
Abstract
Clustered regularly interspaced short palindromic repeat (CRISPR)-based technology has been adapted to achieve a wide range of genome modifications, including transcription regulation. The focus of this review is on the application of CRISPR-based platforms such as nuclease-deficient Cas9 and Cas12a, to achieve targeted gene activation. We review studies to date that have used CRISPR-based activation technology for the elucidation of biological mechanism and disease correction, as well as its application in genetic screens as a powerful tool for high-throughput genotype–phenotype mapping. In addition to our synthesis and critical analysis of published studies, we explore key considerations for the potential clinical translation of CRISPR-based activation technology.
Introduction
An organism's genetic blueprint is brought to life by the coordinated regulation of gene expression, resulting in a dynamic cellular transcription program. Such regulation is achieved via mechanisms that either act to activate or repress expression of genes via binding of proteins or inducing chemical changes to DNA regulatory elements, ultimately controlling how much RNA is created. In this review, we discuss the adaptation of clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 technology for the specific purpose of achieving gene upregulation. Before the emergence of programmable nucleases, targeted gene activation by transcription activator binding or epigenetic remodeling of endogenous loci was not an easy feat, prompting the use of exogenous overexpression gene constructs to increase gene expression as an alternative. Nevertheless, decades of research into the mechanisms of gene regulation have enabled re-engineering of the CRISPR-Cas9 platform to incorporate transcription effectors known to either facilitate assembly of the transcription machinery or catalyze epigenetic remodeling to promote gene activation. Herein, we summarize existing CRISPR-based activation platforms and their various implementations that result in gene activation without modifying the genome sequence. Early uses of the technology began with single-gene applications, but quickly matured to facilitate multiplex-gene activation for genome-scale genetic screens. Once established, these methodologies enabled systematic discoveries across a wide breadth of biological phenomena such as in identifying factors involved in drug resistance, viral infection, and growth rate modulators. A major area of research clearly empowered by CRISPR activation technology is in the understanding of cellular reprogramming and differentiation, with a significant proportion of studies falling under this category. We review these studies and the means by which they use CRISPR activation technology to achieve targeted activation of endogenous factors underlying cell fate decisions, along with advantages of coupling an inducible system. As a step toward clinical translation, we also discuss the in vivo studies that have utilized CRISPR activation technology in the context of genetic compensation to counter a subset of applicable disease states, as well as future areas of improvement. Thus, we aim to convey the versatility of CRISPR activation technologies, as both a research tool and as a form of genetic therapy for future clinical translation.
CRISPR-Cas 9 FOR GENOME MODIFICATIONS
Programmable nucleases for genome engineering have been popularized by the emergence of CRISPR-based technology, capable of RNA-guided homing to targeted regions of the genome to induce desired modifications. CRISPR-Cas9 has eclipsed other genome editing tools such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) due to its increased flexibility and scalability, largely owing to its programmable single-guide RNA (sgRNA or gRNA) component. In contrast to ZFNs and TALENs, which require reengineering of the endonuclease for each new target site, CRISPR-based systems can easily be adapted to new target sites as well as simplifying multiplexed strategies for multilocus editing or transcriptional regulation. The classic class II CRISPR complex originally characterized as part of the adaptive immune system in bacteria was modified for eukaryotic genome engineering and consists of two components: a Cas9 endonuclease and an sgRNA. 1 –3 The sgRNA is a synthetic RNA inclusive of a scaffold sequence for Cas9-binding and a spacer sequence composed of ∼20 nucleotides that define the genomic target through DNA–RNA complementary base pairing. Immediately downstream of the DNA sequence targeted by the Cas9 nuclease is the protospacer adjacent motif (PAM), an ∼2–6 bp sequence that define the Cas9 target site in the genome for editing. The canonical PAM associated with Streptococcus pyogenes is 5′-NGG-3′, where N defines any base, followed by two guanines. Additional PAMs have been associated with Cas9 orthologs isolated from other bacterial species and can serve as alternatives when no suitable SpCas9 PAM sequence is present near the desired genome editing site. 4 Cas9 orthologs also vary in size, for example, Cas9 from Staphylococcus aureus (SaCas9) and Campylobacter jejuni (CjCas9) is significantly smaller in size than SpCas9 and therefore preferable for packaging into adeno-associated viruses (AAV) for in vivo experiments. 5,6
More recently, another RNA-guided endonuclease also belonging to the class II CRISPR/Cas system known as Cpf1 (or Cas12a) was discovered and has since been used in a variety of in vitro and in vivo gene editing applications. 7 –9 Reported to possess lower off-target effects than CRISPR-Cas9, CRISPR-Cpf1 utilizes a single crRNA to recognize a T-rich PAM and results in “sticky end” endonuclease activity at target sites. 10,11
Initial application of the CRISPR-Cas system was to knock out target genes, achieved via induction of double-stranded breaks at targeted genomic loci to initiate an error-prone repair mechanism known as nonhomologous end-joining (NHEJ). 2 NHEJ frequently results in frame shifting indels, introducing premature stop codons and subsequent nonsense-mediated decay of transcripts. Modifications to Cas proteins have since expanded the genome manipulation potential of CRISPR-Cas systems to enable other edits such as targeted gene activation (CRISPRa), gene repression/inhibition (CRISPRi), base-editing, and epigenetic remodeling. 12,13 This review is centered on the engineered CRISPR-Cas system to enable targeted gene activation with a focus on CRISPRa platforms.
GENE ACTIVATION USING CRISPRa PLATFORMS
The Cas9 endonuclease consists of the RuvC and HNH nuclease domains that can be inactivated by point mutations (D10A and H840A in SpCas9), resulting in a nuclease-deactivated Cas9 (dCas9) incapable of cleaving DNA but still retaining its DNA binding ability. 3 The discovery of dCas9 forms the basis for a range of CRISPR-based genome modification technologies that enable RNA-guided control of gene expression. 14 The majority of this review focuses on how dCas9 has been leveraged as a homing machinery to specifically recruit transcriptional activator domains to targeted promoter regions within mammalian genomes. Although less explored, the CRISPR-Cpf1 nuclease has also been engineered for targeted gene activation. Catalytically inactive forms of Cpf1 (dCpf1) have been generated from point mutations in nuclease domains of Cpf1 from Lachnospiraceae bacterium (D832A, E925A) 15,16 and Acidaminococcus sp. (D908A or E993A). 16,17 CRISPRa platforms have also been developed for gene activation in bacterial genomes but are comparatively less flexible than those developed for eukaryotic genomes due to the absence of effective gene activators, limited portability across different bacterial species, and a smaller targetable region upstream of the transcription start site (TSS). 18 –20
First-generation CRISPRa platforms fused dCas9 to multiple copies of the herpes simplex virus transcriptional activator VP16 or a single copy of the NF-kB trans-activating subunit p65, both naturally occurring activation domains. 21 Increasing copies of tandem VP16 activation domains fused to dCas9 (VP48, VP96, VP192) unsurprisingly resulted in higher gene activation, with VP192 generating up to 60- to 70-fold more expression of a target gene than VP48. 22 The most widely used VP16-based transactivator in CRISPRa platforms is VP64, composed of four tandem VP16 domains commonly fused to the C-terminal of dCas9 in most studies. 23 –28 One study reported that the fusion of VP64 domains to both the N- and C-terminus of dCas9 significantly improved the activation capacity when compared with single VP64 fusion to the C- or N-terminus alone. 27 The authors noted that having two VP64 domains increases the probability of transcription factor homing than with just one but did not provide a rationale for why separating the two VP64 domains would perform better than simply placing them in tandem (i.e., VP128).
Driven by the goal of achieving more potent and robust gene upregulation, second-generation activation CRISPRa platforms incorporating hybrid activation complexes were developed, for example, synergic activation mediator (SAM), VPR, and SunTag. The SAM consists of dCas9-VP64 with a modified sgRNA backbone that incorporates two MS2 hairpins (bacteriophage coat proteins) at the tetraloop and stem loop 2. These MS2 hairpins are used to recruit two additional activation domains—p65 (NF-κB trans-activating subunit) and HSF1 (from human heat-shock factor 1), which bind as dimers. A total of four sets of p65-HSF1 dimers are recruited at these loops, and along with VP64, giving a total of twelve activation domains in the SAM system. The VPR system consists of the fusion of VP64, p65, and Rta (Epstein–Barr virus transcription activator) to c-terminal of dCas9, with each domain separated by a short amino acid linker. A total of six activation domains are recruited in the VPR system.
The SunTag system is based on a repeating peptide array, consisting of 10 tandem GCN4 epitopes (derived from the Gcn4 yeast transcription factor), capable of recruiting multiple copies of an antibody-fusion protein, in this case VP64. 29,30 A total of 40 activation domains (10 VP64 units) are recruited to a single locus using the SunTag system. See article by Chavez et al. 31 for graphical representations of first- and second-generation CRISPRa platforms.
A recent study performed a systematic comparison of the most commonly used CRISPRa platforms across different cell types. 31 In this study, second-generation activators (VPR, SAM, SunTag) achieved greater upregulation than the first-generation activators (VP64, P300) when compared across a panel of coding and noncoding genes. The SAM platform achieved highest levels of endogenous gene activation most consistently when tested in HEK293 cells, but notably these levels were always within fivefold difference with respect to the VPR and SunTag platforms. This trend, however, did not hold when the different CRISPRa platforms were tested in other human cell lines (HeLa, U-2 OS, MCF7), where SunTag and VPR platforms were demonstrated to be more potent. These results suggest cell line-specific differences in the activation potential of CRISPRa platforms, which the authors claim were not due to differences in the basal expression of targeted genes, and thus did not affect their overall activation potential across different cell lines. Additional nonhuman cell lines (mouse N2A and 3T3, Drosophila S2R+) were tested and similarly conveyed the superiority of second-generation activators compared with the representative first-generation platform (VP64). However, the potency of second-generation platforms varied on the target gene analyzed, conveying no clear winner. The study also tested the multiplexing potential of second-generation activators to simultaneously upregulate multiple genes and observed that all systems performed comparably. 31 Lastly, the authors also report that the previously described phenomenon of synergistic upregulation using multiple guides in conjunction with VP64 platforms also held true across second-generation platforms; and that their ability to boost activation as aided by multiple guides suggests that these platforms have not yet reached maximal activation potential on their own. Absent from this study is the comparison with dual-VP64 activators, typically composed of VP64 units fused to both the N- and C-terminus of dCas9, reported in other studies to be more potent than single-VP64 activators. 27,32 Despite the increased potency of second-generation platforms, there is still a case to be made for the utility of first-generation platforms for several reasons. First, second-generation activators (e.g., VPR and SAM) are significantly larger in size—at least 10 times larger than VP64 alone (∼150 bp), thus requiring its components to be split across multiple AAV vectors for delivery into in vivo models. Thus, given the requirement for multiple vectors to transduce target tissue to achieve activation, delivery efficiencies are reduced and may counteract the improvements in activation conferred by second-generation systems.
Seeing that there is currently no precedence for dual-vector AAV administration in the clinic, the use of second-generation platforms for gene activation is therefore seen as less clinically translatable. In addition, the highest attainable gene expression level is not always the desired result, given that there are often known phenotypic and molecular consequences associated with genetic overexpression. Thus, depending on the gene of interest and application, the levels of gene activation attainable by first-generation platforms may prove to be sufficient.
GENE ACTIVATION USING CRISPR-BASED EPIGENETIC MODIFIERS
As an alternative to CRISPRa platforms for gene activation, studies have reported the fusion of dCas systems to epigenome-modifying enzymes for targeted modification of epigenetic marks to affect transcriptional regulation. Unlike CRISPRa platforms that rely on recruitment of transcriptional machinery, these approaches rely on the enzymatic modification (e.g., acetylation and methylation) of chromatin state to activate nearby genes. One example is the fusion of dCas9 to the catalytic histone acetyltransferase core domain of the human E1A-associated protein p300 (dCas9–p300). 33 The p300 protein alters chromatin structure via acetylation of histone cores within nucleosomes located in proximity to promoter regions. 34 Nucleosome acetylation is accompanied by increased accessibility of the targeted chromatin region, thereby allowing DNA binding proteins to interact with the exposed regions to initiate transcription. Hilton et al. 33 demonstrated that targeting of dCas9–p300 leads to the acetylation of histone H3 lysine 27 (H3K27ac) at gene regulatory regions, and the subsequent activation of downstream genes. Gene activation resulting from targeting of dCas9–p300 to promoter regions was further shown to be higher with dCas9–p300 than with single-activator domains such as dCas9-VP64. In addition to achieving gene activation from targeting promoters, it was shown that dCas9–p300 can also lead to robust transcriptional activation when targeted to proximal and distal enhancer regions. This was demonstrated when dCas9–p300 was targeted to the distal regulatory region and core enhancer of the MYOD locus, which resulted in significant transcription. whereas dCas9-VP64 did not. Targeting of dCas9–300 to the upstream proximal and distal enhancer regions of OCT4 also showed significant transcriptional activation not observed by dCas9-VP64.
Similar studies have also demonstrated that fusion of dCpf1 with p300 (dCpf1–p300) can also successfully activate gene expression in human cells from targeting the promoter and enhancer regions of selected genes (MYOD, IL1RN, OCT4). 16
Another epigenome marker of active gene transcription is the trimethylation of H3K4 (H3K4me3) at promoter regions. Cano-Rodriguez et al. 35 demonstrated activation of endogenous gene expression by local induction of H3K4me3 using a fusion of dCas9 and the catalytic domain of histone methyltransferase PRDM9 (dCas9-PRDM9). Upregulation via dCas9-PRDM9 was reported to be more subtle than by dCas9-VP64, but was able to achieve more durable epigenetic changes conducive to reduced methylation marks at the target site. 35 A known marker of gene silencing is the presence of multiple methylated CpG sites in promoter regions. Correspondingly, several studies have used a demethylation strategy to achieve transcription activation by targeting the catalytic domain of Tet1 (ten-eleven translocation) fused to dCas9 to regions of hypermethylated genes. 36 –39 In one example, targeted demethylation of the BDNF promoter IV and the MyoD distal enhancer by dCas9-Tet1 achieved robust activation of BDNF expression in postmitotic neurons and MyoD reprogramming of fibroblasts into myoblasts, respectively. 37 These studies collectively establish the utility of epigenome editing strategies to achieve targeted gene activation as an alternative to CRISPRa platforms, however, its in vivo translatability using AAV-delivery methods is hampered due to the significantly larger size of these enzymes compared with the transcription effector domains used in CRISPRa platforms.
GUIDE RNA DESIGN FOR CRISPRa
Numerous studies have investigated aspects of gRNA design properties that have the potential to affect the efficiency of CRISPR-based transcriptional regulation. These properties include length of gRNA, proximity to TSS, strand-bias, and chromatin accessibility. 40 Together, these factors can broadly be categorized as either location- or sequence-based consideration, with each property having a different level of impact depending on the desired CRISPR application.
A key factor in determining target specificity for nuclease-activated Cas9 is the length of gRNA spacer sequence, with the consensus-reported minimum of 17-nt for functionality. 40,41 Both CRISPRi and CRISPRa platforms are observed to function most optimally using a 20-nt gRNA spacer sequence, with reduced efficacy associated with truncated or longer sequences. 14,30,42
Guide RNA targeting relative to a gene's TSS is also known to affect the functionality of transcriptional effectors. Gilbert et al. 30 performed a comprehensive study to investigate positional effects of gRNA's relative to a gene's TSS using both CRISPRi and CRISPRa systems. In this study, a gRNA tiling library was constructed within a 10 kb window surrounding the TSS of 49 genes to identify target considerations for the maximization of transcriptional repression and activation using CRISPRi and CRISPRa, respectively. This study identified that while many gRNA design rules overlap between CRISPRi and CRISPRa systems, such as optimal gRNA length and sequence preferences, a key difference lies in the optimal window for gRNA targeting. 30 The results of this screen revealed that the functional window for CRISPRi is within −50 to +300 bp relative to the TSS of a gene, but most optimally in the ∼50–100 bp region downstream of the TSS. In contrast, the peak of active gRNAs for CRISPRa was observed to be within −400 to −50 bp upstream from the TSS. As typically only a handful of gRNA designs fall in the optimal window, it is not uncommon for all designs to be systemically screened in vitro to determine the best performing sequence. Databases such as Refseq 43 and FANTOM 44 provide continuously updated TSS annotations, which can be used to help define the optimal window for gRNA targeting.
The potential effect of strand-specific bias in CRISPR-based transcriptional regulators has been extensively studied in yeast. 45,46 Farzadfard et al. demonstrated that placing gRNAs at similar positions downstream of the TSS on opposite strands of a promoter conferred similar levels of transcriptional inhibition using CRISPRi systems. 46 Similarly, gRNAs targeted upstream of the TATA box and TSS lead to comparable levels of transcriptional activation using CRISPRa systems. Studies in mammalian cells by Gilbert et al. reported similar findings of no strand-specific bias in their CRISPRi study. 30
The efficiency of CRISPR-based systems is dictated to a large degree by the ability to access and bind target DNA. Therefore, to achieve efficient genomic perturbations in mammalian cells, one should take into account that eukaryotic DNA is typically coiled around histones to form nucleosomes, which in turn affect their accessibility to DNA-binding proteins such as Cas9/dCas9. Correspondingly, several studies have reported chromatin accessibility and nucleosome positioning effects on gRNA efficiency for dCas9-mediated transcriptional regulation. 47,48 These studies concur in their observation of reduced Cas9/dCas9 binding to PAM sites that are located within the nucleosome core, and thus should be avoided.
Several online tools are available to aid in the design of optimal gRNA design specifically for CRISPRa. Some examples include the following: Broad Institute GPP sgRNA designer, E-CRISP, 49 CRISPR-ERA, 50 and CHOPCHOP. 51 Notably, CHOPCHOP v3 incorporates algorithms to calculate nucleosome positioning and accessibility of predicted target sites.
GENETIC SCREENS USING CRISPRa PLATFORMS
High-throughput genetic screens are useful for elucidating phenotype–genotype correlations through unbiased and systematic means. The emergence of CRISPR technology has greatly streamlined the process of performing loss-of-function and gain-of-function genome-scale screens using CRISPRi and CRISPRa libraries, respectively. Before CRISPRa technology, gain-of-function screens were limited to cDNA overexpression libraries, which were expensive to construct, often incomplete in gene isoform representation and achieved nonphysiological overexpression levels. 52 Following the success of CRISPRi platforms for genome-scale knockout screens, 53,54 the CRISPRa platform was subsequently adapted for genome-scale activation screens. 30,55 However, unlike CRISPRi screens, CRISPRa screens have been largely limited to in vitro studies with no in vivo screens reported to date.
A widely used strategy for CRISPRa-based screens is a pooled approach, where a mixed library preparation is applied to a population of cells followed by phenotypic selection. The overall experimental design for in vitro genome-scale activation screens is comparable, regardless of the CRISPRa platform used. Joung et al. provide a detailed guide on how to conduct and analyze both CRISPRi- and CRISPRa-based screens. 52 In brief, a CRISPRa screen begins with establishing a stable cell line that expresses the activation components of a chosen CRISPRa platform (e.g., dCas9-VP64, dCas9-VPR, additional SAM effectors MS2-p65-HSF1). Next, a compatible library of sgRNA plasmids is designed and cloned, or preassembled libraries used in published screens can be acquired from Addgene. Examples of preassembled genome-scale CRISPRa libraries include the CRISPRa-v2, 56 the Calabrese, 57 and SAM libraries 55 —consisting of sgRNA target sequences within 500 bp upstream of the TSS. Pooled plasmid library preps are amplified in bacteria and packaged into lentivirus. The lentivirus library is then transduced into the previously established stable cell line at a low multiplicity of infection to ensure that each cell receives at most one sgRNA construct. A population representing the baseline sgRNA levels is harvested as a reference control sample. Depending on the nature of the screen, a positive, negative, or expression-based selection pressure is applied to the rest of the population (Fig. 1). Positive selection identifies “mutants” that are enriched postselection, whereas negative selection identifies “mutants” that are depleted postselection. Expression-based selection identifies “mutants” that perturb gene or protein expression via application of enrichment assays such as flow cytometry or RNA sequencing. 58,59 Example CRISPRa screens and their respective selective pressures are outlined in Table 1. Cells are harvested at a given time point when the desired phenotypic outcome has been enriched. The sgRNA population is amplified from the reference control and experimental populations. Barcodes may be optionally attached for multiplexing. Samples are analyzed by next-generation sequencing (NGS) followed by statistical analyses to identify candidate genes whose upregulation confers a desired phenotype. Specialized statistical packages such as MAGeCK were designed to assist in the analysis of data from genome-scale CRISPR screens. 60 Candidate genes are usually validated by follow-up experiments such as testing of individual sgRNAs from the library, followed by transcript upregulation analysis via quantitative polymerase chain reaction (qPCR) or protein quantification methods. In choosing candidate genes to prioritize, it is important to keep in mind that false positives and negatives have been noted to occur in CRISPRa screens. 57 False positives can arise from induced activation levels that are not achievable via endogenous means, whereas false negatives can arise from poorly designed sgRNAs that lead to ineffective gene activation, usually resulting from an inaccurately annotated TSS or an inaccessible chromatin region.
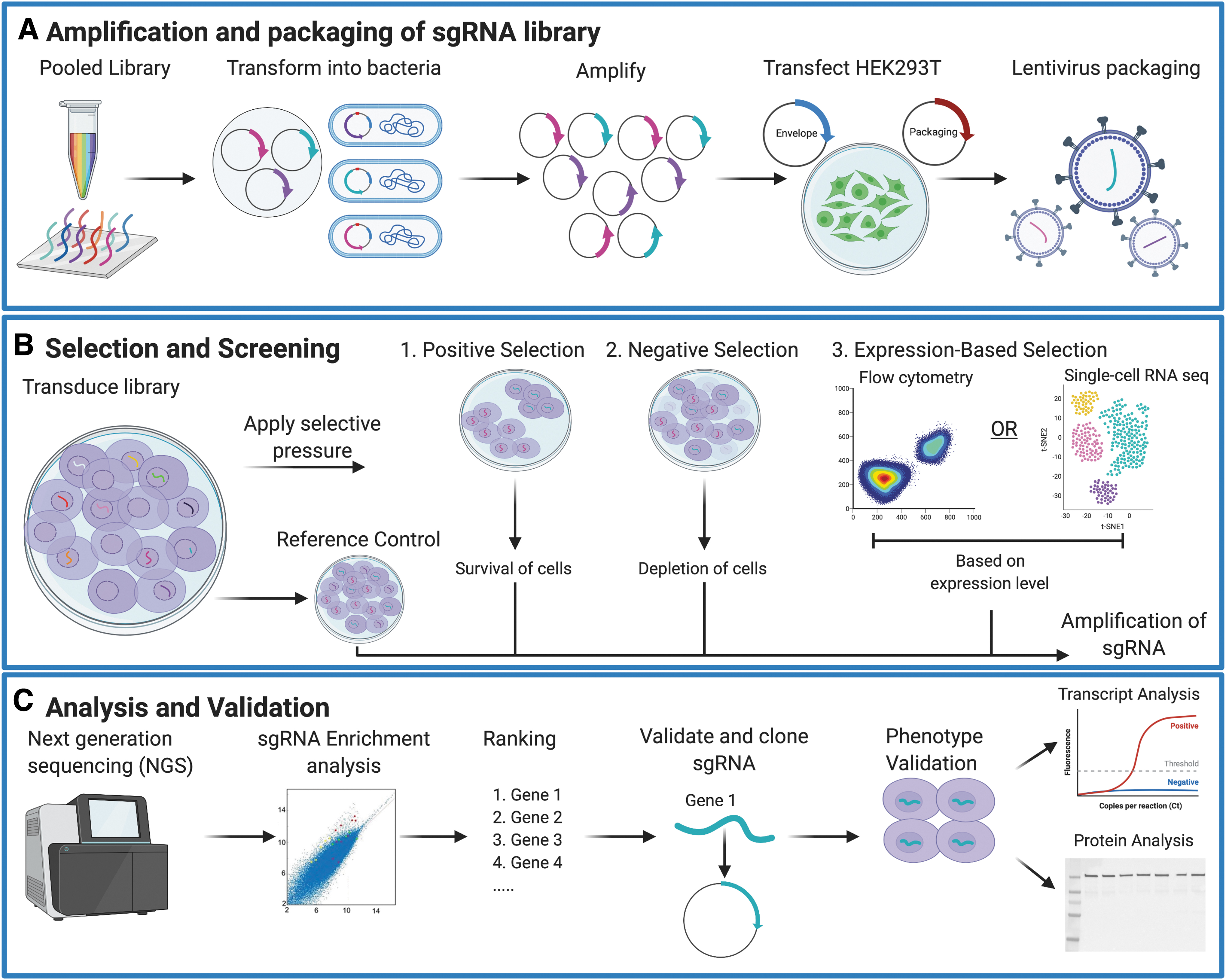
Experimental pipeline for genome-scale CRISPRa screens.
Summary of published in vitro genome-scale CRISPRa screens
dCas9, deactivated Cas9; mESC, mouse embryonic stem cell; ES, embryonic stem; SAM, synergic activation mediator; ZGA, zygotic gene activation; GFP, green fluorescent protein; CRISPR, clustered regularly interspaced short palindromic repeat.
The majority of published CRISPRa screens utilized positive selection strategies, identifying perturbations that confer resistance to selected drugs, 55,57,61 toxins, 30,62 and pathogen infection 63 in resistant cell populations. Results from these positive selection screens all report the discovery of novel genes whose upregulation resulted in rescue from cell death in their respective studies. Importantly, these results point to new drug targets and also serve as potential disease-related biomarkers in response to drug treatment.
Negative selection strategies were used by two published screens that looked at genes whose upregulation caused cells to deplete over time, thus affecting growth rate/proliferation. 30,56 Gene candidates discovered by these studies shown to inhibit growth were reported to be tumor suppressors, transcription factors involved in development and differentiation, and mitosis-related genes.
Examples of incorporating expression-based selection strategies are reported in studies identifying novel factors that lead to cellular reprogramming 58 and differentiation. 64 In the study by Liu et al., 64 mouse embryonic stem cells (mESCs) were differentiated into a neuronal lineage in a positive selection step, followed by a secondary expression-based selection to enrich for neuronal markers via cell sorting. This successful approach was used to derive a genetic interaction map for neuronal fate differentiation, identifying previously unknown factors such as Ezh2 and Mecom. 64 Single-cell RNA sequencing (RNA-seq) is another example of expression-based selection strategy that can be paired with CRISPRa screens. A recent screen on mESCs used an sgRNA library targeting known regulators of maternal zygotic gene activation (ZGA), followed by generation of single-cell transcriptomes using the 10 × genomics platform. 59 From this, a multiomics factor analysis was applied to characterize molecular signatures of ZGA, uncovering 21 novel factors consisting of DNA-binding proteins, chromatin modeling proteins, and transcription factors. Thus, screens using expression-based selection are often useful for elucidating genetic interactions involved in complex biological signaling processes. For additional information on genome-scale CRISPR screens, see reviews by others. 52,65 –67
CELL-TYPE CONVERSION USING CRISPRa
Cellular reprogramming and transdifferentiation of somatic cells are widely used methodologies in biomedical research with applications in disease modeling and drug discovery. The most commonly used strategy thus far has relied on ectopic expression of transgene(s) that encode lineage-specific or pluripotency factors. CRISPRa platforms offer an alternative strategy via activation of endogenous fate-specifying transcription factors to achieve a more “natural” means of cellular reprogramming that are in some cases more efficient, tunable, and durable. For example, transdifferentiation or lineage reprogramming, in which one mature somatic cell is transformed into another mature somatic cell type without an intermediate pluripotent state, can be achieved via single- or multiple-gene activation, and thus amenable to CRISPRa approaches. A single-gene transdifferentiation strategy was demonstrated by Chakraborty et al. who performed direct conversion of primary mouse fibroblasts to skeletal myocytes using a dual-VP64 system to target activation of the endogenous Myod1 gene. 27 The resulting myocytes were induced to fuse into multinucleated myotubes, upregulating myogenic markers such as myosin heavy chain, actin, and desmin both at the transcript and protein levels. In their study, the CRISPRa system was placed under the control of the TetON promoter, with its expression contingent upon doxycycline induction. Interestingly, although doxycycline withdrawal resulted in reduced CRISPRa expression, the endogenous activation of Myod1 was maintained, and the fusion process continued unabated, suggesting stability of the epigenetically remodeled site post-CRISPRa targeting. The study also demonstrated that the sgRNA designed to target either the positive or the negative DNA strand was able to achieve Myod1 upregulation, thus proving that the functionality of the CRISPRa system is independent of the target strand. The overall efficiency of myogenic transdifferentiation was compared between CRISPRa transactivation of endogenous Myod1 and MYOD1 transgene overexpressions. These results conveyed a similar expression level of myogenic markers, with the exception of myogenin, which was shown to be higher in cells with endogenous activation of Myod1. In another study, transdifferentiation of fibroblasts to neuronal cells via multiplex endogenous gene activation was demonstrated by Black et al. 68 via endogenous upregulation of BAM factors (Brn2, Ascl1, Myt1l). In this study, four sgRNAs for each gene were delivered to achieve synergistic gene activation. Although transcriptional upregulation of each endogenous locus was detected, expression of BAM factors via exogenous transgene transfection resulted in higher mRNA encoding each factor. 68 Nevertheless, endogenously activated BAM factors sufficiently resulted in expression of the neuronal markers Tuj1 and Map2, and subsequent neuronal morphologies. The authors also noted that despite depletion of exogenous BAM factors and sgRNAs for endogenous activation after transient transfection, endogenously activated factor genes remained high, suggesting sustained epigenetic remodeling of the targeted genomic loci achieved by the CRISPRa approach. This was further supported by chromatin immunoprecipitation (ChIP) qPCR data showing significant enrichment in H3K27ac and H3k4me3 surrounding the promoter sequences of Brn2 and Ascl1 loci—markers of histone H3 modifications, not observed in BAM factor transgene overexpression.
Another example of cell-type conversion that can be facilitated by CRISPRa technology is cellular reprogramming. Mature somatic cell reprogramming into an induced pluripotent state is achieved by activation of transcription factor genes such as OCT4, SOX2, and MYC. Several studies have utilized CRISPRa strategies to achieve activation of these endogenous genes that have otherwise been achieved through exogenous means. 22,23,69 –71 OCT4 expression was increased up to 70-fold with dCas9-VP192 compared with control, using five simultaneous sgRNAs. 22 Additional pluripotency genes SOX2, NANOG, LIN28, KLF4, and CDH1 were also targeted for upregulation with variable levels observed, in which the authors attribute to either a less permissive chromatin state or a high level of starting basal expression. These results convey that the activation potential indeed varies between genes, and has been confirmed by other studies that observe weakly expressed genes to have higher activation potential than highly expressed genes. 31 Nevertheless, successful derivation of induced pluripotent stem cell (iPSC) from skin fibroblasts was conveyed by expression of additional pluripotency markers (e.g., NANOG, TRA-1-60, TRA-1-81) and was subsequently able to differentiate into three germ line derivatives. Reprogramming efficiency of fibroblasts using endogenous OCT4 and transgene overexpression methods was shown to be comparable via properties pertaining to iPSC colony formation. 22 In a similar study by Weltner et al., robust activation of reprogramming factors OCT4, MYC, KLF4, SOX2, and LIN28A was achieved in HEK293, but only OCT4 and SOX2 activation was successfully activated in human fibroblasts when the same sgRNAs were used. 71 Importantly, this suggests that sgRNA efficiencies associated with CRISPRa systems do not always translate across different cell types.
In several of these studies, authors experimented with fusion of inducible systems such as DHFR and TetON to their CRISPRa construct, linking its expression to the addition of trimethoprim or doxycycline, respectively. 22,71 Despite these systems showing some level of leakiness, expression was significantly increased after addition of both molecules, thus demonstrating their potential to be utilized in combination with CRISPRa platforms to control cellular reprogramming and differentiation.
In addition, cell-type conversion in the form of stem cell differentiation into a more specialized cell type has been successfully demonstrated using a dCpf1 CRISPRa-based system. Choi et al. 17 fused dCpf1 to a VPR system and targeted the proximal region of the BMP4 promoter. Upregulation of BMP4 in mesenchymal stem cells by twofold was sufficient to achieve concomitant activation of osteogenic differentiation factors such as OCN, RUNX2, and COL1. After 3 weeks of endogenous BMP4 activation, cells displayed markers of osteogenic differentiation as assessed by increased calcium deposits in vitro via Alizarin red staining. 17
An advantage of using CRISPRa systems over traditional transgene overexpression methods lies in its multiplexing potential, where multiple sgRNAs can simultaneously target multiple loci for concurrent gene activation. This strategy therefore offers a distinct advantage in scenarios where cell fate decisions are controlled by a network of genes. Concatenation of sgRNA expression cassettes has been described for multiplex-gene activation. 22,24 The small size of sgRNA construct compared with those required for transgenic overexpression methods enables efficient simultaneous targeting of multiple-gene loci. Balboa et al. 22 achieved simultaneous activation of endodermal (FOXA2, SOX17) and pancreatic transcription factors (PDX1, NKX6.1) in human iPSCs to shorten the differentiation protocol typically needed to obtain pancreatic progenitors, from 10 to 3 days. Furthermore, multiplex CRISPRa systems enable relative tuning of individual gene expression levels to achieve a desired stoichiometry by adjusting the relative amounts of sgRNAs used. In their studies, Cheng et al. 23 demonstrated that varying levels of OCT4, IL1RN, SOX2 gene activation could be achieved by altering the precise stoichiometry of sgRNAs—a finding anticipated to be useful for studies of gene regulatory networks and systems biology. Together, these studies demonstrate that both single- and multiplex-gene activations via CRISPRa platforms can be used to drive both reprogramming into pluripotency and directed differentiation into specific lineages in a manner that is both rapid and sustained.
IN VIVO GENE ACTIVATION USING CRISPRa
The ability to turn on genes for genetic compensation provides a promising therapeutic strategy for a subset of genetic conditions, including those that would benefit from expressing an alternate gene isoform that is similar to the mutated gene; or increasing wild-type gene expression where loss-of-function mutations in one gene copy result in reduced protein (haploinsufficiency). Although the delivery of extra copies of the missing/mutated gene often provides a more straightforward solution, many of these disease-causing genes exceed the packaging size limitations of AAVs, thus negating gene replacement approaches for therapy. These various genetic compensation strategies have been explored in vivo using a variety of CRISPRa approaches. In one example, the upregulation of Sim1 or M4cr in mouse models harboring heterozygous loss-of-function mutations in these genes demonstrated that targeting the remaining functional copy was able to rescue the obesity phenotype caused by haploinsufficiency. 72 In another example, activation of Scn1a was able to modestly improve seizures and behavioral phenotypes in Dravet syndrome caused by haploinsufficiency of Scna1. 73,74 CRISPRa can also be applicable in recessive disease where both gene copies are nonfunctional and improvements are achieved through the upregulation of functionally related genes. The upregulation of Lama1 was able to rescue the muscle phenotype in Lama2-deficient mice, 32 while switching on utrophin or follistatin was able to rescue muscular dystrophy in a mouse model of Duchenne muscular dystrophy. 75 Conversely, CRISPRa can also be used to induce genetic diseases, where upregulation of Mef2d or Klf15 produces models of cardiac hypertrophy. 76 This can be important for studying diseases where there are no reliable in vivo models currently available. Lastly, upregulation of genes may also confer benefits in nongenetic disease conditions. Examples include reduction of neuronal excitability and decreased seizures in an acquired mouse model of epilepsy in response to overexpression of Kcna1, 77 reduction in kidney fibrosis in Rasa1 mouse models upon activation of Rasal1 and Klotho, 78 and reduction in serum cholesterol levels upon upregulation of Apoa1. 79
CRISPRa platforms have been successfully used in conjunction with viral delivery methods to improve a range of disease phenotypes in mouse models (Table 2). These studies serve to demonstrate the versatility of AAV-CRISPRa strategies in terms of gene targets and tissue specificity, as achieved via the combinatorial use of tissue-specific promoters and AAV serotypes. The majority of these studies have similar overall design strategies involving the use of dCas9 fused to activator proteins, sgRNA targeting upstream of the TSS, tissue-specific regulatory cassettes, and viral-based delivery. Earlier approaches were constrained to packaging SpCas9 and activator proteins within the ∼4.7 kb size limitation of AAV. Such strategies involved splitting SpCas9 at the disordered linker region to be combined post-translationally via inteins 80 or using MS2 RNA loop structures adjacent to the sgRNA to promote binding of transcriptional activators. 75 In addition, these pioneering approaches did not use a dCas9, instead using a truncated sgRNA sequence to reduce unwanted Cas9-induced cutting. More successful approaches transitioned to utilizing dCas9 and a smaller Cas9 species (e.g., SaCas9) to enable addition of regulatory elements and sgRNA for a single-vector design. Due to the resource-intensive nature of mouse experiments, most studies were limited to testing one sgRNA targeting the gene of interest. Selection of sgRNA was typically based on in vitro experimental data where candidate sgRNAs were screened and the one resulting in the highest RNA expression of their target gene was selected. Only one study explicitly considered off-target effects in terms of potential to activate untargeted genes in their sgRNA selection process, 77 whereas the majority used off-target assessment by transcriptomic analysis after in vivo experimentation. Future sgRNA decisions may benefit from factoring protein expression in addition to RNA levels of target genes, as well as assessment of target gene expression durability. In vivo CRISPRa studies would also benefit from additional exploration of multiple sgRNA designs. The varying levels of expression associated with different sgRNA designs can serve as a tunable system to achieve the desired level of gene activation that may not be recapitulated in in vitro models. Notably, one of the studies took advantage of the synergistic effects associated with using multiple different sgRNAs to achieve greater overall activation potential of the Lama1 gene. 32 The cooperative increase in endogenous gene activation using multiple sgRNAs targeted to a gene promoter is a finding that has been demonstrated in vitro for many genes 23,25,28 but remains largely unexplored in vivo. Furthermore, considerations pertaining to expression regulation also extend to the promoter selection upstream of dCas9. In these studies, the use of promoters driving Cas9 expression has varied and included ubiquitous tissue specificity and one study having no promoter linked to Cas9 expression. 79 In contrast, all in vivo studies used the same ubiquitous (is this human?) U6 promoter to drive sgRNA expression. Other RNA polymerase III promoters (mouse U6, 7SK, H1) capable of driving gRNA expression have not been explored in conjunction with in vivo gene activation.
Summary of in vivo CRISPRa experiments
Finally, the majority of the studies utilized a dual-AAV design due to the size of the CRISPRa system and/or the desire to control levels of the sgRNA. Only one study involving the delivery of dCas9-TET3CD used a lentivirus, 78 which is associated with a larger packaging limit to AAV, but at the cost of reduced safety due to its propensity for genomic integration.
OFF-TARGET ANALYSIS OF CRISPRa PLATFORMS
An overall concern with CRISPR technology is the introduction of “off-target” or unintended genetic modifications. The utility of dCas9 in CRISPRa platforms omits concerns of nonspecific cleavage, with the exception of early-generation CRISPRa, which used active Cas9 coupled to truncated sgRNA that leads to insertions and deletions. Instead, off-target consequences associated with CRISPRa technology pertain to changes in expression of nontarget genes triggered by either nonspecific binding of dCas9 or the activator to regions of the genome. Several strategies have been used in CRISPRa studies to identify both primary and secondary off-target effects.
Primary off-target effects relate to the nonspecific binding of the dCas9-activator complex to an unintended region of the genome to activate a gene and can be determined using ChIP-seq to identify regions of DNA bound to the dCas9-activator complex. Binding to unintended regions of the genome can either occur via guide-dependent or guide-independent means. Guide-dependent off-targets occur as a result of sgRNA-mediated binding of dCas9 to DNA, whereas guide-independent off-targets occur as a result of direct or indirect binding of the transcriptional activator (e.g., VP64) to DNA and/or DNA-bound proteins (Fig. 2). Computational prediction methods have been used to account for potential secondary sgRNA binding sites in the genome bearing a degree of similarity to the spacer sequence that may result in guide-dependent off-target binding. Such methods identify potential binding sites and assess whether nearby genes have been inadvertently activated. One associated pitfall of computational prediction is that it utilizes the human genome reference sequence, which does not take into account that each human genome differs in over 3 million sites, thus potentially impacting the performance of these prediction approaches. 81 Primary off-target effects can also be assessed with respect to chromatin remodeling changes that reflect the specificity of genome activation tools. Polstein et al. assessed off-target chromatin remodeling across the genome by DNase-seq, comparing DNase I hypersensitive regions before and after CRISPRa gene activation. 82 As expected, their results conveyed an increase in chromatin accessibility at targeted gene promoter regions, but an unexpected result was that these changes occurred both in the presence and absence of VP64 fusion to dCas9. Another aspect explored in this study is the head-to-head comparison between Cas9- and TALE-based activation technologies in targeting IL1RN and HBG1/HBG2 genes. Although there was an increased expression achieved by TALE, there were no significant differences in terms of off-target gene activation, DNA binding sites, and changes to chromatin structure between Cas9 and TALE approaches. 82 Further studies involving different regions of the genome will be required to make a general conclusion regarding differences in off-target effects in the context of different gene activation technologies.
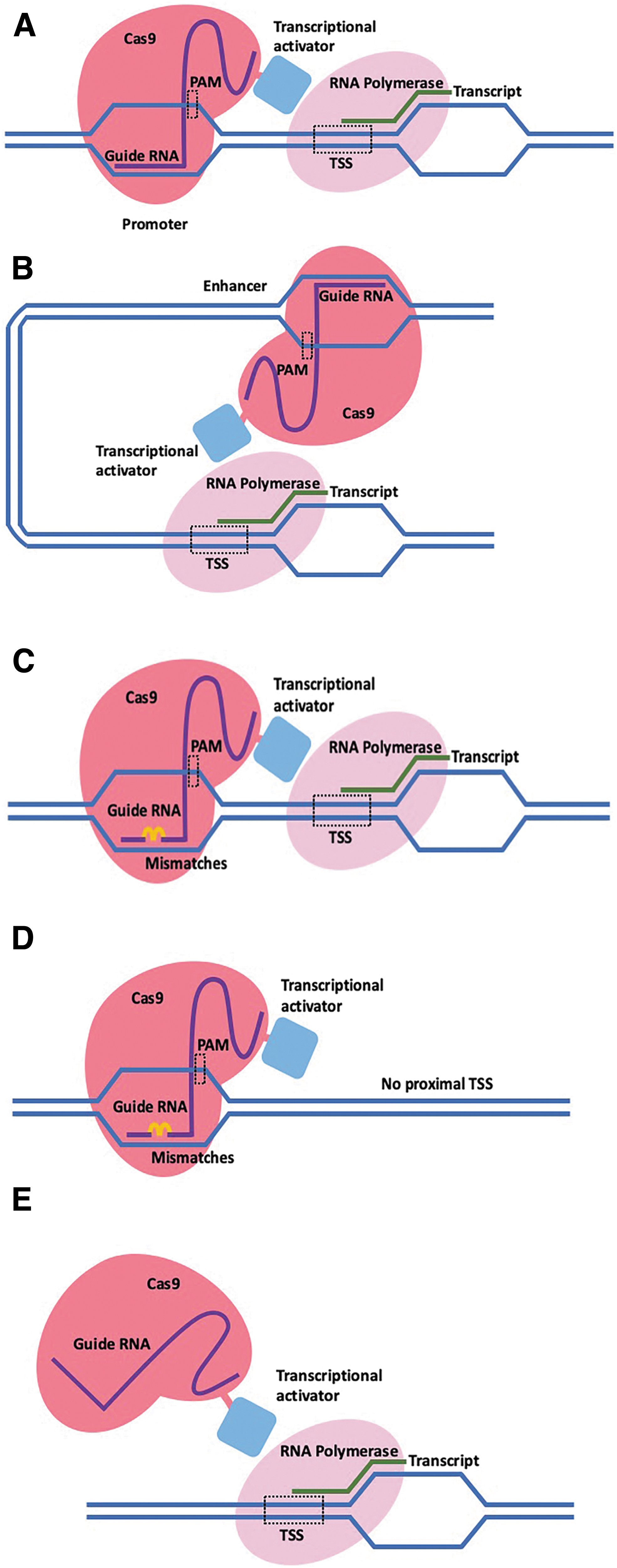
Example scenarios of on-target and off-target CRISPRa binding. Example of dCas9-activator complex on-target binding to
Secondary off-target effects concern the nonspecific activation of transcriptional networks that results as a consequence of expressing the target gene. These manifest as differentially expressed genes and can be detected by RNA-seq. The use of RNA-seq has been the most widely used strategy to broadly detect off-target gene activation effects, and is easily adapted using standard RNA preparation protocols for NGS and analysis using STAR 83 or Bowtie 84 alignment tools. Expression levels of each transcript are determined before statistical packages such as DESeq285 are used to identify differentially expressed genes and filter results by false discovery rate or p-value after adjusting for multiple test correction (i.e., genome-wide significance). There is currently no consensus on statistical significance thresholds for the classification of off-target genes, resulting in a wide variability of reported off-target genes. This challenge is compounded by the lack of understanding on whether off-target gene activation and their associated levels can potentially result in biologically significant changes.
Although the majority of studies explored off-target effects using the methods discussed above, only one study discussed the consequences of their findings in the context of interpreting their novel findings and clinical safety. In this study, dCas9-TET3CD was used to demethylate the promoter region of Rasal1, and subsequently used ChIP-seq to identify 159 off-target binding sites within 59 genes, some of which have known profibrotic functional association. 78 The authors conclude that these unexpected findings may in fact counteract the desired antifibrotic effects through the upregulation of Rasal1. This finding was used as a motivation to fuse TET3CD to a high-fidelity (HF) SpCas986 that required the deactivation of its endonuclease activity. The resulting use of HF SpCas9 reduced the off-target genes to eight and is thought to underlie the improved 50% reduction in fibrosis in the kidney fibrosis mouse model. Thus, directly addressing off-target effects can not only improve safety but also provide insight into improving desired outcomes.
Very few studies explored gene activation beyond the standard study design consisting of one guide to upregulate one target gene. The use of different guides to activate a target gene(s) can offer opportunities to identify secondary transcriptional effects that are related to transitioning between a disease and healthy biological state through identifying common differentially expressed genes. For example, activation of Sim1 through targeting upstream of the TSS or a distant hypothalamic enhancer yielded similar differentially expressed genes from RNA-seq, interpreted to be a shift in transcriptional profile to a healthy state. 72 Another example of elucidating secondary transcriptional effects is through the use of guides targeting different genes that result in a similar phenotype change. Examples include the upregulation of Klotho or Rasal1, 78 which both result in a desired reduction in kidney fibrosis; and similarly, the upregulation of Fst or Utrn, which both improve the muscle weakness associated with Dmd mice 75 —in both these cases, the opportunity to identify common differentially expressed genes (i.e., secondary off-target effects) was not explored.
A final consideration regarding off-target effects with CRISPRa technology relates to tissue and cell line specificity. This is more than likely given the unique transcriptional profiles and differences in promoter and enhancer accessibility associated with different cell types—a phenomenon echoed by differences in sgRNA targeting efficiencies across different cell lines. Thus, the relevant cell or tissue type should always be considered for off-target analysis. In vivo studies in particular should perform off-target analysis in the liver—a tissue with high tropism for all AAV serotypes.
CONSIDERATIONS FOR CRISPRa TRANSLATION IN HUMANS
The collective results on preclinical models that use CRISPRa approaches have demonstrated promising proof-of-concept for genetic compensation strategies but will require additional development and optimization before clinical translation. In comparison with traditional “cutting” CRISPR platforms, the nuclease-deficient property of CRISPRa is seen as more conducive to human translation, circumventing safety issues such as unpredictable outcomes associated with NHEJ, and rAAV integration at cleavage sites as a consequence of nuclease activity. 87,88
In this section, we discuss issues of consideration pertaining to CRISPRa in in vivo studies that hinder its capacity for direct human translation. First and foremost is the need for consideration that CRISPR-induced genome targeting is species specific. That is, the sgRNA designed and evaluated in these studies have sequences specific to the mouse genome, which are unlikely to be identical in the human genome. In the case where they may be identical, their position relative to the TSS and other important regulatory elements are likely to be different in humans, thus requiring a new set of sgRNAs to be designed and validated in human cell models. Ideally, in vivo CRISPRa studies would benefit from testing on humanized mouse models, through which the human gene of interest (including introns and flanking intergenic sequences) has been inserted into the genome to allow for testing of human-specific sgRNAs. 89,90 Another aspect pertaining to species specificity is the assessment of off-target effects. CRISPRa in vivo studies typically use RNA-sequencing and/or ChIP to assess off-target activation using differential gene expression analysis in mouse tissue. Given the established differences in genetic regulation between species, it is therefore important that off-target assessment be performed in a relevant human cell line to accurately model any potentially unwanted genetic perturbations. Next is the consideration that the aforementioned studies have opted for ubiquitous expression promoters such as cytomegalovirus to drive dCas9 expression. Given that systemic AAV delivery results in high liver transduction and has been linked to toxicity, 91 –93 it is therefore important to utilize tissue-specific promoters to limit dCas9 expression in only tissues of interest. Epitope and/or fluorescent tags in CRISPRa constructs used in these studies also impede on the translatability of their vectors in humans. Furthermore, the majority of studies used a dual-vector AAV delivery approach, likely due to the packaging constraints of using a single-vector approach. However, there is currently no precedence for a dual-vector AAV approach in human gene therapy, largely because treatment of patients with high doses of two viral vectors poses safety concerns. Unless CRISPRa platforms can be packaged into a single vector, AAV-based human translation will be challenging. An alternative approach explored for other CRISPR-based technologies are nonviral-based delivery methods such as lipid- or polymer-based nanocarriers for delivery of Cas9/sgRNA ribonucleoprotein complexes. 94 Such platforms do not typically result in long-lived expression and subsequently may not be suitable for CRISPRa strategies that rely on sustained expression of gene activating components to achieve a therapeutic benefit.
Another issue of consideration relating to clinical translatability is the compatibility of AAV with CRISPRa cassettes in relation to its potential impact on vector genome structure. CRISPRa systems incorporate several unique system elements such as gRNA hairpins, VP16 repeats, and adapter loop structures that may impact on the secondary structure of the resultant rAAV genome. Strong secondary structures incorporated into AAV genomes have been known to interfere with replication and lead to an increase in nonfunctional vector genomes. For example, Xie et al. demonstrated that AAVs carrying small interfering RNA cassettes can form DNA hairpin structures, which can lead to the formation of truncated genomes. 95 Another study discovered that construct designs incorporating dual sgRNA expression cassettes orientated in a head-to-head or tail-to-tail manner can also form strong secondary structures that result in high-frequency truncation and reduced abundance of functional vector produced. 96 These studies collectively demonstrate that further investigation into AAV-compatibility of CRISPRa cassettes is needed to determine the feasibility of generating functional clinical-grade AAV preparations at titers that will enable safe and effective translation in humans.
Additional aspects that warrant further consideration pertain to long-term durability and immunogenicity of CRISPRa platforms in different human tissues, 88 as well as the implications of tissue turnover in regenerative organs. Further studies need to be conducted to investigate the sustainability of CRISPRa platforms after systemic delivery, both in terms of sustained expression of the CRISPRa machinery and its ability to sustain targeted gene activation long term. Although all studies reviewed here reported a degree of success in conferring functional benefit with their selected dose, it will undoubtedly be a difficult task to establish the minimal efficacious dose required for genetic compensation that will translate from mouse to human.
Conclusions
CRISPR-based gene activation technology offers a robust and easily implementable modality for the interrogation of genetic mechanisms in single-gene or genome-scale applications. The tools and resources enabling the implementation of various CRISPRa platforms have been made widely available through commercial means (e.g., Addgene) and are compatible with existing CRISPR modules, such as sgRNA plasmids and libraries typically used in conjunction with standard CRISPR-Cas9 or CRISPRi platforms. The emergence of CRISPRa technology and its widespread availability have culminated in a wealth of publications in the field of genetics, cell biology, and disease research. In addition, its versatility across different in vitro and in vivo systems has made it an attractive platform to researchers in both basic science and translational fields. Future translation of CRISPRa technology in humans will require additional work to address delivery modality, safety concerns, and long-term sustainability of expression in targeted tissues. Although some of these considerations are unique to CRISPRa technology, the majority are common across all CRISPR-based technologies, where solutions will likely be translatable to CRISPRa applications.
Footnotes
Authors' Contributions
A.L., M.L., K.G.W., and K.M. contributed to the conception and preparation of the article. All authors have reviewed and approved this article before submission.
Author Disclosure
No competing financial interests exist.
Funding Information
Work on clustered regularly interspaced short palindromic repeat activation technologies in the Lek laboratory has been supported by the Cure Rare Disease foundation.