colony and housed according to the Animal Care and Use Committee guidelines. All macaques tested negative for AAV1 neutralizing antibodies. The experimental design was to deliver the recombinant AAV1 vector sequentially to determine whether re-exposure to vector inhibited subsequent gene transfer and expression.
Samples of blood and sera were taken before each vector administration. Bronchoalveolar lavage (BAL) samples were taken before each dose. The animals were autopsied for detection of DNA and protein expression as well as pathologic analysis. Lungs were cut into regions as defined previously. 32 A portion of each lung region was flash-frozen, and another was fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned and mounted on slides for analysis of vector transduction as discussed below.
Vector delivery
Animals were anesthetized with intramuscular ketamine hydrochloride (10–15 mg/kg), and blood samples were obtained. An endotracheal tube (ET) was chosen that was appropriate for the size of the animal; the tube was at least 4 mm in diameter to accommodate the bronchoscope. Before ET insertion, a polypropylene suction catheter (Covidien llc) was passed through the ET tube until 1–2 cm protruded from the end. The catheter was marked so that it could be reinserted. The ET tube was inserted into the trachea through the larynx and secured with an umbilical tape tied around the back of the neck. A side arm was affixed so that isoflurane anesthetic could be administered through the tube and the bronchoscope could be passed down the ET tube. An Olympus BF Type 3C20 bronchoscope (3.7 mm diameter) was passed down the ET tube to just anterior to the bronchial bifurcation. BAL samples were collected via multiple sterile saline washes and aspiration. The bronchoscope was then removed, and the polypropylene suction catheter was inserted into the ET tube up to the marked point. Four milliliters of sterile saline or PBS containing AAV was injected through the catheter into the upper airway, followed by 1 mL of air. Isoflurane was then discontinued, and the monkey was allowed to awaken to a point at which the ET tube could be removed. The monkey was then returned to its home cage.
Real-time DNA analysis
Quantitative PCR with LightCycler (Applied Biosystems 7500, Waltham, MA) was used to test the various lung regions for CFTR- and GFP-specific DNA. Genomic DNA samples were prepared using a PureLink Genomic DNA Mini Kit (#K1820-02; Invitrogen). Vector genomes per microgram of lung DNA were measured using the qRT-PCR/Fast SYBR Green Master Mix (#4385612; Applied Biosystems) in genomic DNA obtained from various parts of the body (lung: trachea, bronchus, carina, cranial, middle, and caudal lobes; heart, liver, kidney, and spleen). Sample values were calculated using a standard curve of arbitrary fluorescence units plotted versus serial dilutions of plasmid DNA ranging from 200 to 0.002 ng. The number of fluorescence units obtained from the untreated tissue samples was subtracted to obtain specific gene expression. The primer sequences used are as follows: CFTR-AAV-forward: GATACAAGGCTTTAGAGAGC and reverse: CTCCATCACTAGGGGTTCCT; GFP-AAV (UF11) forward: GTGCAGGAGAGAACCATCT and reverse: GCCATTCTTTTACTTGTCGGC.
Immunoprecipitation experiments
The protocol was modified from a previous publication. 36 Samples (20 mg) of flash-frozen lung tissue were cryopulverized with liquid nitrogen using a mortar and pestle and then suspended in 1 mL of NP40 lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 2 mM EGTA, 1.0% Nonidet P40) with complete protease inhibitor (Roche Diagnostics, Mannheim, Germany). Lysate samples were allowed to incubate on a rotator for 1 h at 4°C. Supernatants were collected by centrifugation at 15,000 g for 10 min. Samples of 100 μL were set aside for protein assays of total lysate, and 50 μg of primary antibody was added to the remaining supernatants before incubation at 4°C for 4 h. Immune complexes were collected using a 3,000-μg sample of protein A/G plus agarose. The beads were washed five times in lysis buffer containing protease inhibitors and then prepared for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting.
Immunoblotting
The supernatants described above (30–50 μg of proteins) were subjected to 4–15% SDS-PAGE and immunoblotting, followed by enhanced chemiluminescence (SuperSignal West Dura Extended Duration Substrate, Catalog No. 34075; Thermo Scientific). The chemiluminescent signal on the polyvinylidene difluoride membrane (Bio-Rad) was directly captured by an Amersham imager 600, GE system with a cooled CCD camera. CFTR protein was detected with monoclonal anti-human CFTR (217; 1:1,000) antibody provided by Dr. J. Riordan, Department of Biochemistry and Biophysics and Cystic Fibrosis Center of North Carolina. β-Actin, used as a loading control, was detected with anti-β-actin monoclonal antibody (1:10,000; Santa Cruz Biotechnology).
Confocal microscopy
Monkey tissues were fixed with paraformaldehyde and embedded in paraffin blocks. Deparaffinized tissue sections were blocked with 4% serum for 1 h at 25°C; antigen retrieval was accomplished by heat mediation in a citrate buffer (#S1699; Dako). Samples were incubated with primary and secondary antibodies as detailed in the figure legends. Images were captured using a Zeiss LSM 880 confocal microscope with a 63 × oil-immersion objective (NIH Grant #S10OD016374). For quantification, intensity measurements were made using Zen 2012 software.
Antibodies
For Western blotting, monoclonal antibodies against CFTR (217) (from Dr. J. Riordan, Department of Biochemistry and Biophysics and Cystic Fibrosis Center of North Carolina), GFP (#11814460001; Roche), and β-actin (Sc#47778; Santa Cruz Biotechnology) were used. For confocal studies, monoclonal antibodies against CFTR (596) (also from Dr. J. Riordan), GFP (#11814460001; Roche), and cytokeratin5 (#ab52635; Abcam); anti-mouse-Alexa Fluor 594 (#A11032; Life Technologies), anti-mouse Alexa Fluor 488 (#A11001; Life Technologies), anti-rabbit Alexa Fluor 488 (#A11008; Life Technologies), and anti-rabbit Alexa Fluor 594 (#A11012; Life Technologies) were used.
Neutralizing antibody experiments
Neutralizing antibodies were evaluated using AAV1-LacZ as reported previously. 32,37 The neutralizing antibody titer was reported as the highest serum dilution that inhibited AAV1LacZ transduction by 50%, when compared with its own AAV1 vector-positive/serum-negative control.
ELISPOT assays
T cell responses to AAV1 were determined by evaluating the release of interferon-gamma (IFN-γ) after stimulation of T cells with transgene antigens from AAV or CFTR using an enzyme-linked immunoblot (ELISPOT) assay according to previously described protocols. 37 In brief, transgenic antigens were generated from peptide libraries composed of 15-amino acid constructs (15mers), each having a 10-mer overlap of the AAV1 VP1 capsid or Δ27-264 CFTR protein sequence with the preceding peptide (Mimotopes, Minneapolis, MN). For AAV1, the peptides were divided into three groups, and for CFTR into five groups. Blood samples were collected from the monkeys before and after the administration of AAV1-CFTR through venipuncture (with heparin as anticoagulant), and then shipped overnight to the testing laboratory at the University of Pennsylvania. The peripheral blood mononuclear cells (PBMCs) were analyzed using the IFN-γ ELISPOT assay by stimulation with the respective AAV1 capsid or CFTR pools or with medium alone (no peptide). PBMCs were added at a density of 2 × 105 cells/well and stimulated for 18–20 h at 37°C with peptide libraries derived from the AAV1-VP1 or Δ27-264 CFTR protein at 2 μg/mL. Medium alone served as a negative control, and phytohemagglutinin as a positive control. The AAV1-VP1 peptide library was divided into three pools, such that pool 1A contained the first 50 peptides of AAV1 VP1, pool 1B contained peptides 51–100, and pool 1C contained peptides 101–146. The Δ27-264 CFTR peptide library contained a total of 249 peptides. The final pool was divided into 5 pools of 50 peptides each. Spots were counted with an ELISPOT reader (AID, San Diego, CA). Peptide-specific cells were recorded as spot-forming cells per 106 PBMCs. A positive response to any peptide pool was arbitrarily defined as three or more times the background (medium-alone control) level, or ≥55 spot-forming cells per 106 PBMCs.
Results
Body weights and clinical observations
All the animals maintained their body weight throughout the experimental period (Supplementary Fig. S1). The complete results for the clinical chemistry analyses are provided in Supplementary Table S1. All the clinical values remained within their normal range throughout the study, with one exception: one animal (32A) had elevated aspartate aminotransferase (AST) following the second dose of the virus. All other samples for AST before and following this one sample were normal. In summary, there were no clinical observations attributable to vector administration.
AAV vector genomes
Samples were taken from six different lung regions (trachea, carina, bronchus, cranial, middle, and caudal, Fig. 1A), similar to what was done previously. 32 The samples were taken from animals 90 days after the first instillation of virus (Fig. 1B).
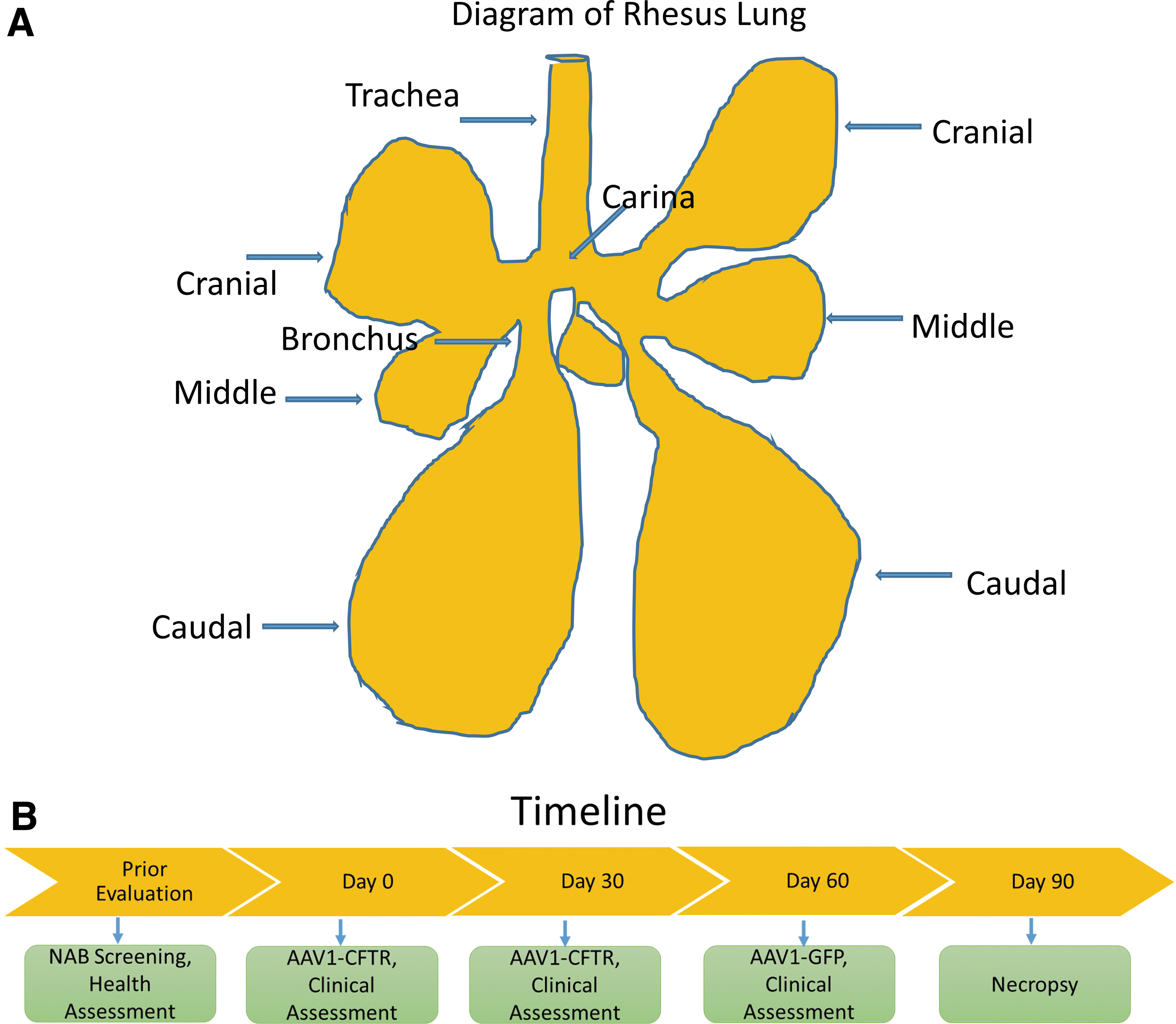
Figure 2A shows the vector genomes measured in each monkey, in each lung region at necropsy. AAV1-CBΔ27-264CFTR and GFP vector DNA was detectable in all lung tissues in all four vector-treated animals. Note that the vector was widely distributed throughout the lung. The number of particles was higher after two doses of AAV1-CBΔ27-264CFTR, compared with the results for GFP. Figure 2B also shows that AAV1-CBΔ27-264CFTR and GFP could be detected in several organs throughout the body, including the heart, liver, and spleen. This result is not surprising because we have shown previously 32 that after lung delivery, AAV1 vector genomes readily enter the blood, peaking at ∼3 h after treatment. By 48 h, the vector genomes are no longer detectable, suggesting (as we show here) that the vector genomes have entered the organs, particularly the liver and kidneys, and have been cleared from the blood.

AAV vector transduction
To assess protein expression from the transgenes, we performed immunofluorescent staining for CFTR and GFP in the various lung sections and compared the results with those for an untreated animal sacrificed for nonrelevant reasons. Sections were chosen to highlight the conducting airways, namely the trachea, carina, and bronchus and the alveoli selected from the cranial, middle, and caudal regions of the monkey lung (Fig. 1A and Supplementary Fig. S2). Figure 3 shows the confocal images for CFTR. All conducting airways including the trachea, carina, and bronchus showed higher fluorescent staining for CFTR than did those from the control animal, indicating that CFTR had been expressed from the transgene. Less expression was noted in the alveoli, particularly in the middle and caudal lung regions. This pattern is not surprising given that the vector was instilled in the trachea.
CFTR colocalization in the lung. Representative images of treated monkeys versus a nontreated control monkey. The figures depict sections stained with an antibody against CFTR whose epitope is within NBD2 (Antibody 596, University of North Carolina, Jack Riordan) and secondary antibody (Alexa Fluor 594 [red]; Lifetech A11032) in the trachea, carina, bronchus, cranial, middle, and caudal sections. The scale bar is 10 μm. Four or five images were obtained from each section of each animal, and the red intensity was measured using Zen software (Zeiss Microscopy). Data for four treated animals were analyzed and compared with those for one nontreated control. Data represented in this and the two subsequent figures were analyzed with statistical significance indicated as follows: *p < 0.05; **p < 0.01; ****p < 0.0001. CFTR is expressed in the untreated monkey lung, and thus, the sections from the untreated monkey are stained red. Higher levels of red staining were measured in the conducting airway of the treated animals than in the nontreated animal, with the highest levels in the trachea, where the virus was administered. In all the figures, arrows are added to emphasize positive staining. N = 4 treated +1 untreated. One-sample t-test was conducted with n = 4 treated animals compared with the one control. In this analysis, the control measurements are averaged to a single and compared with all of the treated animal experiments. Thus, the control measurements do not have error bars.
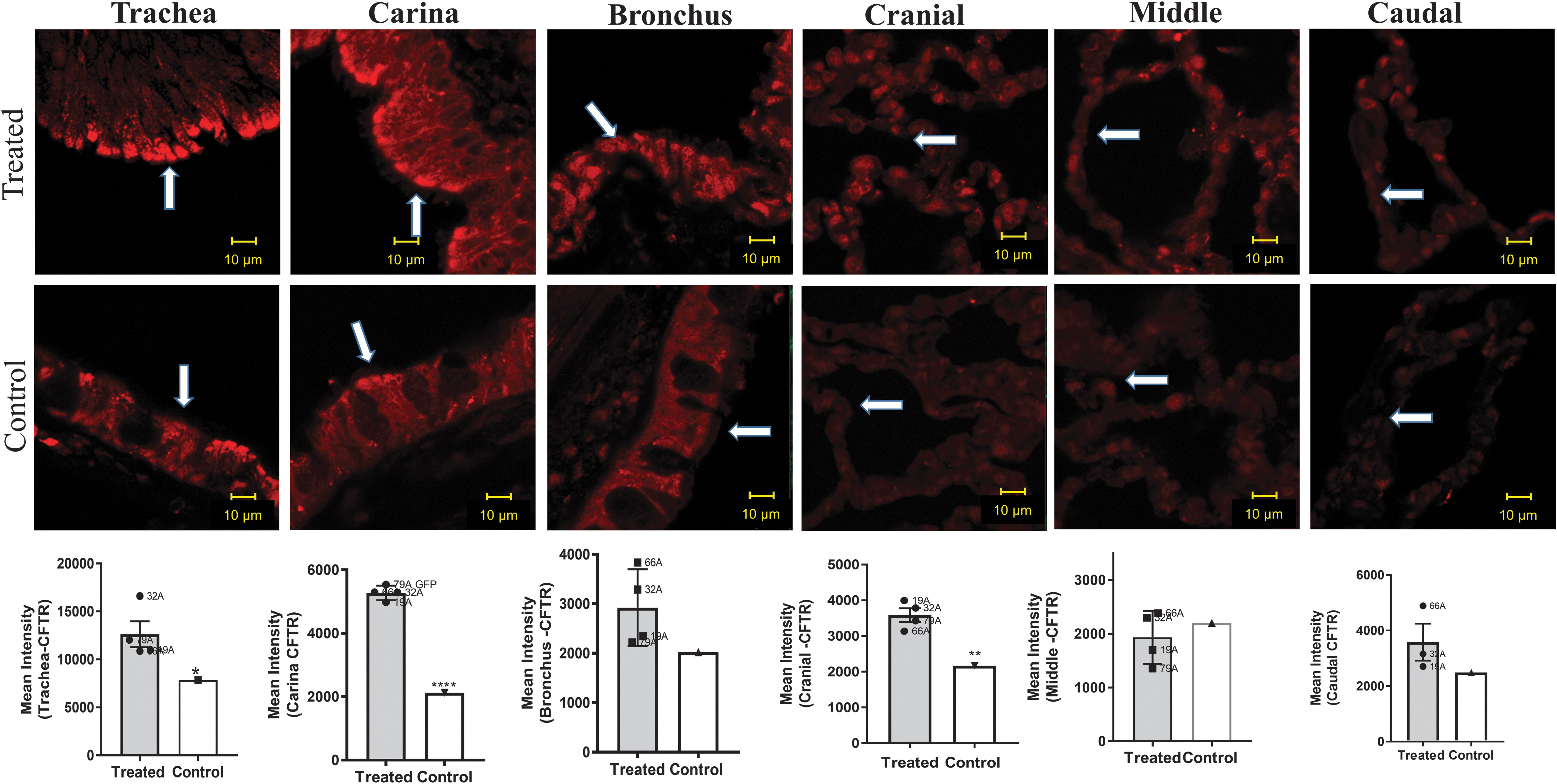
GFP protein expression was detected in all the lung sections (Fig. 4). The tissue sample did exhibit some autofluorescence at the same wavelength as GFP, however, positive staining for GFP was detected by a signal at least threefold higher than the background in all lung sections.
GFP and DAPI localization in the lung. Representative images of treated monkeys versus a nontreated control. The figures depict sections stained for GFP (Roche 1181446001 antibody), followed by staining with a secondary antibody Alexa Fluor 488 (Lifetech A11001 [green]) and counterstaining with DAPI (blue) in the trachea, carina, bronchus, cranial, middle, and caudal sections. See Fig. 3 for further details. GFP is not expressed in untreated monkey lung, and thus, the untreated animal sections represent background fluorescence. Higher levels of GFP staining were measured in all the sections from the treated monkeys than in those from the nontreated monkey. DAPI staining shows the location of the nuclei. See Fig. 3 for details. DAPI, 4′,6-diamidino-2-phenylindole.
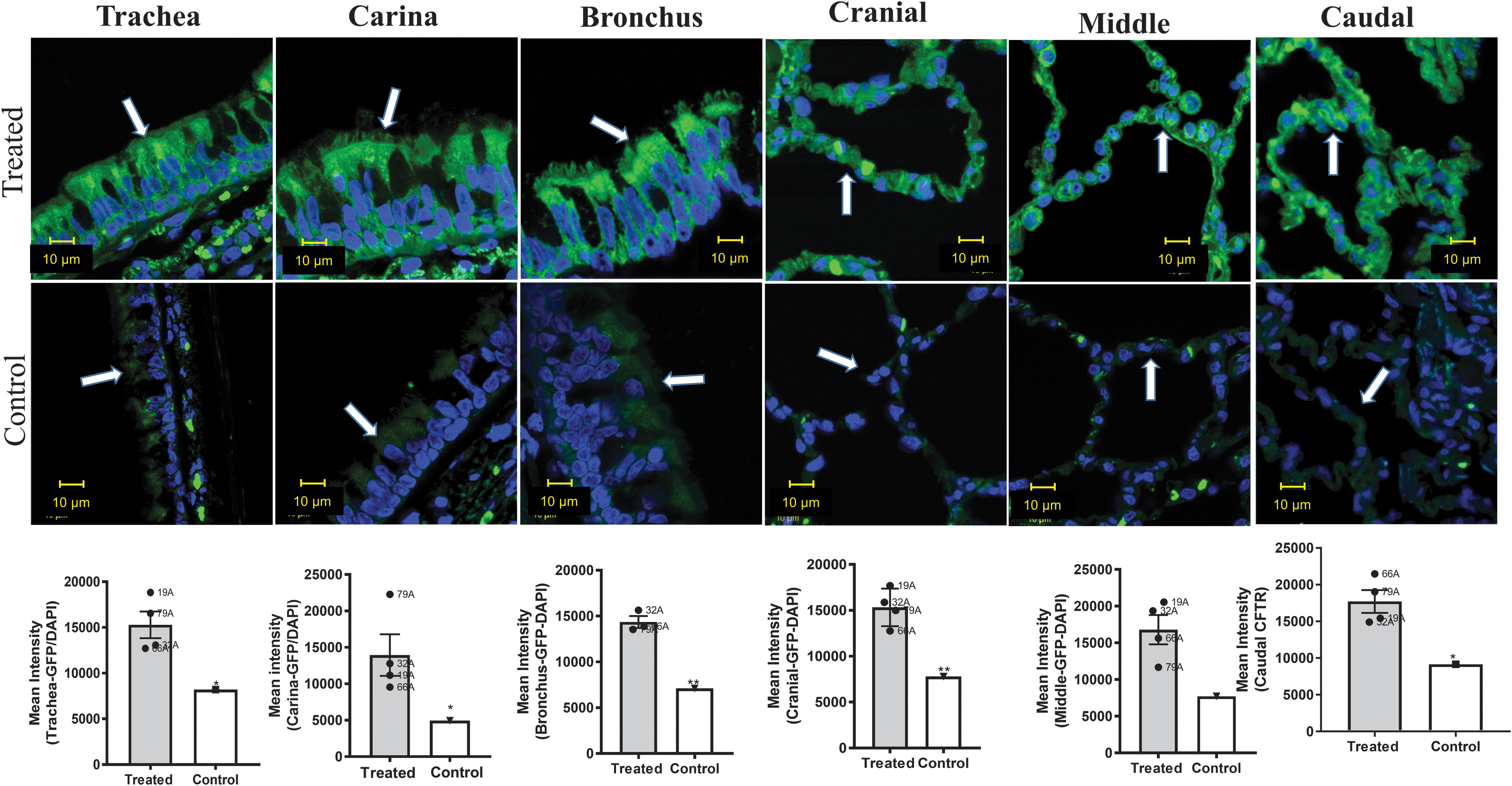
Figures 3 and 4 demonstrate transduction primarily of the surface epithelium of the conducting airways, including the trachea, carina, and bronchus. The surface epithelial cells in the conducting airways are terminally differentiated and turn over slowly unless challenged (about every 6 months as measured in healthy mice 38 ). Regeneration of the surface epithelium of the airway, particularly after cell injury, 39 occurs via the basal keratin 5/14-expressing cells. 40,41 Thus, one way to extend the half-life of CFTR transduction of the surface epithelium would be to target the basal cells. To determine whether CFTR is expressed in the basal cells, we stained the tissue samples with antibody detecting keratin 5, and then counterstained with either CFTR or GFP. As expected, K5-expressing cells could clearly be seen along the basement membrane of the trachea, carina, and bronchus. CFTR expression was higher within the basal cells of the transduced monkeys than in the nontreated monkey (Fig. 5). Likewise, we also saw positive expression of GFP within the K5-positive cells when compared with untreated cells (Fig. 6). These data indicate that AAV1 does indeed transduce basal cells, a necessary goal for extending the efficacy of gene therapy for the lung; however, transduction of the basal cells must be carried out safely because of their importance in regeneration after injury. 40,41

CFTR and keratin 5 colocalization in basal cells. Representative images from treated monkeys versus a nontreated control. The figures show sections stained for CFTR (red) and K5 (green, Krt ab52635) in the trachea, carina, and bronchus. CFTR is located within the K5-positive cells that are located near the basal membrane. To quantify CFTR, the K5-positive regions were outlined using the Zen software and CFTR quantified within those regions. Insets represent magnified regions remarked by dotted squares. Statistics as in Fig. 2.
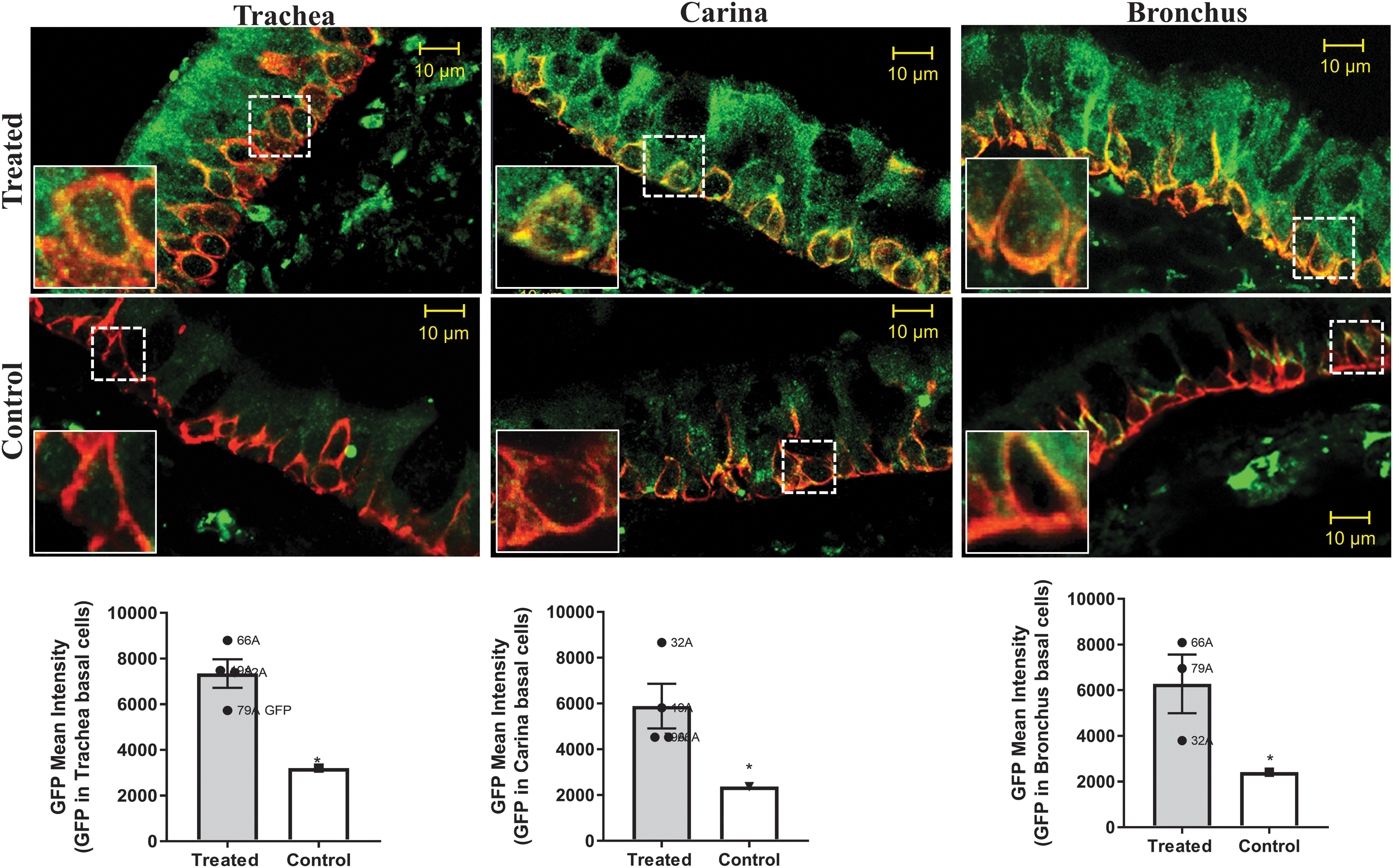
GFP and keratin 5 colocalization. Representative confocal images from treated monkeys versus a nontreated control. The figures depict sections stained for GFP (green) and K5 (red) in the trachea, carina, and bronchus. See Figs. 2 and 4 for additional details. GFP is located within the K5-positive cells that are located near the basal membrane. Insets represent magnified regions remarked by dotted squares.
To validate the immunofluorescence studies, we conducted Western blotting experiments for both CFTR and GFP. The results confirmed that there is indeed a higher expression of CFTR in the treated monkeys than in the control (Fig. 7). GFP expression was also detected only in the treated animals. Data for all the experimental animals are given in Supplementary Figs. S3 and S4. Taken together, these data suggest that repeat dosing of AAV1 leading to transduction of the transgene is feasible.
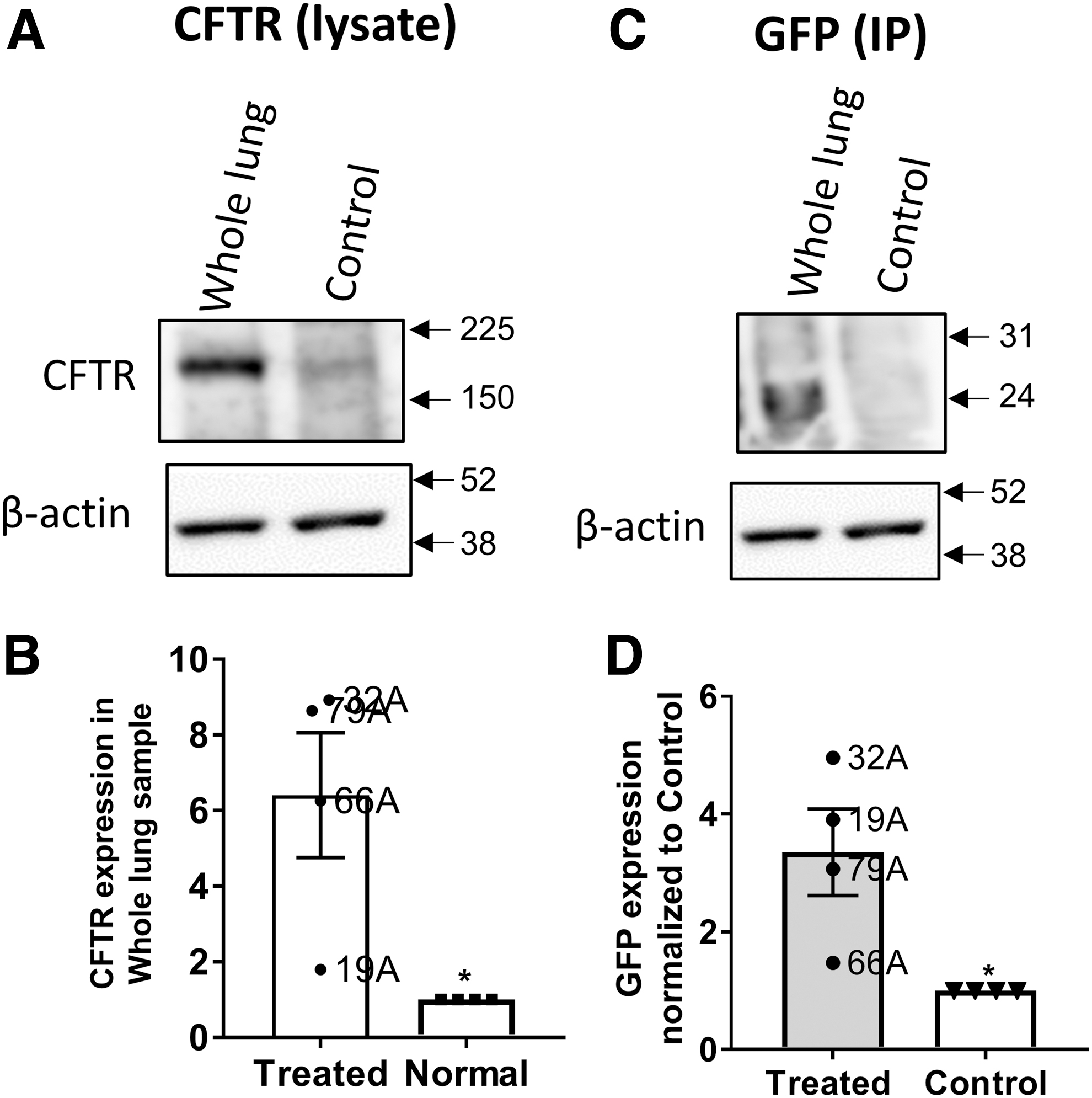
Protein expression of CFTR and GFP in whole-lung samples.
Neutralizing antibodies
The results of an evaluation of anti-AAV antibody are shown in Fig. 8. All animals used in the study had low serum neutralizing antibody titers (<5 reciprocal dilution) at the beginning of the study. The serum levels increased dramatically in all animals between the prestudy time points and the time of necropsy on study day 90. BAL fluid was also positive for neutralizing antibodies at necropsy, but the levels were lower than for serum. The neutralizing antibody titer for the first dose was comparable with the titer we observed previously in a single-dose study of AAV1. 32 Interestingly, after the third dose of AAV1, the neutralizing antibody titer in the present study was still less than what we observed after we treated monkeys with a single dose of AAV5. 30
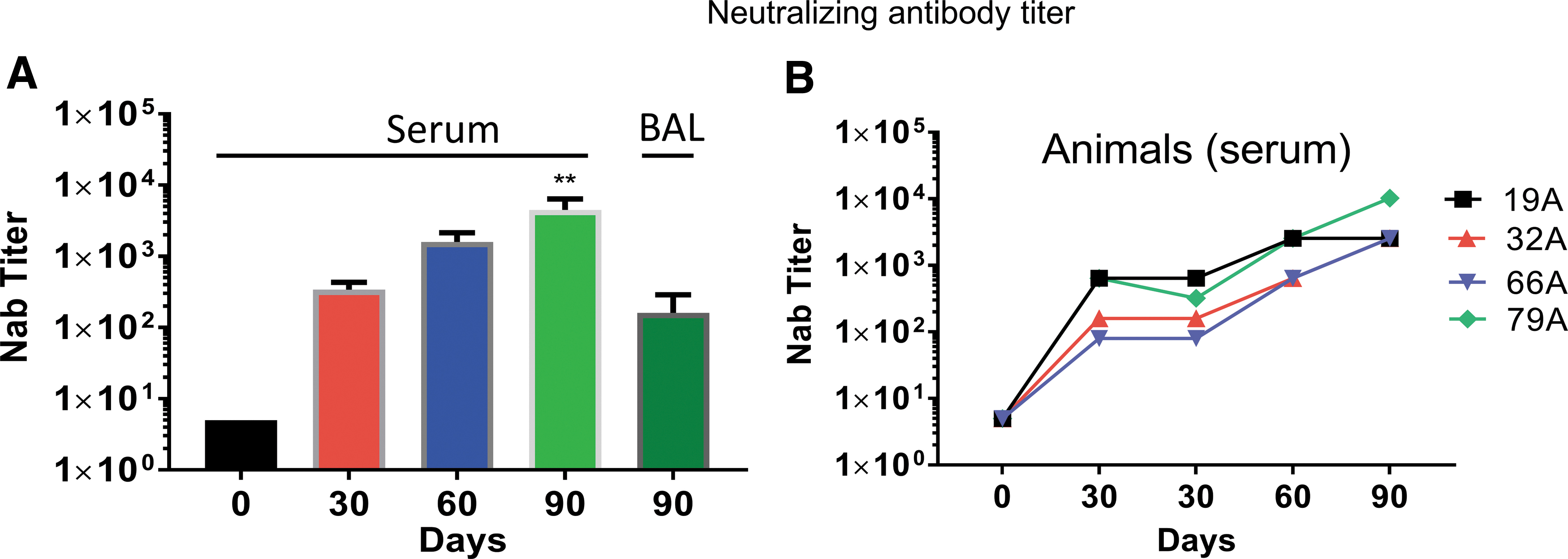
Neutralizing antibody: the neutralizing antibody titer increased significantly after the first vector instillation and continued to increase thereafter.
Immune responses to AAV capsid components and CFTR
To determine the immune responses to the AAV1 capsid proteins and CFTR, we performed IFN-γ ELISPOT assays on PBMCs using previously described protocols. 37 Three animals (19A, 32A, and 79A) elicited a significant ELISPOT response to AAV1 peptide pools B and C by the third dose of AAV1 (Fig. 9). Interestingly, there was no response in animal 66A. To determine whether there any immunogenicity to the CFTR transgene was present, we also performed an IFN-γ ELISPOT using a CFTR peptide library (Fig. 9). Whereas the AAV1 capsid elicited a positive response, there was no response to the CFTR transgene.

Ex vivo IFN-γ ELISPOT assays. For AAV1, representative raw data are shown for each of the monkeys. VP1 capsids were divided into three groups, A–C. Data are reported as the number of SFCs per million PBMCs at each of the time points indicated. Each point represents PBMCs from a single monkey stimulated with an AAV1 peptide pool. Each graph depicts the results from each peptide pool. PBMCs were assessed using an IFN-γ ELISPOT assay (see the Materials and Methods section) by stimulation with the respective AAV capsid pools or medium alone (no AAV = NS). A positive result was considered to be >55, as indicated by the dotted line. Note that three of the four monkeys had a positive response to AAV1 VP1 peptides B and C after the third dose. Only monkey 66A did not respond to any VP1 peptide. For CFTR, representative raw data are shown for each of the monkeys. The CFTR protein was divided into five sections, A–E. Note that there was no response to CFTR. Two samples were taken at 30 days on the day of the vector administration and on the day after. This was done to look for an immediate response of vector instillation. IFN-γ, interferon-gamma NS, no stimulation; P2BMC, peripheral blood mononuclear cell; SFCs, spot-forming cells.
Discussion
Previous human studies on AAV gene therapy for CF using serotype 21 were limited by the fact that low expression of the transgene did not allow clinical relevance to be achieved. One problem was the inefficiency of AAV2 in transducing human airway cells. 1,42 In our recent studies, we tested known AAV serotypes such as AAV5 and AAV1. With these serotypes, we conducted studies in rhesus macaques 32,43 and a nonlethal study in chimpanzees, 30 the closest genetic relative to Homo sapiens, to find a more efficiently transducing serotype than AAV2. Our results showed that in the primary human airway cells grown in tissue culture and in nonhuman primates, AAV1 is more effective in transduction than is either AAV5 or AAV2.
Understanding that repeated dosing would be necessary for a successful gene therapy for CF, we undertook the present study to test the feasibility of repeat dosing with AAV1. At necropsy, we detected vector genomes in the lungs of all four treated animals, ranging from 1 to 4 × 106 for CFTR in the trachea, bronchi, and carina, and ∼105 vector genomes/μg of lung DNA for GFP. In our previous single-dose study using a similar amount of AAV1 LacZ, we found an average of 1 × 106 vg/μg of lung DNA remaining after 45 days of treatment. 32 In addition to the lung, we also detected vector genomes in the heart, liver, kidney, and spleen.
Although we recorded the number of vector genomes in the lungs at necropsy, one remaining question was whether the vector genomes present in the lung actually lead to transduction. We approached this question by using confocal microscopy and Western blotting. Both techniques gave similar results, higher expression of CFTR and GFP in the treated versus the nontreated monkey.
We found that CFTR is expressed at higher levels in the monkeys treated with CBΔ27-264CFTR than in the untreated monkey. Δ27-264CFTR, contains a truncated form of wild-type CFTR missing its first four transmembrane domains. 28 We have shown previously that AAV1-CBΔ27-264CFTR can increase the protein levels of wild-type CFTR via transcomplementation in both monkey lungs and in cells in tissue culture. 33,43,44 Thus, the increased expression of CFTR noted in the treated animals is most likely the result of increased protein levels of wild-type monkey CFTR induced by transcomplementation via AAV1-CBΔ27-264 CFTR. 28
Neutralizing antibodies are a barrier to repeated dosing with AAV vectors (reviewed by Mingozzi and High 45 ). All of the monkeys had increasing titers of neutralizing antibodies in serum following each dose and at necropsy. Much lower levels were detected in the BAL fluid. The serum titer of <104 at necropsy was higher than what we previously observed with a single dose of AAV1. 32 It is interesting that the titers after three doses of AAV1 were still less than what we observed previously with a single dose of AAV5, 30 suggesting that AAV1 generates less neutralizing antibody than does AAV5.
We have observed detectable GFP expression both in lung and peripheral organs following instillation of a third dose of AAV1 despite increasing titers of AAV1 anti-capsid neutralizing antibodies. Thus, although the titers of neutralizing antibodies did increase, they apparently were not large enough to block transduction of the airway cells or prevent the virus from entering cells in peripheral organs, indicating that such antibodies many not represent an impediment to gene transduction, at least when three doses are used.
It has been noted in human studies of hemophilia that the CD8+ T cell response that resulted in transaminase elevation after several weeks was not predicted from previous animal studies, and it occurred after a single dose of an AAV2 virus expressing factor IX under the control of a liver-specific promoter. 45 Analysis of PBMCs by IFN-γ ELISPOT assay showed a T cell response to the vector capsid, but not to the transgene. It was concluded that the introduction of AAV2 may have activated memory T cells, which had developed as the result of prior infection with wild-type AAV2. In our previous experiments on chimpanzees, we saw no ELISPOT response to a single dose of AAV1, whereas AAV5 did elicit a response in three of four chimpanzees. 30 This elicitation of an antibody response probably occurred because the animals were prescreened for the absence of prior exposure to AAV1, and therefore, they would not have been expected to have activated memory T cells. However, in the present experiment in which three doses of AAV1 were administered, we did see a positive ELISPOT response to AAV1 peptides, particularly after the third dose. Interestingly, although all four monkeys consistently produced neutralizing antibodies, the ELISPOT response was quite variable from animal to animal. For example, animal 19A appeared to be the most responsive, particularly when compared with 66A, which showed no response to any of the doses. This result did not correlate with the number of vector genomes detected in the lung, with 66A having much high numbers of vector genomes than did 19A. Thus, the differences in response were most likely systemic and intrinsic to each animal. Despite the ELISPOT responses, the positive animals displayed no changes in clinical parameters throughout the study.
We have previously shown that AAV1 vector genomes can be detected in the blood shortly after AAV1 vector instillation into the lung 32 ; thus, it is not surprising that in this study, AAV1 vector genomes were detected in organs throughout the body. Given that CF is a multiorgan disease, some systemic delivery of AAV1 applied to the lung may be beneficial. Here we also demonstrate basal cell transduction following three doses of AAV1. Given that all the monkeys readily mounted a neutralizing antibody response to AAV1, transduction of basal cells would also be beneficial, because these cells give rise to the surface epithelial cells. 41 Thus, transduction of basal cells may prolong any beneficial effects of vector transduction, lengthening the time necessary between administrations of the vector. Prolonging the time between administrations would also be expected to reduce the neutralizing antibody titer.
It was noted several years ago that although some small amount of integration does occur, recombinant AAV vectors to a large extent do not integrate but instead persist as episomes. 46 Since surface airway cells turn over thereby diluting the presence of AAV instilled for therapeutic purposes, it is beneficial to transduce basal cells with AAV vectors to replenish those lost during airway cell turnover. To this endpoint, Cooney et al. 47 developed an AAV vector containing an integrating AAV-based vector containing a piggyBac transposon, which rescues function in a pig model of CF. The goal is to transduce progenitor cells, which would induce lifelong expression of functioning CFTR. Despite the challenges that still exist with the safety of integration, these experiments and those described here point to the potential of more long-lasting therapies for CF using AAV-based vectors.
One common conclusion from both the earlier chimpanzee and macaque studies is that a single-dose delivery of AAV1 or AAV5 is generally safe. The present study shows that a triple dose is also safe and is still effective in inducing vector transduction. It is likely that repeated dosing of AAV1 beyond what we show here will lead to reduced transduction induced by an increasing neutralizing antibody titer. However, if perhaps the neutralizing antibody titer reaches a plateau, it still may be possible to have enough transduction to be therapeutic. It should be mentioned that AAV gene therapy still has a number of challenges to overcome (recently reviewed in Guggino et al. 1 ); however, given this body of promising data, it appears that further development of AAV with the goal of resumption of gene therapy clinical trials is certainly warranted.
Footnotes
Acknowledgments
The authors specially thank Dr. Robert Adams for providing the monkeys, for anesthetizing the monkeys, drawing blood samples, and for instilling the virus. They thank the Comparison Medicine Primate Facilities at JHU for the toxicology services for this study. They also acknowledge Dr. Jessica A. Chichester, PhD, Director of the Immunology Core, Gene Therapy Program, Perelman School of Medicine, University of Pennsylvania, for the neutralizing antibody and ELISPOT assays, which were conducted by the core. In addition, the authors acknowledge Mark Potter, PhD, of the Vector Core Laboratory, Powell Gene Therapy Center (PGTC), University of Florida, Gainesville for manufacturing the virus, and Deborah McClellan, PhD, for editorial assistance.
Author Disclosure
L.C. has a license agreement with RA Capital, and W.B.G. is a consultant for Vertex and Sarepta Pharmaceuticals in areas that are unrelated to this study.
Funding Information
The work was funded by an NIH grant R01 HL122267 to Liudmila Cebotaru and William B. Guggino.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.