
Abstract
Dendritic cell (DC)-based vaccines have shown some degree of success for the treatment of prostate cancer (PC). However, the highly immunosuppressive tumor microenvironment leads to DC dysfunction, which has limited the effectiveness of these vaccines. We hypothesized that use of a fully serotype 3 oncolytic adenovirus (Ad3-hTERT-CMV-hCD40L; TILT-234) could stimulate DCs in the prostate tumor microenvironment by expressing CD40L. Activated DCs would then activate cytotoxic T cells against the tumor, resulting in therapeutic immune responses. Oncolytic cell killing due to cancer cell-specific virus replication adds to antitumor effects but also enhances the immunological effect by releasing tumor epitopes for sampling by DC, in the presence of danger signals. In this study, we evaluated the companion effect of Ad3-hTERT-CMV-hCD40L and DC-therapy in a humanized mouse model and PC histocultures. Treatment with Ad3-hTERT-CMV-hCD40L and DC resulted in enhanced antitumor responses in vivo. Treatment of established histocultures with Ad3-hTERT-CMV-hCD40L induced DC maturation and notable increase in proinflammatory cytokines. In conclusion, Ad3-hTERT-CMV-hCD40L is able to modulate an immunosuppressive prostate tumor microenvironment and improve the effectiveness of DC vaccination in PC models and patient histocultures, setting the stage for clinical translation.
INTRODUCTION
Prostate cancer (PC) is the most common cancer in men. 1,2 There are currently no curative treatments for metastatic or hormone refractory disease. 3 After recurrence, androgen-deprivation therapy is often an effective treatment initially, but the disease eventually progresses. 4 –6 Treatments used in castration-resistant disease include chemotherapeutics such as taxanes and second-generation antiandrogen therapies, including enzalutamide and abiraterone. 7 Radium therapy is also approved. However, none of the above are curative and can lead to significant adverse events. 6,8
Sipuleucel-T (ProvengeR) was the first cell therapy shown effective in the field of oncology. It was approved for the treatment of PC by the FDA in 2010, which generated interest in using immunotherapy for treatment of this disease. 8 Several randomized trials demonstrated increased overall survival, 9 –11 even if cures were not seen. Also, puzzlingly, tumor or prostate-specific antigen (PSA) responses are not seen with this approach, and the mechanism of action is unclear. Nevertheless, this prototype drug validated the concept that immunotherapy can be useful in the treatment of PC. Notably, results with sipuleucel-T appear to suggest that antigen-presenting cells could be useful in the treatment of PC, and therefore dendritic cells (DCs) have become interesting in this setting.
Oncolytic adenoviruses are an attractive immunotherapy approach for the treatment of cancer, due to good safety and signs of efficacy. 12 Tumor-restricted replication of oncolytic adenoviruses leads to the release of tumor-associated antigens (TAAs), thus reactivation of antitumor immune responses. 13 Therefore, it can be regarded as a personalized anticancer vaccine, which spontaneously activates in tumors by replication followed by lysis. Epitopes relevant for each individual tumor are released in the presence of immunological danger signaling, resulting in conditions compatible with therapeutic antitumor immunity. 14,15 To overcome the immunosuppressive tumor microenvironment, oncolytic adenovirus can be armed. 14
In the context of DC therapy, a promising arming strategy is CD40L, which is a potent stimulator of DCs. 16,17 Viruses such as Ad3-hTERT-CMV-hCD40L are able to deliver the payload to the tumor, where tumor-selective replication amplifies the virus genome with the transgene, resulting in high-level local production of the transgene product. TAAs are also locally released due to tumor-specific replication (leading to lysis of the cell), thus maximizing the immunotherapeutic effect while reducing systemic exposure. 14,18
Systemic administration of recombinant CD40L has been safe up to moderate concentrations, after which off-tumor toxicity becomes limiting, before sufficient concentration can be reached in tumors. 19 This generates the rationale for local production through an oncolytic virus platform.
Previously, we have shown that adenovirus based on serotype 3 (Ad3) armed with CD40L, Ad3-hTERT-CMV-hCD40L, is able to transduce tumors after intravenous administration. 16,17 This is a major advantage in the context of disseminated tumors, for which local delivery might be challenging. The only oncolytic virus currently approved in the United States and EU is talimogene laherparepvec, which is used only intratumorally, and does not disseminate from lesion to lesion.
Of note, a similar but unarmed virus, Ad3-hTERT-E1A, was safely used in treatment of cancer patients refractory to other therapies, with promising signs of efficacy. 20,21 Ad3-hTERT-CMV-hCD40L, also known as TILT-234, is the hCD40L variant or Ad3-hTERT-E1A. This armed virus exhibited strong antitumor efficacy through its lytic ability along with expression of functional CD40L in tumors 17 and enabled DC therapy in lung cancer A549 xenograft in humanized mice model. 16 Previously, preclinical studies and human data have shown the ability of virally expressed CD40L to stimulate DCs. 14,21 –24
In this study, we explore the therapeutic benefits of Ad3-hTERT-CMV-hCD40L as an enabler of DC therapy in a clinically relevant humanized model of PC and histocultures obtained fresh from patients.
MATERIALS AND METHODS
Cell lines
PC-3 PC cells were obtained from American Type Culture Collection (ATCC; LGS standards). PC PC-3MM2 cell line was gifted by Isaiah J. Fidler, M.D. Anderson Cancer Center. All the cell lines were cultured in Roswell Park Memorial Institute medium (RPMI 1640; Sigma). All the cell lines were maintained under a humidified 5% CO2 atmosphere at 37°C, and growth media were supplemented with 1% penicillin/streptomycin (P/S), 1%
Viruses
Oncolytic adenovirus serotype 3 (Ad3); Ad3-hTERT-E1A; and Ad3-hTERT-CMV-CD40L were used in this study. Construction of these viruses has been described. 17,20
Cytotoxicity assay
PC-3 and PC-3MM2 cells were used for cytotoxicity studies. 10,000 cells/well were plated in 2% growth media. Twenty-four hours later cells were infected with either Ad3, Ad3-hTERT-E1A, or Ad3-hTERT-CMVhCD40L in concentrations from 1 to 1,000 viral particles (VPs)/cell. The assay was done in triplicate. Cell viability was determined on days 3, 5, and 7 with MTS cytotoxicity assay according to manufacturers' instructions (Cell Titer 96 AQueous One Solution Cell Proliferation Assay; Promega [G3582], Madison, WI).
Generation of human DCs
Human DCs were generated according to the previously reported protocol. 16,17 In brief, buffy coat of healthy donor was obtained from Red Cross Blood Service (Helsinki, Finland). Lymphoprep (StemCell Technologies) was used to isolate peripheral blood mononuclear cells (PBMCs) from buffy coat through density gradient centrifugation. PBMCs obtained were washed with phosphate buffered saline (PBS) followed by 4–5 min incubation at room temperature with ACK lysis buffer to remove red blood cells; then washed again with PBS. HLA-matched PBMCs were used in this study.
CD14+ magnetic beads (130-0505-201; Miltenyi Biotec) were used (according to the manufactures' instructions) to isolate CD14+ cells from isolated PBMCs.
To get immature DCs, 4.5 × 10e6 separated CD14+ cells were cultured in 10 mL of 10% RPMI supplemented with 1,000 U GMCSF and 20 ng IL-4 for 5 days in T25 flasks at 37°C. These immature DCs were incubated with 50 μg/mL of tumor lysate for 24 h, followed by incubation with lipopolysaccharide (100 ng, LPS) for 24 h.
Animal experiments
The Provincial Government of Southern Finland and the experimental animal committee of the University of Helsinki (Helsinki, Finland) approved all the protocols used for animal experiments (animal experiments have been permitted under permission: ESAVI/28404/2019).
Immunodeficient SCID mice (5 weeks old) were implanted subcutaneously with 2 × 10e6 PC-3MM2 cells. When the tumors become injectable that is after 14 days of implantation, mice were randomized into groups (n = 10/group). All mice received 10 × 10e6 HLA-matched PBMCs on day 0 intravenously. Mice received either only viruses (10e8 VP/tumor) administered on days 1, 3, and 5, or followed by 1 × 10e6 DCs on days 2, 4, and 6 intratumorally. Tumors were measured every other day using electronic caliper until day 25 and followed until day 93 for survival. When tumors reached the maximum limit of 18 mm, mice were euthanized. Tumor ulceration was considered as an exclusion criterion.
Due to animal regulations, mice that developed tumor ulceration were euthanized and are therefore censored from analysis of tumor-specific survival. Animal regulations require this even though tumor necrosis is a goal of therapy. In cancer-specific survival, only animals that die or are killed because of tumor progression result in changes in the curve.
Patient material
For patient material research ethics committee case number is HUS/850/2017.
PC samples were collected from five patients undergoing surgical resection. All patients have signed an informed consent. All tumor histologies were confirmed by a pathologist at Helsinki University Hospital. A local ethics committee has evaluated and approved the project (HUS/850/2017).
Tumor histocultures
Fresh PC tumors were digested into single cells using the protocol described in a previous study.
25
In brief, PC tumors were cut into small pieces, and were enzymatically digested in media (RPMI 1640; Sigma) supplemented with 1%
Flow cytometry
Above-mentioned single cell suspensions, on day 3, were treated with either Ad3-hTERT-E1A or Ad3-hTERT-CMV-hCD40L (100 VP/cell), and were stained with fluorochrome-labeled antibodies according to the manufacturers' instructions and acquired through LSR Fortessa (BD). Flowjo software v10 was used to analyze the data.
Cytokine analysis
On day 3, supernatants from the treated PC histocultures were collected and stored at −80°C. The presence of TGF-b1, TNFa, IL-2, CD40L, IL-10, IFNg, granzyme B, and IL-12 cytokines was determined through CBA Flex set cytokine beads (BD). Accuri C6 flow cytometry (BD) and FCAP array software was used to measure samples and analyze the samples, respectively.
Statistics
Generalized estimation equations (GEEs) were used to assess tumor growth in time in mouse populations. The quasi information criterion (QIC) was used to compare different working correlation structures in GEE models, and to determine if tumor growth development was either linear or quadratic over time. In addition, analysis of variance (ANOVA) was used to compare models with and without interaction term between treatment group and time. It appears to be that the first-order autoregressive correlation structure had the smallest QIC value, and there was no need for quadratic term for time in any models, but there was need for interaction term between treatment groups and time in all models. The log rank-test and Kaplan–Meier curves were used in survival analyses of the animal experiment.
Human patient samples were analyzed using ANOVA, and if the assumptions of ANOVA did not hold, then the nonparametric Kruskal–Wallis test was used to compare groups. If the ANOVA or Kruskal–Wallis test was statistically significant, then post hoc analyses for pairwise comparison of groups were done (either Tukey's HSD or Dunn's test with Holm multiple testing correction method). p-Value <0.05 was considered as statistically significant. Graph Pad Prism 6 (Graph Pad Prism Software Inc., San Diego, CA), and R statistical software (R Core Team, 2019) was used to make figures and statistical analyses. GEE analyses were carried out using geepack-package. 26
RESULTS
Oncolytic adenovirus therapy kills prostate tumor cells in vitro
To study the cytotoxic ability of viruses in PC cells, we performed a cell killing assay on PC-3 and PC3-MM2 cells. PC-3MM2 is an aggressive subline of PC-3, 27 and both are CD40 negative, 28,29 and thus the armed virus is not expected to be superior to the unarmed virus, as has been published for CD40+ cells (in which there is additional cell killing due to the proapoptotic effect of CD40L). 14 By days 3 and 5, we saw effective cell killing (Fig. 1A, B). By day 7, we observed complete cell killing with the highest dose (i.e., 1,000) VP/cell (Fig. 1A, B). Both of the oncolytic adenoviruses used in this study feature a fully serotype 3 capsid. These types of viruses enter cells through desmoglein 2. 21,30 Since there is no difference in the virus capsid (the difference being in the arming device), no differences in virus entry or postentry replication are expected. Treatment with Ad3-hTERT-CMV-hCD40L exerted antitumor killing profiles similar to the control viruses. Thus, our results show that the presence of a transgene does not negatively impact the oncolytic potency of Ad3-hTERT-CMV-hCD40L.
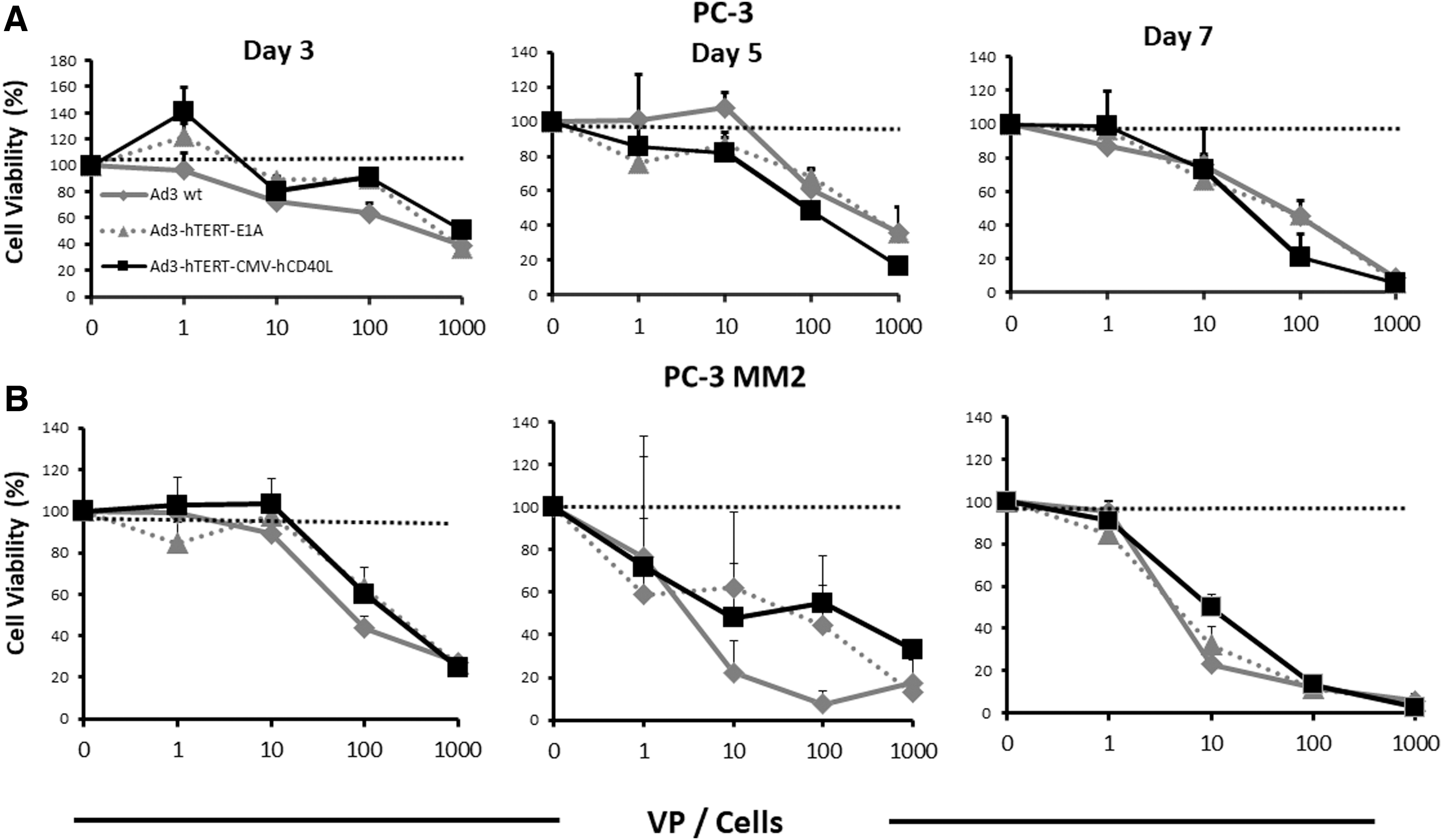
Efficient tumor cells killing ex vivo: oncolytic potency of Ad3-hTERT-CMVhCD40L in PC-3
Antitumor efficacy of oncolytic Ad3 viruses in vivo
To study the oncolytic efficacy of Ad3 viruses, immunodeficient SCID mice were subcutaneously engrafted with PC-3MM2 PC cells. When tumors become measurable and injectable, mice received human PBMCs intravenously on day 0.
After PBMC administration, mice either received Ad3-hTERT-CMV-hCD40L or Ad3-hTERT-E1A or PBS intratumorally on days 1, 3, and 5. Tumor growth was followed until day 25. Our results showed that control groups, that is, mock (PBS) and PBMCs alone, had minimal inhibitory effect on tumor growth, and therefore all mice in these groups were euthanized by day 17. Of note, in this part of experiment, mice did not receive DC because the goal was to study the antitumor effect of oncolysis.
Mice treated with PBMCs plus Ad3-hTERT-E1A showed statistically significant reduced tumor growth as compared with mock (p = 0.004) and PBMCs (p < 0.001) only groups (Fig. 2A). There is no significant difference between PBMCs plus Ad3-hTERT-E1A and PBMCs plus Ad3-hTERT-CMV-hCD40L. Thus, the oncolytic Ad3-hTERT-CMV-hCD40L virus was as potent as the unarmed control virus Ad3-hTERT-E1A in humanized mice bearing PC. Moreover, Ad3-hTERT-CMV-hCD40L and Ad3-hTERT-E1A increased mouse survival (p = 0.071, p = 0.034, respectively) as compared with the PBMCs only group. Thus, oncolytic Ad3 viruses were shown to improve cancer-specific survival (Fig. 2B).
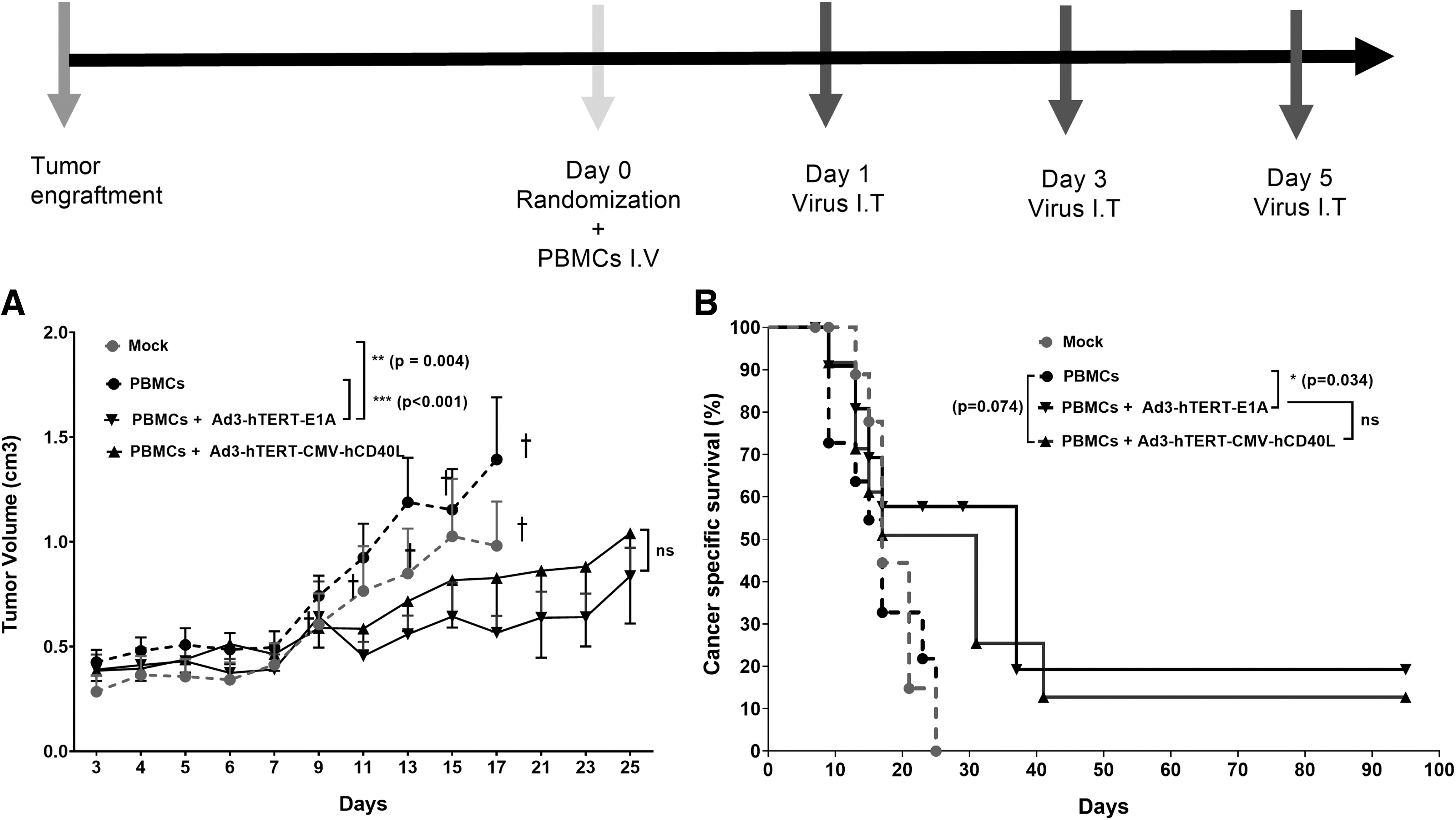
Antitumor effects of oncolytic viruses without DC therapy: schematic presentation. Antitumor efficacy
Unarmed Ad3-hTERT-E1A does not enhance the antitumor efficacy of adoptively transferred DCs in vivo
To evaluate the impact of unarmed virus Ad3-hTERT-E1A on DC therapy, in the second part of the same animal experiment, humanized mice were treated with either PBS or Ad3-hTERT-E1A or DCs or with both Ad3-hTERT-E1A and DCs intratumorally on alternative days. Treatment with Ad3-hTERT-E1A statistically significantly enhanced antitumor effect as compared with mock (p = 0.002) and PBMCs plus DCs group (p = 0.003). However, we did not find any significant difference between the groups treated with Ad3-hTERT-E1A with or without DCs (Fig. 3A). Therefore, unarmed virus did not enhance DC therapy. Nevertheless, due to oncolytic effects, cancer-specific survival data showed somewhat prolonged survival in the group receiving Ad3-hTERT-E1A compared with the PBMCs plus DCs group (Fig. 3B, p = 0.057). However, we did not find significant difference between groups receiving Ad3-hTERT-E1A with or without DCs. Thus, our results show that unarmed Ad3-hTERT-E1A does not enhance the antitumor efficacy of adoptively transferred DCs, but it does have oncolytic potency resulting in antitumor effects.
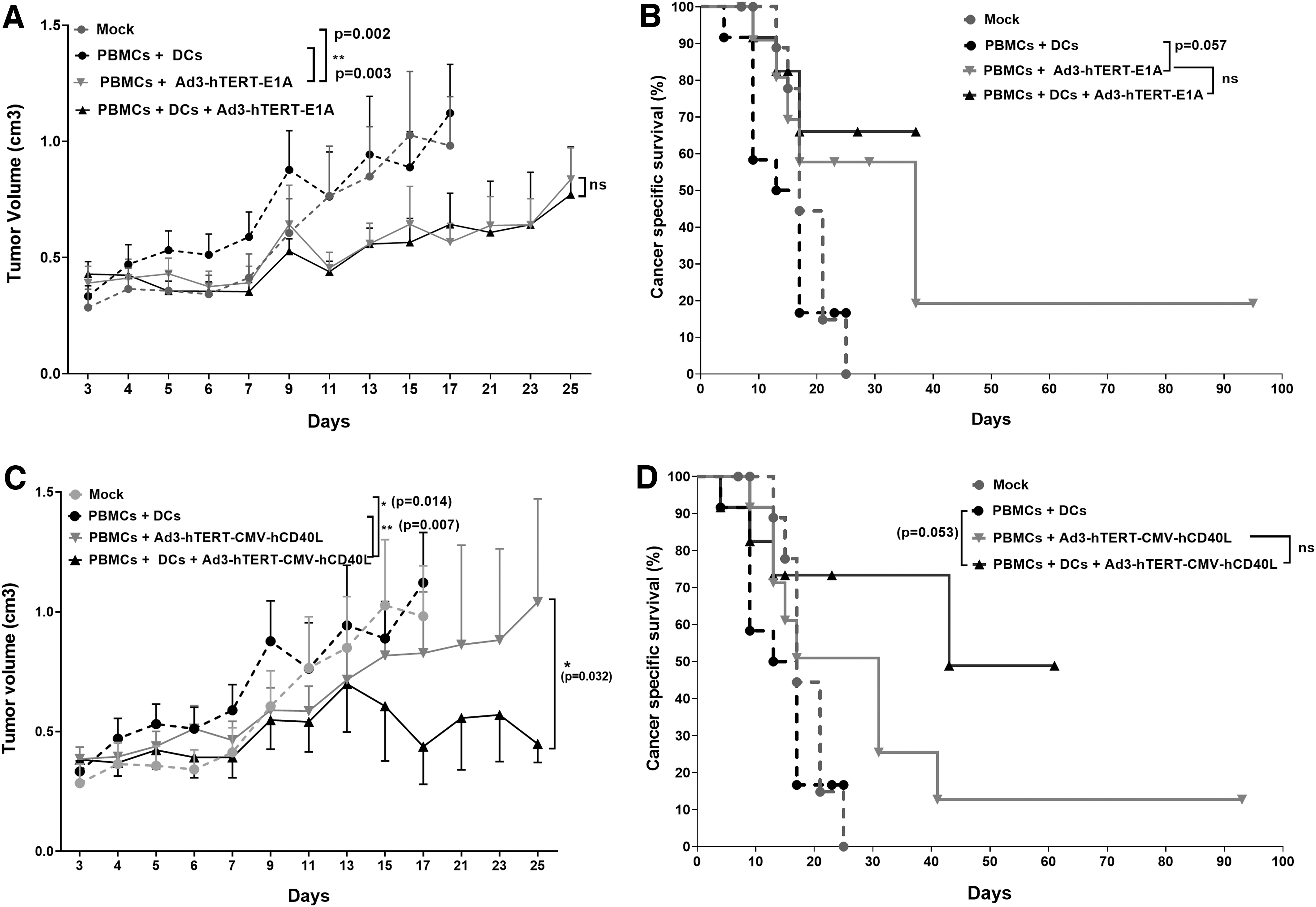
Enhanced antitumor effects and survival in mice: schematic presentation. Antitumor efficacy and cancer-specific survival of humanized mice receiving injections of the unarmed control virus Ad3-hTERT-E1A
CD40L-armed Ad3 enhances the antitumor efficacy of adoptively transferred DCs in vivo
To study the ability of Ad3 armed with CD40L (Ad3-hTERT-CMV-hCD40L) to impact DC therapy, we used the same conditions as in the second part of the same animal experiment, except that instead of the unarmed Ad3-hTERT-E1A virus, we used the armed Ad3-hTERT-CMV-hCD40L virus. The control group treated with PBMCs plus DCs showed minimal tumor growth control. The group treated with PBMCs plus Ad3-hTERT-CMV-hCD40L plus DCs showed significant antitumor efficacy as compared with all other groups, that is, mock (p = 0.014), PBMCs plus DCs (p = 0.007), and PBMCs plus Ad3-hTERT-CMV-hCD40L (p = 0.032) group (Fig. 3C). The average of tumor volumes of PBMCs plus Ad3-hTERT-CMV-hCD40L group is higher than the average of the group treated with Ad3-hTERT-CMV-hCD40L plus DCs plus PBMCs in whole follow-up (data not shown).
Moreover, cancer-specific survival data showed prolonged survival in the group with PBMCs plus Ad3-hTERT-CMV-hCD40L plus DCs compared with PBMCs plus DCs (p = 0.053) (Fig. 3D). We saw improved survival in the groups treated with Ad3-hTERT-CMV-hCD40L, but we did not see significant differences between the PBMCs plus Ad3-hTERT-CMV-hCD40L and PBMCs plus Ad3-hTERT-CMV-hCD40L plus DCs (p = 0.221).
Ad3-hTERT-CMV-hCD40L induces maturation of DCs in the tumor microenvironment of human PC samples
An immunosuppressive tumor microenvironment can be overcome if appropriate stimuli are introduced. Five human PC samples were available to study this (Table 1). As usual in humans, some differences were seen between samples. However, it appeared that treatment with Ad3-hTERT-CMV-hCD40L, as compared with mock and Ad3-hTERT-E1A, statistically significantly induced changes in DC maturation markers CD83 (p = 0.0057, p = 0.0194, respectively), CD80 (p = 0.0001, p = 0.0288, respectively), and CD86 (p = 0.0009, p = 0.1706, respectively) (Fig. 4B–D). We saw robust upregulation of DC maturation markers in the group treated with Ad3-hTERT-CMV-hCD40L as compared with other groups in four of five samples on day 3. In the group treated with Ad3-hTERT-E1A, we observed some increase in DC maturation. Our results showed that Ad3-hTERT-CMV-hCD40L infection in tumor microenvironment leads to the expression of virally expressed CD40L, which in turn induces DC maturation.

Virally expressed hCD40L induces DC maturation in tumor histocultures: Patient histocultures were treated either with viruses or left uninfected
Characteristics of prostate cancer patients
PSA, prostate-specific antigen; RALP, robotic-assisted laparoscopic prostatectomy; TNM, pathological tumor-node metastasis.
Ad3-hTERT-CMV-hCD40L induces immunostimulatory modulation in the tumor microenvironment
PC patient histocultures were treated with Ad3-hTERT-CMV-hCD40L or Ad3-hTERT-E1A. These adenoviruses alter the immunological status of the tumor microenvironment, which is reflected also in the production of cytokines (Fig. 5). Ad3-hTERT-CMV-hCD40L therapy, as compared with mock and Ad3-hTERT-E1A, induced production of proinflammatory cytokines IL-2 (p = 0.0172, p = 0.354, respectively), TNF-a (p = 0.0079, p = 0.120, respectively), IL-12 (p < 0.0001, p < 0.01, respectively), granzyme B (p < 0.002, p = 0.0285, respectively), IFNg (p < 0.0001, p < 0.01, respectively), and CD40L (p < 0.0001, p < 0.001, respectively) (Fig. 5A). Because CD40L-armed virus is capable of inducing the production of CD40L, the level of CD40L is very high on day 3 as compared with the mock or Ad3-hTERT-E1A treated groups (Fig. 5A).
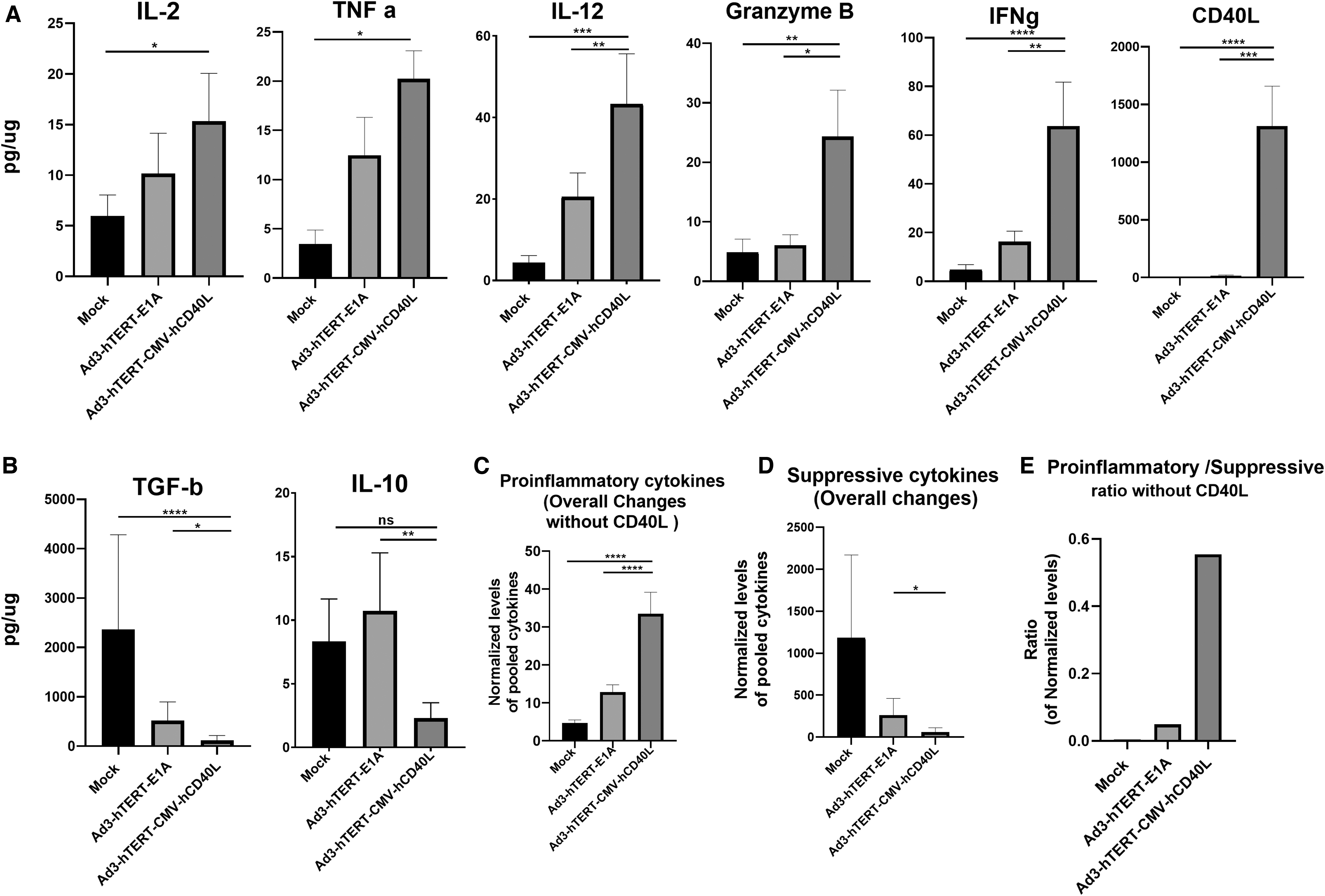
Changes in cytokine profile in tumor histocultures. Patient histocultures were treated either with viruses or left uninfected. Supernatants were collected from histocultures on day 3 for analysis of IL-2, TNFa, IL-12, granzyme B, IFNg and CD40L, *normalized pooled proinflammatory cytokines
In comparison with mock (untreated) and Ad3-hTERT-E1A, a trend for significantly reduced presence of immunosuppressive cytokines IL-10 (p = 0.183 (non-sig); p = 0.0074, respectively) and TGF-b1 (p < 0.0001, p < 0.013, respectively) was found in histocultures treated with Ad3-hTERT-CMV-hCD40L on day 3 (Fig. 5B).
The overall presence of proinflammatory (excluding CD40L as Ad3-hTERT-CMV-CD40L upon replication express CD40L) cytokines in patient tumor histocultures treated with Ad3-hTERT-CMV-hCD40L differed significantly from the mock and Ad3-hTERT-E1A (p < 0.0001) on day 3 (Fig. 5C). The overall presence of suppressive cytokines in the same histocultures treated with Ad3-hTERT-CMV-hCD40L differed significantly from the Ad3-hTERT-E1A (p < 0.01) on day 3 (Fig. 5D). In group treated with Ad3-hTERT-CMV-hCD40L, these immune modulations led to a high proinflammatory to suppressive ratio as compared with other groups (data not shown). Even upon exclusion of CD40L (produced by the virus), there were clear changes (Fig. 5E).
DISCUSSION
DCs are professional antigen-presenting cells, which can be activated in situ, or they can be activated and amplified ex vivo and returned to the patient as an adoptive cell therapy. DC therapy is an attractive immunotherapeutic approach due to the unique capacity of DCs to activate and regulate T cell responses through antigen presentation. 31 –33 Thus, they play an important role in inducing innate and adoptive immune responses.
The safety and immunogenicity of DC therapy have been demonstrated in many clinical trials. Of note, proof-of-concept efficacy has also been seen, using DCs loaded with prostate antigens (either messenger RNA [mRNA], peptides, or proteins). 34 –38 However, efficacy has usually been transient and seen only in a proportion of patients. Therefore, while the approach has been safe, there is much room for improvement in efficacy.
There are number of factors that affect the efficacy of a DC-based vaccine. For example, reduced expression of TAAs by tumors contributes to cancer cell immune evasion. Moreover, overexpression of molecules such as immune checkpoints PD-L1, PD-1, CTLA-4, and cellular subsets, such as myeloid-derived suppressor cells and Treg, contributes to immunosuppression in the tumor microenvironment. 39,40 Furthermore, impaired DC function such as insufficient presentation of antigens, migratory capacity, and cytokine release facilitates immune evasion and tumor progression. 41 –45 Also ex vivo culturing conditions influence the behavior of DCs. Thus, despite the achievement of some therapeutic benefits, additional strategies are needed to elevate the efficacy of DC therapy to a level compatible with clinical use.
In this regard, oncolytic adenovirus Ad3-hTERT-CMV-hCD40L is an attractive approach to facilitate DC therapy. 16,17 Our previous work has shown the capability of Ad3-hTERT-CMV-CD40L and virally expressed CD40L 14,24 to induce maturation of DCs. 16,17 Moreover, we have demonstrated the ability of these matured DCs to activate cytotoxic T cells ex vivo and in vivo. 16,17 As human CD40L is not active in mice, 14,24 in some studies we used nonreplicating adenovirus expressing murine CD40L to study immunological aspects. 14,17 We have shown that expression of CD40L in the tumor microenvironment recruits DCs and is able to stimulate them, resulting in induction of antitumor immune responses. 17
In a study in a humanized mouse model engrafted with human lung cancer A549, Ad3-hTERT-CMV-hCD40L significantly activated DCs, which were then able to activate cytotoxic T cells and Th1 immune responses, resulting in 100% survival. 16 In this study, we have demonstrated the ability of Ad3-hTERT-CMV-CD40L to enhance DC therapy for the treatment of PC. Our preclinical data provide proof of principle for Ad3-hTERT-CMV-hCD40L (also known as TILT-234) as a promising candidate for clinical translation in PC patients receiving DC therapy.
In prostate tumor histocultures, oncolytic replication of Ad3-hTERT-CMV-hCD40L and concomitant hCD40L expression from infected cells induced DC maturation and production of proinflammatory cytokines. By extension, our studies predict that this would happen also in the tumor microenvironment of prostate cancer patients'. As seen with our histocultures, the production of proinflammatory cytokines coincided with a decrease in immunosuppressive cytokines, and a dramatic increase in the ratio of proinflammatory to immunosuppressive cytokines in the tumor microenvironment.
The histoculture system used here utilized tumor explants obtained fresh from patients. They were immediately subjected to analysis without allowing them to transform into a cell line. While a cell line would have some practical advantages, such as unlimited growth and the feasibility of freezing the cells down for future use, the selection process associated with cell line generation would have lost important aspects inherent to fresh tumors. For example, our samples had also the tumor microenvironment present, with its various cell types. Thus, the histocultures represent a clinically relevant model system.
The ability of adenoviruses to selectively replicate in a wide variety of tumors, and to modulate the tumor microenvironment through the expression of immunostimulatory transgene at the tumor site, makes them an attractive platform for cancer immunotherapy. 46 –48 Moreover, oncolytic adenoviruses have demonstrated a good safety profile in clinical use. 20,49 –52 Of note, it has been seen that oncolytic adenoviruses are able to induce antitumor immune responses in humans. 47,53 –58
Human data have also indicated that the Ad3 capsid may be different from the ubiquitous Ad5, in that it allows intravenous delivery, resulting in efficacy even without arming. 21 Adenovirus serotype 3 binds to desmoglein 2, which is expressed abundantly in advanced tumors. 59 Patients treated with Ad3 did not show serious adverse events leading to patient hospitalization. Systemic administration is a clinically attractive approach for the treatment of disseminated tumors. As reported by Hemminki et al. in 2012, a total of 25 patients were treated with Ad3 virus. Virus was administered intravenously only (with no intratumoral administration) to seven patients. The treatment was well tolerated, and in six of seven patients efficacy was seen. 21 A drawback of systemic delivery is induction of high neutralizing antibodies (Nabs) that would affect the additional systemic treatments with the same virus. However, despite the presence of Nabs, oncolytic adenovirus has been seen to reach tumors upon systemic administration, because of the virus' ability to hitchhike on cells to avoid Nabs. 60,61 Therefore, Nabs may not present an absolute obstacle for systemic delivery.
Taken together with promising preclinical and clinical data on CD40L in the context of oncolytic adenovirus, 11,17 these human data sets led us to construct Ad3-hTERT-CMVh-CD40L. This virus allows intravenous delivery in humans and is particularly appealing for enabling DC therapy in PC patients.
In summary, our findings provide rationale for clinical trials using Ad3-hTERT-CMVh-CD40L for DC vaccination in PC patients with currently incurable tumors.
Footnotes
ACKNOWLEDGMENTS
We thank Minna Oksanen and Susanna Grönberg-Vähä-Koskela for their expert assistance.
AUTHOR DISCLOSURE
A.H. and O.H. are shareholders in TILT Biotherapeutics Ltd. A.H. is shareholder in Targovax ASA, and A.H., R.H., J.S., V.C.-C., and R.K. are employees of TILT Biotherapeutics Ltd. The other authors declare that they have no competing financial interest.
FUNDING INFORMATION
This study was supported by KLTO (Doctoral School for Clinical Research programme at the University of Helsinki), by Jane and Aatos Erkko Foundation, HUCH Research Funds (VTR), Sigrid Juselius Foundation, Finnish Cancer Organizations, University of Helsinki, Novo Nordisk Foundation, Päivikki and Sakari Sohlberg Foundation, and TILT Biotherapeutics Ltd. We thank Albert Ehrnrooth and Karl Fazer for research support.