
Abstract
Pulmonary hypertension (PH) is a proliferative disease characterized by pulmonary arterial remodeling (PAR). SAM and SH3 domain containing 1 (SASH1) is a novel tumor suppressor gene whose biological function in PH is unclear. In this study, a hypoxia-induced pulmonary hypertension (HPH) rat model was constructed to explore the role of SASH1 in PAR. Histopathological changes in the lung tissue and hemodynamic alteration were detected in SASH1-knockdown rats through adeno-associated virus type-1 (AAV1) infection. In vitro, primary human pulmonary arterial smooth muscle cells (HPASMCs) were transfected with SASH1siRNA to investigate the effects of SASH1 on hypoxia-induced proliferation and migration. The molecular mechanisms associated with SASH1 were explored through knockdown and overexpression approaches. We found that SASH1 expression was significantly increased in rat pulmonary arteries and HPASMCs after hypoxia exposure. In vivo, silencing the SASH1 gene expression improved HPH in rats. The SASH1 downregulation inhibited proliferation and migration of hypoxia-induced HPASMCs. The protein expression of phospho-AKT (known as protein kinase B), proliferating cell nuclear antigen, and matrix metalloproteinase 9 (MMP9) in HPASMCs were increased after SASH1 overexpression, whereas these effects were inhibited by SASH1 knockdown. In conclusion, SASH1 downregulation improved hypoxia-induced PAR and PH. SASH1 may be a novel target for PH gene therapy in the era of precision medicine.
Introduction
Pulmonary hypertension (PH) refers to a mean pulmonary arterial pressure (mPAP) of >20 mmHg at rest. 1 The chronic progressive disease is characterized by pulmonary arterial remodeling (PAR), leading to right heart failure and, ultimately, death. 2 –4 It is mainly caused by exposure to hypoxia resulting from certain chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD). 5 According to previous diagnostic criteria (mPAP ≥25 mmHg) for PH, 30–70% of patients with COPD 6 and 32–84% of patients with advanced ILD 7 develop PH. Excessive proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs) are the main cytopathology of PAR in hypoxia-induced PH (HPH). 8,9 To date, medication and long-term oxygen therapy for patients with HPH can only improve pulmonary vascular resistance, but these cannot reverse PAR. 10,11
Gene therapy is commonly defined as the introduction of genetic material into human cells to treat disease. In recent years, studies on the different categories of PH have suggested that PAR caused by PH can be improved by gene therapy. Adenovirus-mediated BMPR212 overexpression or pulmonary endothelial Tph113 silencing can attenuate PAR in rats with hypoxic PH. More recently, gene transfer of Ca2+-ATPase 2a (SERCA2a) via intratracheal delivery of
As such, PH is considered a chronic inflammatory and vascular proliferative disease. Multiple inflammatory factors 16,17 (IL-6, IL-1β, TNF-α) and tumor-suppressor genes (P53, 18 P21 19 ) are implicated in PH pathogenesis. The SAM and SH3 domain containing 1 (SASH1) belongs to the SYL family of signal adapter protein. 20 Previous studies have identified that SASH1 is a novel tumor suppressor candidate 20 –23 and that it mediates the inflammatory process. 24 Chen et al. reported that SASH1 expression was decreased in lung cancer and that overexpression of the SASH1 gene suppressed the growth and proliferation of A549 cells. 25 In hepatocarcinoma cells and thyroid cancer cells, SASH1 downregulates the PI3K-AKT pathway, leading to the inhibition of proliferation and metastasis. 26,27 Moreover, SASH1 has been identified as a scaffold protein downstream of TLR4 and enhanced lipopolysaccharide-induced NF-κB, JNK activation, culminating in the increase of pro-inflammatory factors and endothelial migration. 24 Thus, we propose that SASH1 could be involved in the occurrence and development of PH.
In this article, we explored the expression of SASH1 in PAs of HPH rats. The SASH1shRNA.AAV1 vector was constructed to investigate the therapeutic effect of SASH1 on HPH.
Materials and Methods
Experimental animal models
Eight-week-old male Sprague-Dawley rats were acquired from British SIPPR/BK Lab Animal Ltd. (Shanghai, China). The animals were randomly allocated to the normoxia or the hypoxia group (n = 10 each). They were exposed to room air (FiO2 of 0.21) or a hypoxic environment (FiO2 of 0.1) in a normobaric chamber, according to a previously described method. 28 All animals had free access to food and water. The experimental animal protocols were approved by the Animal Ethics Community of Qingdao Municipal Hospital.
SASH1 downregulation in PAs of HPH rats
Another 30 rats were randomly allocated to the normoxia (n = 10) or hypoxia group (n = 20). After exposure to the hypoxia environment as described earlier, the rats were subcategorized into two groups of 10 rats each. The two groups were, respectively, infected with adeno-associated virus type-1 (AAV1; Vigene Biosciences, China) expressing green fluorescent protein (GFP.AAV1) or AAV1 containing rat SASH1shRNA (SASH1shRNA.AAV1) via tracheal injection (1 × 1011 viral genomes/rat), according to a previously described method. 29 All the rats were euthanized by a sodium pentobarbital injection at 4 weeks after infection.
Analysis of hemodynamic and histology
The right heart catheter method was used to detect hemodynamic changes. 30 After measurement of the right ventricular systolic pressure (RVSP) with a 3F polyethylene catheter and PowerLab data acquisition equipment (AD Instruments, Australia), the animals were euthanized, and the hearts were separated into the right ventricle (RV) and left ventricle plus septum (LV + S). The RV-to-LV + S weight ratio was defined as the RV hypertrophy index. Left lung tissues were collected, fixed, and cut to 4-μm slices. The slices were then stained with hematoxylin and eosin (H&E). The PAs with an outside diameter of 50–150 μm were chosen, and the wall thickness was measured as previously described. 31
Immunofluorescence and immunohistochemistry
The rat lung sections were deparaffinized and incubated with anti-α-smooth muscle actin (Thermo Fisher Scientific). Next, they were fluorescent-labeled with goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (Thermo Fisher Scientific). For immunohistochemical analysis, the primary antibody against SASH1 (Thermo Fisher Scientific) and p-AKT (Abcam) were used. The observation of tissue sections was done in a double-blind manner.
PASMC culture and transfection
The primary PASMCs were prepared from normoxic rats according to a previously described method.
32
Human PASMCs (HPASMCs) purchased from ATCC were cultured under hypoxic (2% O2, 5% CO2) or normoxic (21% O2, 5% CO2) conditions separately in the Dulbecco's modified Eagle medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; Life Technologies) and penicillin/streptomycin (Gibco) at 37°C. According to the instructions of lipofectamine 2000 transfection reagent, SASH1-siRNA (FulenGen, China) was transfected into PASMCs, and the cells were harvested after 24 h. The following sequences were used: human SASH1-si: Forward: 5′-GCAGCAGUAUGC-AGAUUAUTT-3′, Reverse: 5′-AUAAUCUGCAUACUGCUGCTT-3′. Rat SASH1-si: Forward: 5′-GGAUCCAGCUAAUGAUGAUTT-3′, Reverse: 5′-AUCAUCAUUAG CUGGAUCCTT-3′. NC-si: Forward: 5′- UUCUCCGAACGUGUCACGUTT-3′, Reverse: 5′-ACGUGACACGUUCGGAGAATT-3′. The SASH1 overexpression adenovirus vector (
Western blot analysis
Total protein was extracted from PASMCs or rat PAs to perform Western blot analysis, as previously described. 33 We used antibodies targeting SASH1 (Absin Bioscience, China), phospho-AKT (Ser473; Cell Signaling Technology), AKT (Cell Signaling Technology), proliferating cell nuclear antigen (PCNA; Proteintech Group, China), and matrix metalloproteinase 9 (MMP9; Abcam) for this assay. The gray value of each protein was analyzed by Image J software.
Real-time polymerase chain reaction
Total RNA of PASMCs and rat PAs was extracted by TRIzol (Thermo Fisher Scientific). The first cDNA was synthesized by using the RevertAid First-Strand cDNA Synthesis kit (Takara, Japan), and real-time PCR was performed on ABI 7500 System by using the SYBR® Premix-Ex-Taq™ kit (Takara). The following primer sequences were used: β-actin (human): Forward: 5′-GAAAATCTGGCACCA-CACCT-3′, Reverse: 5′-GATAGCACAGCCTGGATAGCA-3′; SASH1 (human): Forward: 5′-CGGGAAAGCGTCAAGTCGGA-3′, Reverse: 5′-ATCTCCTTTCTTGA-GCTTGAG-3′; β-actin (rat): Forward: 5′- CGTAAAGACCTCTATGCCAACA-3′, Reverse: 5′-CGGACTCATCGTACTCCTGCT-3′; SASH1 (rat): Forward: 5′-AAATA-CAGCAGC CCCGTCAC-3′, Reverse: 5′-TCCGTGGTGCTCACAGTATG-3′. The ratio for the mRNA of the target was normalized by β-actin.
Cell proliferation analysis
First, to determine the role of SASH1 in HPASMC proliferation, we treated HPASMCs with SASH1 siRNA or NC siRNA. Next, the HPASMCs were incubated with 21% O2 or 2% O2 for another 48 h. Cell Counting Kit-8 (CCK8; Dojindo, Japan) and cell cycle detection kit (KeyGEN, China) were used for cell viability and proliferation analysis, respectively. The operations were in accordance with the manufacturer's instructions. The assay was, respectively, performed by a microplate reader (PerkinElmer) and flow cytometer (Becton Dickinson and Company).
Migration assay
For the PASMC migration assay, 24-well transwell plates with 8-μm pores in membranes were used. After transfection with SASH1-si and NC-si for 24 h, the PASMCs were added to the upper chamber. Another 24 h was needed for adherence. The upper chamber liquid was replaced with 200 μL DMEM containing 1% FBS, and the lower compartment was inserted in 600 μL DMEM with 10% FBS. The cells were incubated under 21% O2 or 2% O2 conditions. The migrated cells were fixed and stained with 0.1% crystal violet. The staining images were observed under an optical microscope.
Statistical analysis
All statistical analyses were performed with the IBM SPSS Statistics software (SPSS 19.0).) and presented as means ± SD. The Student t-test was used for comparisons between two groups and one-way analysis of variance followed by a Tukey test for multiple-group comparisons. A p-value of <0.05 indicated statistical significance.
Results
Hypoxia-induced PH in rats
The HPH rat model was developed by exposure to chronic hypoxia (10% O2). The wall thickness of pulmonary arterioles was significantly increased in the hypoxia group than in the normoxia group (Fig. 1A, B). The RVSP was higher in the hypoxia group (38.43 ± 2.664 mmHg vs. 20.02 ± 1.721 mmHg; Fig. 1C, D).
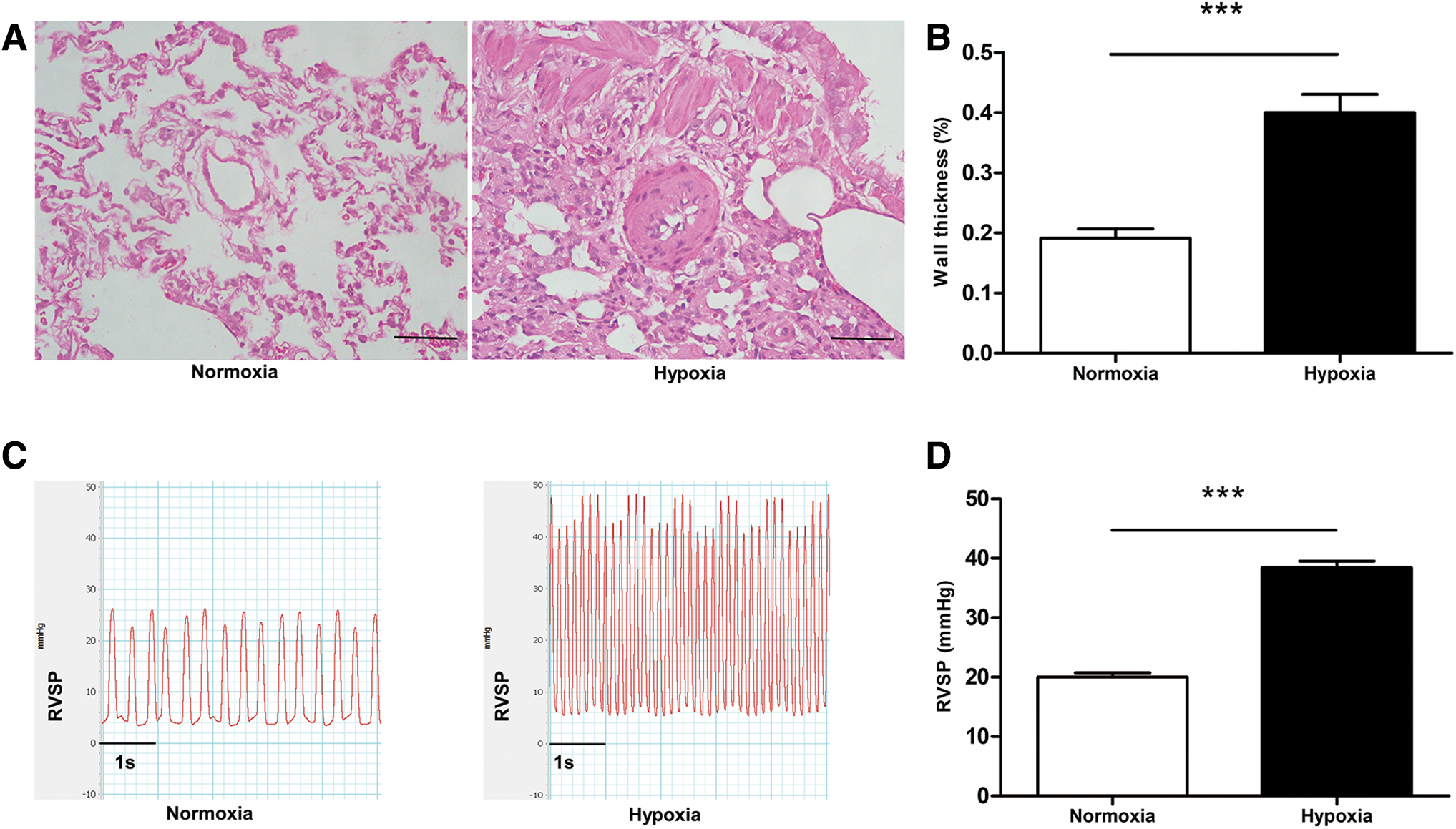
Histopathology and hemodynamic changes in HPH rats.
SASH1 expression increased after hypoxia
To determine the effect of hypoxia on SASH1 expression in PASMCs, both in vivo and in vitro experiments were conducted. SASH1-positive stained cells localized in the rat PAs were markedly increased in the hypoxia than in the normoxia group (Fig. 2A). The SASH1 protein level in rat PAs was also higher in the hypoxia group (Fig. 2B, C). Moreover, both SASH1 protein and mRNA expression were increased in the 2% O2 group than in the 21% O2 group in HPASMCs (Fig. 2D–F).
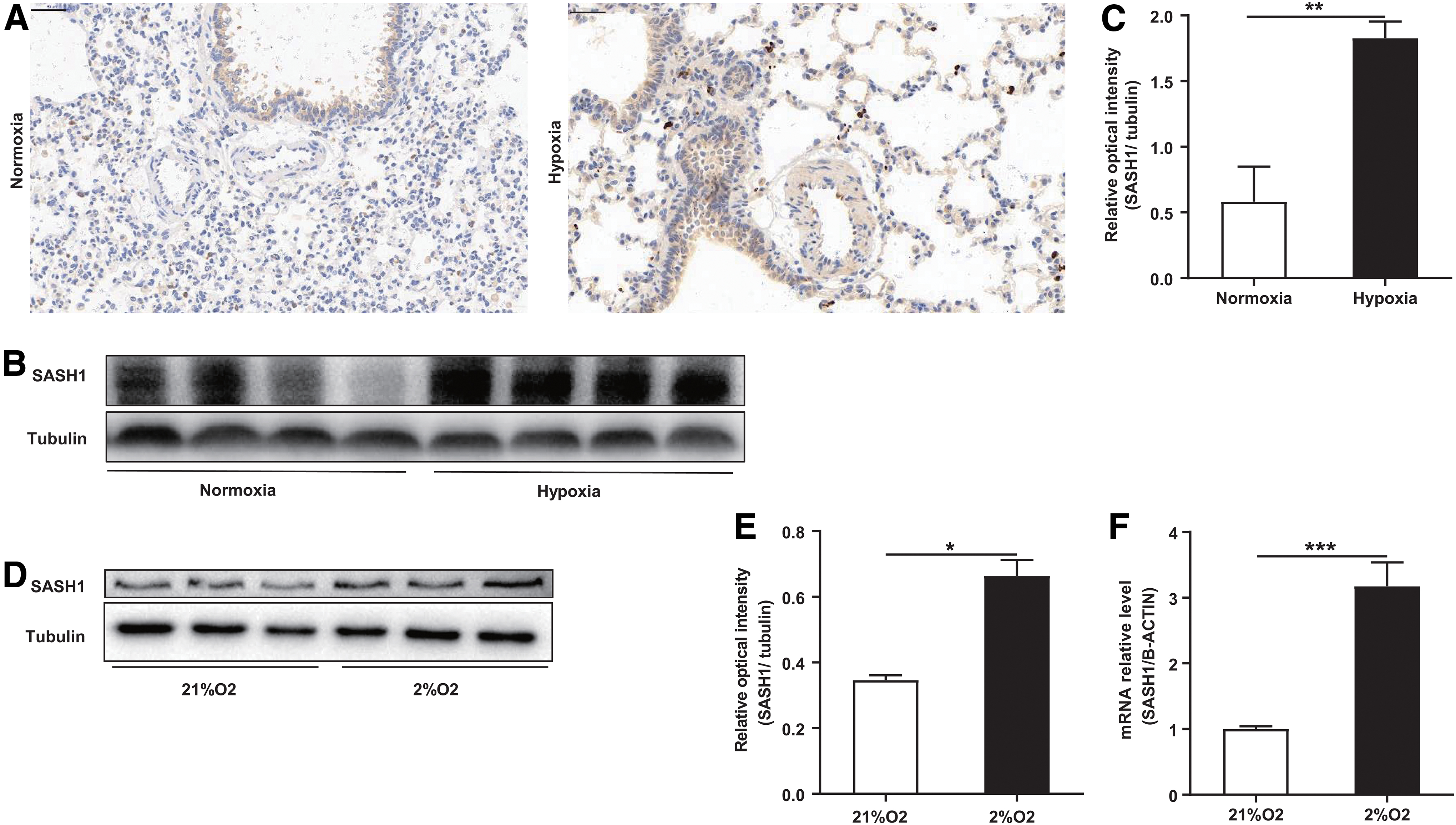
Effects of hypoxia on SASH1 expression in rat pulmonary arteries and HPASMCs.
SASH1sh.AAV1 attenuates HPH and pulmonary artery remodeling in rats
Rat SASH1siRNA and SASH1sh.AAV1 were synthesized for the functional study of SASH1. The SASH1-si successfully decreased the expression of SASH1 in rat PASMCs (Supplementary Fig. S1E–G). Immunofluorescence results revealed that rat PA smooth muscle cells were successfully infected by GFP.AAV1 (Supplementary Fig. S2). Moreover, in rat PAs, SASH1sh.AAV1 was able to reverse the hypoxia-induced SASH1 upregulation (Fig. 3B–D). Immunohistochemical analyses also revealed a similar result (Fig. 3E).

SASH1sh.AAV1 suppressed hypoxia-induced SASH1 upregulation in rat PAs.
Further, the hemodynamics and cardiopulmonary pathology were evaluated. Compared with the hypoxia-PH group, the RVSP and the degree of RV hypertrophy were significantly reduced in the SASH1sh.AAV1 treatment group (Fig. 4A, B, E, F). The H&E staining revealed that the PA wall thickness was thinner in the SASH1shAAV1 group than in the PH model group (Fig. 4C, D).

SASH1sh.AAV1 attenuated HPH in rats.
SASH1 knockdown suppressed the proliferation and migration of HPASMCs
In vitro, the role of SASH1-si in the biological behavior of HPASMCs was examined. The cell viability was markedly enhanced in the 2% O2 group than in the 21% O2 group, but SASH1-si reversed the effect caused by hypoxia (Fig. 5A). The abnormal cell cycle distribution (increased S phase, G2/M phase, and reduced G1 phase) caused by hypoxia was restored by SASH1-si (Fig. 5B, C). The transwell assay revealed that SASH1-si inhibited hypoxia-induced cell migration (Fig. 5D, E).
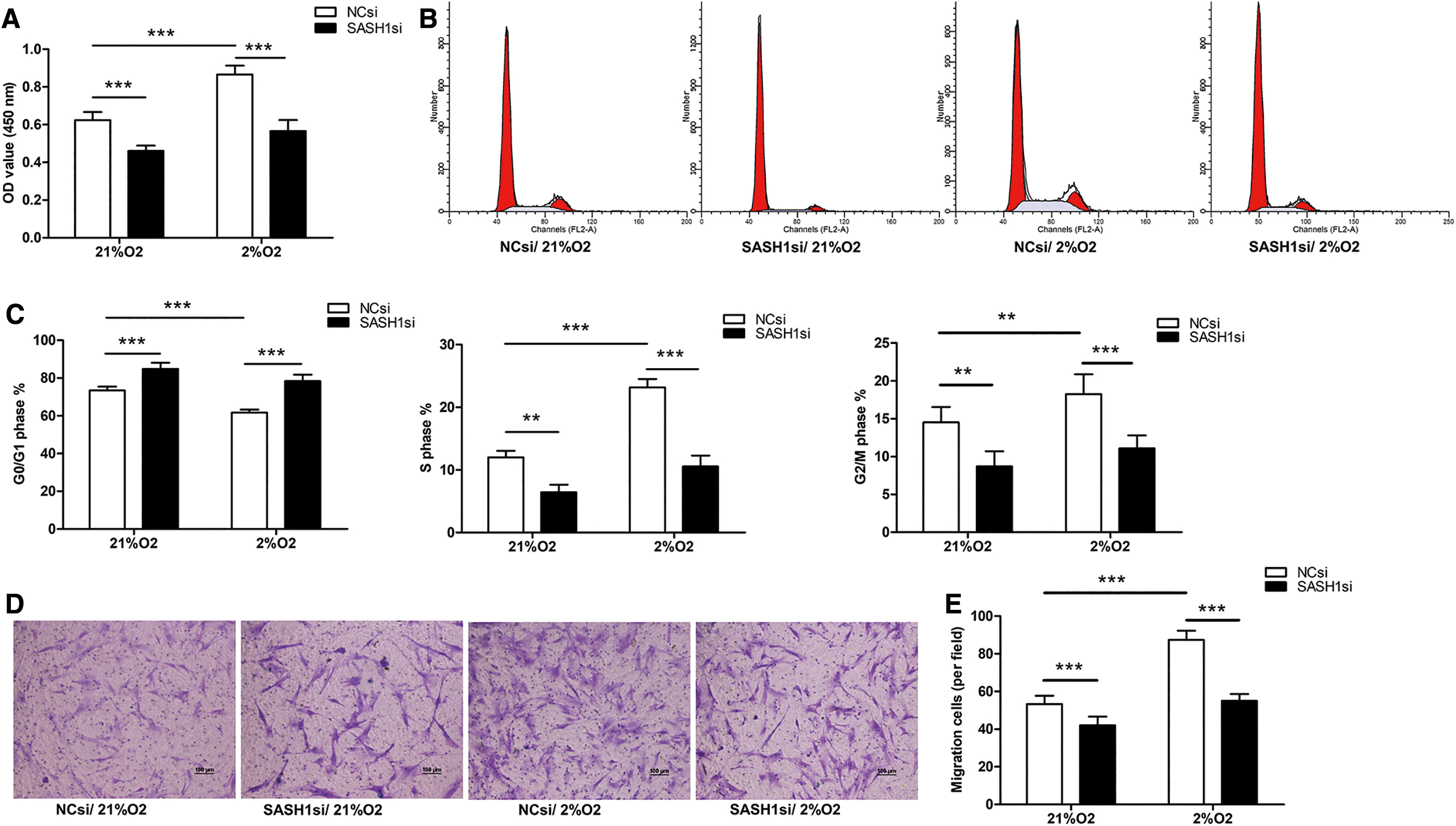
Effects of SASH1 siRNA on hypoxia-induced HPASMCs proliferation and migration.
The level of SASH1 impacted the activation of AKT signal
AKT signal is a vital participant in regulating the proliferation and migration of vascular SMCs. SASH1 up- or downregulation (Supplementary Fig. S1A–D) affected the degree of AKT activation in HPASMCs.
In vivo, more p-AKT-positive PASMCs were observed in the HPH group than in the normoxia group, and SASH1sh.AAV1 was able to inhibit the hypoxia-induced upregulation of p-AKT (Ser473) (Fig. 6A). In HPASMCs, SASH1 knockdown reduced the protein expression of p-AKT and PAR-related molecules (PCNA and MMP9) (Fig. 6B), whereas SASH1 overexpression increased this effect (Fig. 6C).
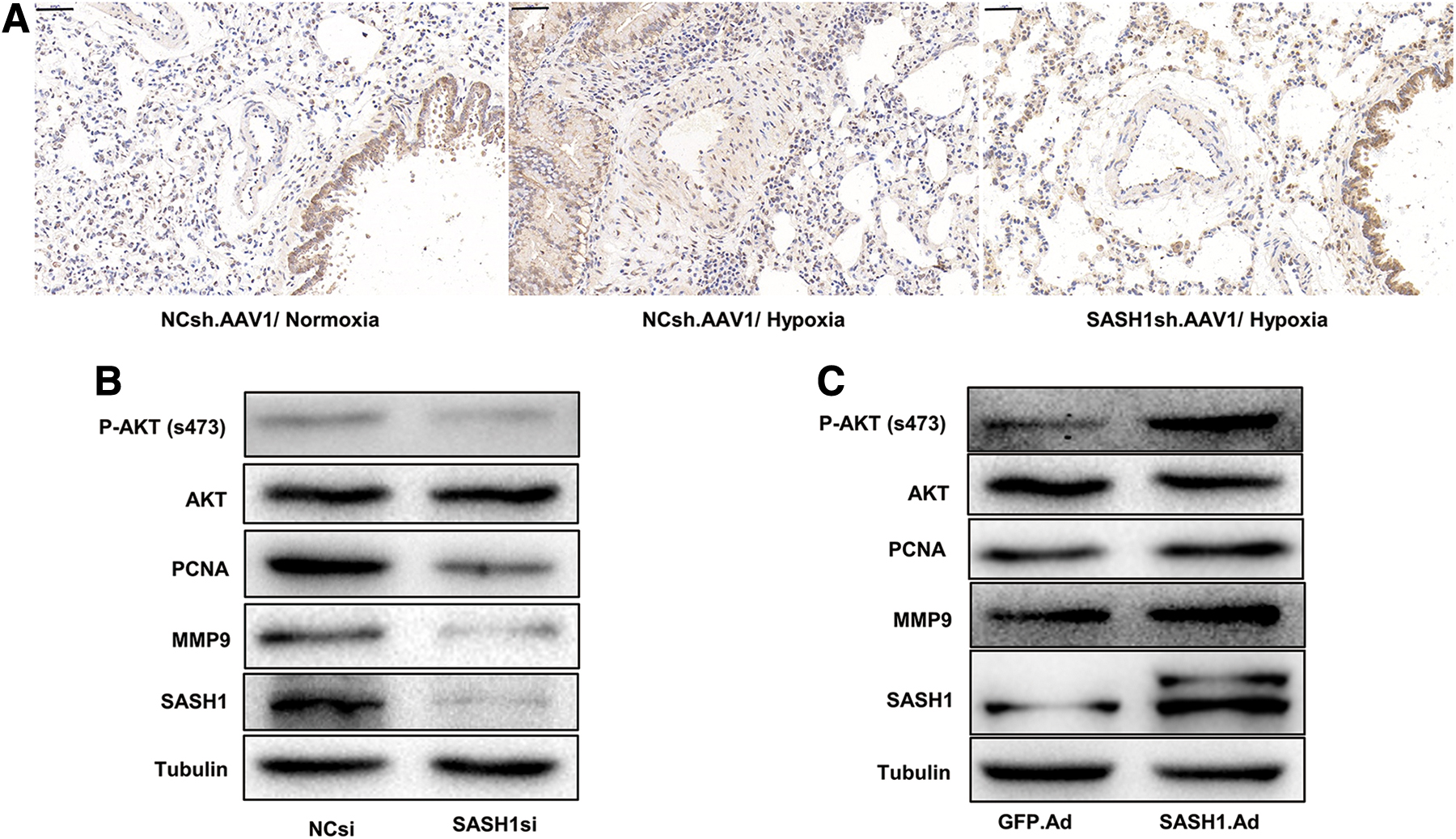
Effects of SASH1 overexpression or downregulation on AKT activation in HPASMCs.
Discussion
In our study, both in vivo and in vitro experiments were performed to determine the role of SASH1 in the initiation and progression of HPH. In the HPH rat model, an increase in SASH1 protein was associated with PA remodeling. Silencing the SASH1 gene with SASH1shRNA.AAV1 ameliorated pulmonary vascular remodeling and attenuated PH in HPH rats. Moreover, we confirmed that SASH1 downregulation was able to suppress the proliferation and migration of HPASMCs. Interestingly, overexpression and inhibition of SASH1 altered the activated state of AKT signals. In summary, this study is the first to demonstrate the possible mechanism of SASH1 in HPH pathogenesis.
PH is a multifactorial disease with complex pathological mechanisms. 34 Arterial vasoconstriction, thrombosis, and remodeling of the small pulmonary arterioles are the three typical pathogenetic features of the disease. 35 –38 Since 1976, chronic hypoxia has been proven to increase pulmonary pressure in rodent models, and the model is widely used in HPH studies. 39 –41 In our study, RVSP was increased with the obvious thickening of PAs in the HPH rat model. PAR is known to be characterized by abnormal proliferation and migration of PASMCs, and this feature is similar between PH and neoplasms. 42 –44
SASH1 is considered a novel tumor suppressor, 25 and its upregulation seems to inhibit proliferation and invasion of lung cancer, hepatocarcinoma, melanoma, and colon cancer cells. 22,45,46 Moreover, Coulombe et al. reported that SASH1 promotes Akt phosphorylation and drives mouse lung maturation. 47 To date, there are no reports regarding the effect of SASH1 on PAR or PH. In this study, we demonstrated that the level of SASH1 was increased in PAs of HPH rats. Further, SASH1 knockdown by SASH1sh.AAV1 with intratracheal injection attenuated RV hypertrophy and RVSP, accompanied by suppressed PAR, in HPH rats. Silencing of the SASH1 gene led to inhibition of hypoxia-induced migration and proliferation of HPASMCs. We can, thus, conclude about the involvement of SASH1 in hypoxia-induced PAR.
The role of SASH1 in the proliferation and migration of PASMCs is contradictory to its biological function in tumor cells. However, our results are consistent with some studies on non-neoplastic diseases. Zhou et al. reported that a mutation in the SASH1 gene upregulates SASH1 and promotes the migration of transepithelial melanocytes by decreasing E-cadherin expression in A375 cells. 48 Meanwhile, a study of the lentiginous phenotype found SASH1 to promote the proliferation of melanocytes and epidermal cells in the skin. 49 Together with the findings of the previous studies, we can conclude that SASH1 may play particular but different biological functions in different organs and tissues. Our study enriches the data on the role of SASH1 in the proliferation and migration of various types of cells.
Several studies have demonstrated the essential role of AKT signaling in angiogenesis 50 and vascular remodeling. 9,51 Baicalin improved RV dysfunction indexes and attenuated PH in monocrotaline-induced PH rats by regulating the AKT/eNOS signaling. 52 Moreover, SASH1 suppressed the proliferation, invasion, and metastasis of numerous tumors through the AKT signaling pathway. 21,26,27,53 However, it is unclear whether SASH1 interferes with the HPH process through the AKT cascade. In our study, the protein levels of p-AKT, PCNA, and MMP9 were decreased in HPASMCs on silencing the SASH1 gene expression, whereas SASH1 overexpression was associated with increased expression of these proteins. In addition, several previous studies have confirmed that the expression of PCNA 54,55 and MMP9 30,56 is regulated by the AKT signaling pathway. Therefore, our study findings illustrated that SASH1 facilitates pulmonary vasculature remodeling, possibly through AKT signal regulation.
Admittedly, several limitations exist in our study. We explored the role of SASH1 in HPH pathogenesis in rats. However, the SASH1 expression level in patients with PH was not evaluated due to the difficulty in obtaining human PA specimens. Since the pathogenesis of PH is complicated, we only explored the regulatory function of SASH1 in HPH. And further refinement of SASH1 function in other types of PH is needed.
Conclusion
SASH1/AKT plays an important role in hypoxia-induced PAR. Our research adds to the evidence that could help broaden gene therapy for HPH.
Footnotes
Authors' Contributions
Q.L. and W.H. created the study and the hypotheses; Q.L., W.H., and H.L. designed the study; H.L. and N.W. performed the experiments; H.L., W.W., and J.L. analyzed the experimental data; and H.L., Q.L., and N.W. wrote the article. All authors read the authorship agreement of the journal and agreed to the final article.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 81900048, No. 81973012).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.