
Abstract
Mucopolysaccharidosis type IIIA (MPS IIIA, Sanfilippo A syndrome) is a single gene (SGSH) childhood onset neurodegenerative disease for which gene therapy is in clinical trial. Theoretically, the transfer of a working gene should enable functional expression of the defective protein and rescue the phenotype when administered before the onset of irreversible disease. Recombinant adeno-associated virus (AAV) is being used as a vehicle for a number of gene therapy applications and the neurotropism of serotype 9 affords utility for monogenetic neurological disorders. To assess the efficacy of restoring the underlying biochemistry in the MPS IIIA brain, tail vein injections of self-complementary AAV9 human N-sulfoglucosamine sulfohydrolase (scAAV9.U1A.hSGSH) at 3 × 1013 vg/kg were administered to 6- and 16-week-old MPS IIIA mice. Heparan sulfate (HS) and GM2 and GM3 gangliosides were cleared from the cortex, hippocampus and subcortex with residual storage remaining in the brain stem and cerebellum. SGSH activity increased in the brain of the MPS IIIA-treated mice, but remained significantly reduced compared with wild-type. Motor activity as assessed in an open-field arena, and gait length, improved in MPS IIIA mice treated at both 6 and 16 weeks of age. However, functional assessment of cognition in the water cross-maze test, as well as gait width, normalized in mice treated at 6 weeks of age only, with mice treated at 16 weeks performing similar to untreated MPS IIIA mice. Astrogliosis was reduced in mice treated at 6 and 16 weeks of age compared to untreated MPS IIIA mice. These results demonstrate that the gene product is actively clearing primary HS and secondary ganglioside accumulation in MPS IIIA mice, but in older mice, neurocognitive impairments remain. This is likely due to secondary downstream consequences of HS affecting neurological functions that are not reversible upon substrate clearance.
Introduction
Mucopolysaccharidosis type IIIA (MPS IIIA), often referred to as Sanfilippo A syndrome, is an autosomal recessive lysosomal storage disorder arising from molecular defects in SGSH. This results in a deficiency of the gene product, N-sulfoglucosamine sulfohydrolase (SGSH, EC 3.10.1.1), an enzyme involved in the sequential degradation of the glycosaminoglycan (GAG) heparan sulfate (HS) which consequently accumulates. 1 Lysosomal accumulation of HS causes progressive neurological decline, not evident in the neonatal period, but typically manifesting as autistic-like behaviors from around 3 years of age. 2
No clinically approved treatment exists for MPS IIIA and the greatest hurdle in developing therapies rests not only with access to the brain but also correction of pathology therein. Gene therapy is considered the cornerstone for single-gene disorders like MPS IIIA, by delivering a normal copy of the malfunctioning gene. Targeting the brain can be achieved by administering gene therapy vectors through different routes, and such approaches have been evaluated in animal models of disease. 3 Viral vectors have generally been employed for this purpose and although all have advantages and disadvantages, adeno-associated virus (AAV) has shown particular promise. This is because AAV is not known to be associated with any disease in humans and is capable of long-term transduction in a variety of cell types. 4,5 Despite the generalized tissue tropism of all AAV serotypes, specific capsid amino acid differences facilitate targeting of certain cell types, with AAV9 showing neurotropism for the brain. 6,7
SGSH is encoded in 1,509 bp, cDNA that is small enough to be accommodated in a self-complementary AAV9 vector. Intravenous administration of scAAV9.hSGSH has been successfully targeted to the brain in a mouse model of MPS IIIA. 8 Widespread delivery of the therapeutic vector to the brain was reported along with significant reductions in GAG accumulation and prolonged lifespan of MPS IIIA mice treated at 8 weeks of age and followed until 10 months of age. A subsequent study revealed that the therapeutic impact of scAAV9.U1a.hSGSH (transgene under the control of a murine small nuclear RNA promoter, U1a) was dose dependent with MPS IIIA mice treated at a higher dose of 5 × 1012 vg/kg, returning greater amounts of transgene expression, larger reductions in GAG, and improved neurocognitive function compared with mice treated with 1 × 1012 vg/kg. 9 Furthermore, this study included the treatment of mice at an advanced stage of disease, which is particularly important, given that the majority of MPS IIIA patients are diagnosed from 4 years of age. Realistically, this means that any treatment approach for MPS IIIA will be challenged with ameliorating an undefined preexisting disease burden. Fu et al. 9 reported that at the higher dose of 5 × 1012 vg/kg, MPS IIIA mice treated up to 3 months of age showed improvements in neurocognitive function, increased lifespan, and a depletion of GAG storage throughout the brain. Although mice treated at later stages of disease (6 and 9 months) displayed no correction in behavioral performance, there was clearance of GAG. The success of the effectiveness of intravenous delivery of scAAV9.U1a.hSGSH in MPS IIIA mice has since provided the foundation for a phase I/II clinical trial in MPS IIIA patients (NCT02716246).
No study to date has systematically evaluated the primary or secondary storage burden in specific areas of the brain or examined whether biomarkers measurable in accessible fluids in the human trial do indeed provide a suitable assessment of biochemical correction within the brain itself. To address these questions, we treated MPS IIIA mice with a higher dose than previously investigated (3 × 1013/kg as in cohort 3 of NCT02716246) and evaluated the biochemistry in the brain at the study endpoint of 9 months. Given a concomitant trial (NCT04088734) is also enrolling MPS IIIA patients at an advanced phase of disease, we attempted to mimic this in the MPS IIIA model by including the treatment of older mice in our study.
Materials and Methods
Mice and scAAV9.U1a.hSGSH administration
The spontaneous mutant MPS IIIA mouse model (B6.Cg-Sgshmps3a) was purchased from The Jackson Laboratory, Bar Harbor, MA, and has been characterized. 10 This model contains a single point mutation at position 91 of SGSH, producing an amino acid change from aspartic acid to asparagine, which impacts the catalytic domain. This results in residual enzyme activity, thereby faithfully recapitulating the human disease as opposed to knockout models that have a complete absence of enzyme activity. Both MPS IIIA mice and unaffected littermates were bred from heterozygous pairs and offspring were genotyped as described previously. 11 The mice were group housed (3–5 mice per cage) in an enriched environment with food and water ad libitum, on a 14-h light/10-h dark cycle in a temperature-controlled facility (22°C). Eight 6-week-old MPS IIIA mice (four female and four male) were administered 3 × 1013 vg/kg of scAAV9.U1a.hSGSH (clinical grade preparation was gifted by Abeona Therapeutics) intravenously through the tail vein. The vector genome consists of the AAV2 terminal repeats, the murine small nuclear RNA promoter U1a, and a codon-optimized hSGSH sequence with an SV40 polyadenylation signal. 9 An additional nine MPS IIIA (five female and four male) mice were administered the same dose of scAAV9.U1a.hSGSH at 16 weeks of age. Eight wild-type (WT; 4 female and 4 male) and 10 MPS IIIA (4 female and 6 male) mice were used as controls. All mice were weighed weekly. Urine was collected by individually housing the mice in metabolic cages for 24 h. At 9 months of age (endpoint of the study), cerebrospinal fluid (CSF) was collected as previously described, 12 mice were euthanized, and the liver, bladder, and brains removed. The brain stem, cerebellum, cortex, hippocampus, and subcortex were dissected and, along with the CSF and urine, all samples were stored at −70°C until analysis. This study was approved by the Institutional Animal Ethics Committee (AEC 1084/12/20) and the Biosafety Committee (B150).
Behavior testing
Mouse activity was monitored using an automated activity monitoring system consisting of infrared sensors that detect horizontal and vertical crosses (Harvard apparatus, Holliston, MA) to generate data on distance travelled and vertical activity (rearing) for each mouse that was left to roam freely in an open-field arena (40 × 40 cm) for 3 min as previously described. 13 Mice were tested at 4, 6, and 8 months of age. Learning ability of the mice was determined using the water cross-maze test at 8 months of age. Visual cues were placed around a circular pool containing a submerged platform concealed by water clouded with milk powder. For six consecutive days and for six trials per day, the number of times the mouse swam from the release point to the platform (correct entries) was recorded. 13 Walking patterns were assessed in the mice by determining gait length and width at 8 months of age. The hind feet of each mouse was dipped into black food coloring before being released at the brightly lit end of a paper-lined runway terminating at an escape box. Two sets of prints were collected for each mouse consisting of 3 gait lengths from 4 consecutive left and right footprints totaling 12 individual gait lengths. Gait width was determined from the distance between the middle of 1 left footprint to the perpendicular gait length of the adjacent 2 right footprints and vice versa, for 5 measurements totaling 10 individual gait widths.
Biochemical analyses of brain tissue, CSF, and urine
Samples were thawed on ice before analyses and homogenates for each of the five brain regions were prepared by sonication in 0.02 M Tris (pH 7) containing 0.5 M NaCl and 0.1% Nonidet P-40. Total protein was estimated by the method of Lowry et al. 14 The HS-disaccharide [HN-UA(1S)] was derivatized with 1-phenyl-3-methyl-5-pyrazolone in 0.1 mg protein for tissue homogenate, 7–10 μL of CSF and 0.2 μmol of creatinine in urine, and quantified by mass spectrometry as described previously. 15 Gangliosides were extracted from 0.1 mg protein of brain homogenate as earlier described 16 with the inclusion of 10 pmol N-omega-CD3-octadecanoyl disialoganglioside GD3 as internal standard for GD2 and GD3. Total GM2, GM3, GD2, and GD3 were determined by summing the concentration of the individual isoforms.
SGSH activity in the brain homogenates was measured in 30 μg of protein combined with 10 μL of 20 mM 4MU-GlcNS in Michaelis' barbital CH3COONa buffer (29 mM sodium barbital, 29 mM CH3COONa, and 0.68% (w/v) NaCl; pH 6.5) and 10 μL of Pefabloc (0.45 mg/mL in Michaelis' barbital CH3COONa buffer). Samples were incubated at 47°C for 17 h before addition of 6 μL of McIlvaine's phosphate/citrate buffer, pH 6.7, and 10 μL α-glucosidase (10 U/mL in 0.2% bovine serum albumin). Samples were incubated for a further 24 h at 37°C before the reaction was stopped with the addition of 0.2 mL of glycine buffer (0.2 M glycine and 0.125 M Na2CO3; 0.16 M NaOH; pH 10.7). The samples were aliquoted into black microtiter plates and fluorescence determined on a Wallac Victor2 1420 multilabel plate reader (PerkinElmer, Waltham, MA) with an excitation wavelength of 355 nm and an emission wavelength of 535 nm. Enzyme activity was determined by relating the fluorescence of the sample to that of a known concentration of 4MU and expressed in nmol/17 h/mg protein. 17 β-hexosaminidase activity was measured in 1 μL brain homogenates as previously described. 18 Samples were incubated at 37°C for 30 min before the reaction was stopped with the addition of 1.2 mL of glycine buffer and enzyme activity determined as described above for SGSH.
Analysis of glial fibrillary acidic protein astrogliosis in brain by immunohistochemistry and gene expression
Sections of cortex were removed postmortem and fixed in 10% (v/v) formalin for 24 h and then stored in 70% ethanol. Sections were processed and embedded in paraffin and 6 μm sections cut and stained for immunohistochemical analysis of glial fibrillary acidic protein (GFAP). 19 Sections were visualized at 100 × magnification and the degree of astrocytic reactive gliosis detected in the cortex of mice was assessed independently by a pathologist, blinded to experimental design.
The expression levels of Gfap were determined and normalized to cyclophilin A (Cypa) using the 2−ΔΔCt method to calculate the fold change in gene expression. 20
Quantitative real-time PCR
Total genomic DNA was extracted from the brain stem, cortex, cerebellum, and subcortex (100 μg protein) using a Promega Wizard Genomic extraction kit as per manufacturer's instructions (Promega, Madison, WI). The gDNA was then analyzed by qRT-PCR using TaqMan (Applied Biosystems, Foster City, CA) and Applied Biosystems 7300 Real-Time PCR system, following the procedures recommended by the manufacturer. TaqMan primers specific for native hSGSH were used to detect rAAV vector genomes; forward; AAGTCAGCGAGGCCTACGT, reverse; GATGGTCTTCGAGCCAAAGAT, and probe 6FAM-CCTCCTAGA/ZEN/CCTCACGCCCACC-MGBNFQ (non-codon-optimized sequence) to quantitate ds copies of SGSH from a standard curve and codon-optimized hSGSH in treated rAAV samples; forward; GCCTCTGCTTCTGTCTCAAG, reverse; GATGTTTCTGCCCACTTGC, and probe 6FAM-ACCCCTTCGATTTCGCCTACACC-Zen/IBFQ (codon-optimized sequence). Genomic DNA was quantified in parallel samples using mouse transferrin primers; forward; AAGCAGCCAAATTAGCATGTTGAC, reverse; GGTCTGATTCTCTGTTTAGCTGACA, and probe 6FAM-CTGGCCTGAGCTCCT-NFQ. Genomic DNA from tissues of untreated (WT and MPS IIIA) mice served as controls.
Statistics
Results are reported as mean ± standard error of mean. Statistical analyses were performed using GraphPad Prism V. 6 (GraphPad Software, La Jolla, CA), with a p < 0.05 considered significant. Differences between groups were analyzed for significance using a two-way analysis of variance (ANOVA) with a Tukey HSD post-hoc test.
Results
scAAV9.U1a.hSGSH corrects biochemical abnormalities in the urine, CSF, and brain
In mice treated at 6 and 16 weeks, the urine HS-disaccharide decreased approximately 3-fold within 1 week of treatment, reducing to WT levels (below the limit of quantification) by 8 months of age (Fig. 1A). In the CSF, the disaccharide also normalized (Fig. 1B) and was almost 50-fold lower than untreated MPS IIIA mice. In the cerebellum, hippocampus and subcortex scAAV9.U1a.hSGSH administered at both 6 and 16 weeks completely cleared HS storage analogous to WT (Fig. 1C). Although residual storage remained in the brain stem and cerebellum, it was still 100-fold less than that in untreated MPS IIIA mice, and no difference was apparent in mice treated at 6 or 16 weeks.
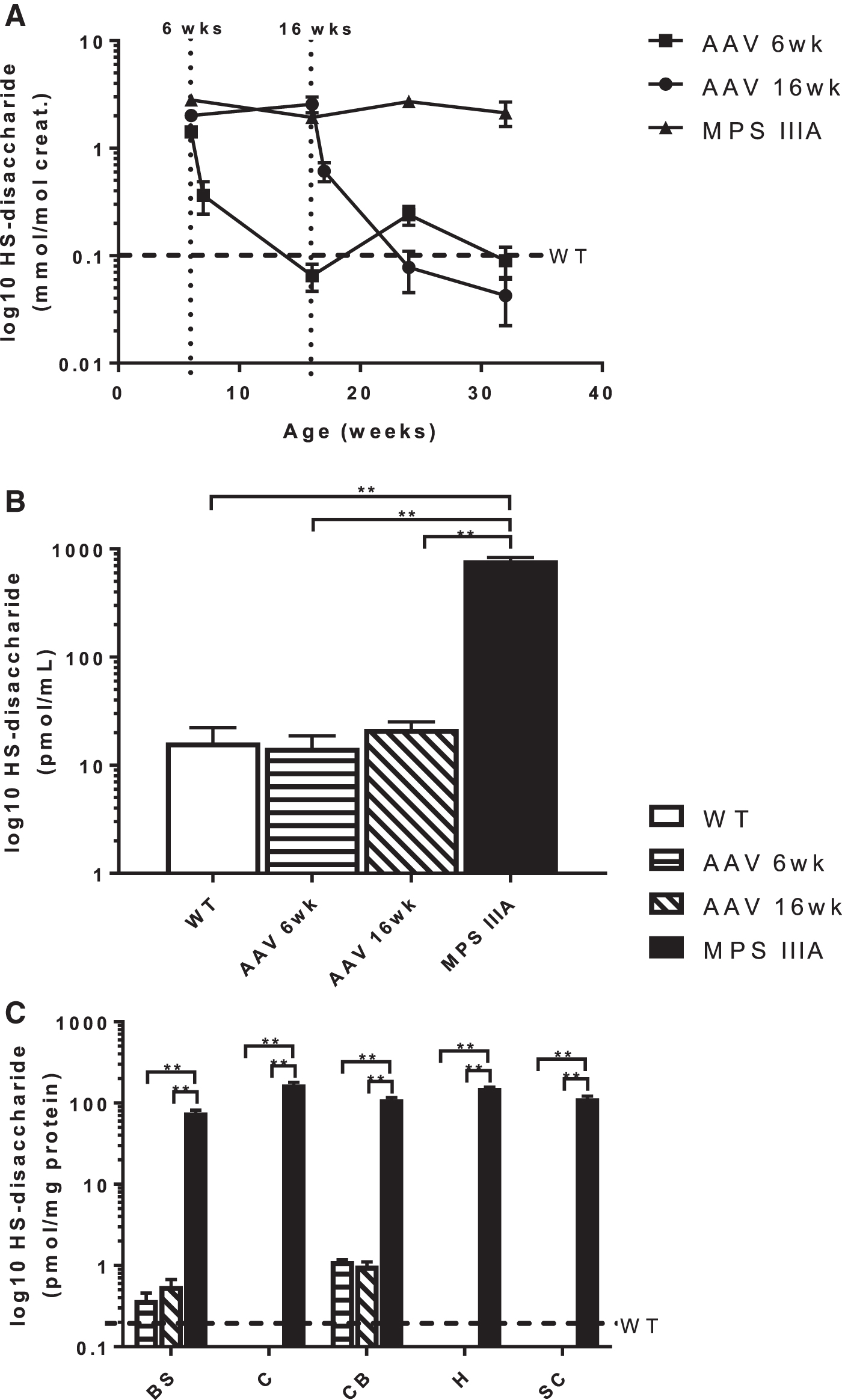
Accumulation of HS-disaccharide in MPS IIIA mice following treatment. Concentration in
SGSH activity in treated mice did not normalize in any of the brain regions and remained significantly reduced compared with WT mice, with no difference between mice treated at 6 or 16 weeks (Fig. 2A). β-hexosaminidase activity, measured as a biomarker of primary HS storage, was increased more than 2-fold in the brain regions of the MPS IIIA mice when compared to WT, whereas mice treated at both 6 and 16 weeks had normal activity (Fig. 2B).
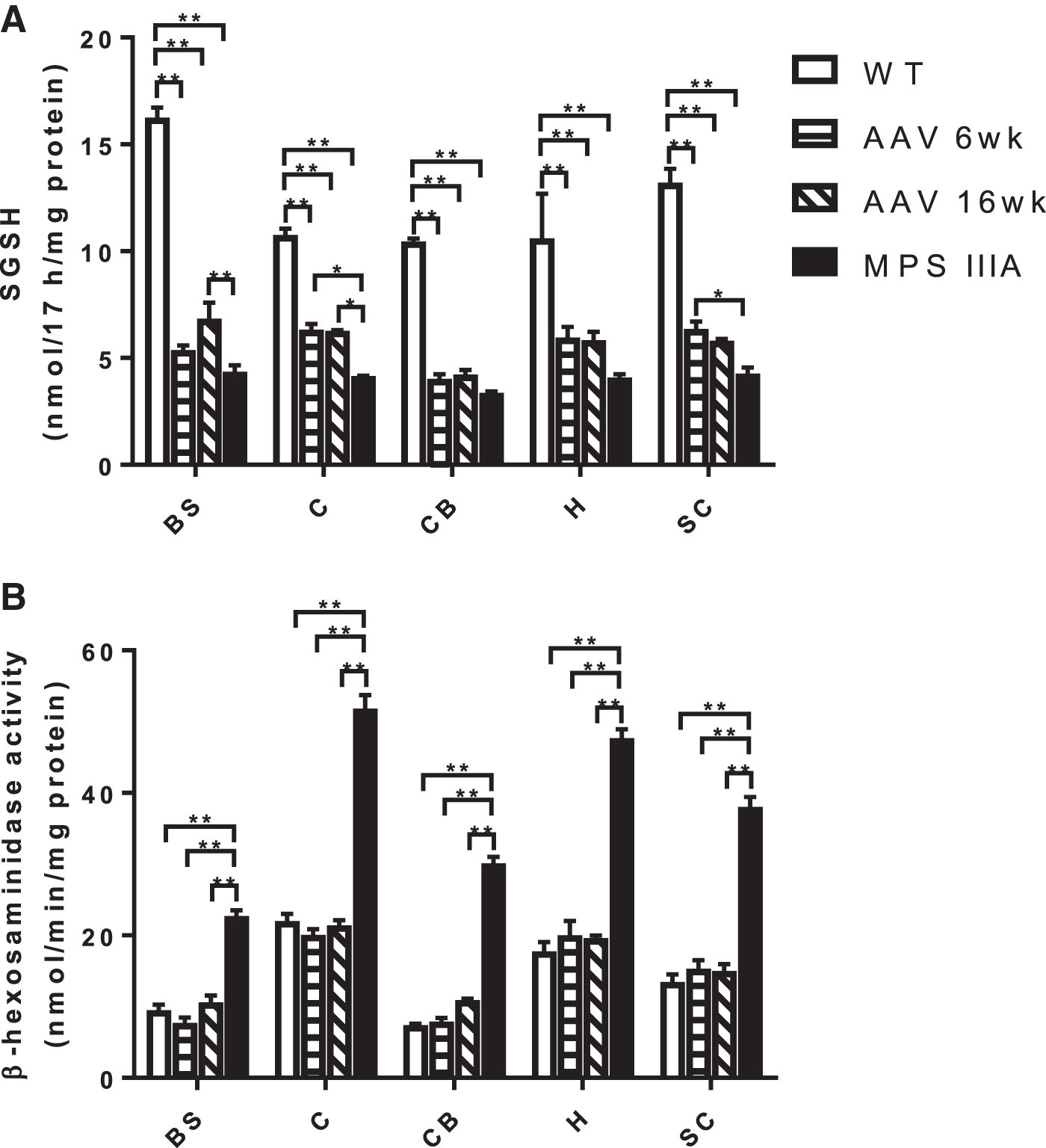
Enzyme activity in the brain regions of MPS IIIA mice following treatment.
Ganglioside accumulation in the five brain regions was alleviated in response to scAAV9.U1a.hSGSH delivered at 6 and 16 weeks of age. In all regions, except the cerebellum, the concentrations of GM2 and GM3 were the same as WT in the treated MPS IIIA mice (Fig. 3). Although the concentrations of GM2 and GM3 in the cerebellum reduced in the treated mice, it was not to the same extent as the other regions. There was no difference in GD2 and GD3 between WT and treated mice in any of the brain regions (Supplementary Fig. S1).

Concentration of GM2 and GM3 gangliosides in the brain regions of wildtype, MPS IIIA, and treated mice. Total concentrations were determined by summing the individual species within each subclass and data are presented as mean ± SEM (n = 4 for all groups). WT, wild-type; AAV 6wk, MPS IIIA mice treated at 6 weeks of age; AAV 16wk, MPS IIIA mice treated at 16 weeks of age; BS, brain stem; C, cortex; CB, cerebellum; H, hippocampus; SC, subcortex. **p < 0.01.
Physiological improvements in scAAV9.U1a.hSGSH-treated mice
scAAV9.U1a.hSGSH delivery in mice was well tolerated with no apparent adverse impact on general health. Mice were weighed daily for the first week following vector administration and no weight loss or changes in behaviour or body condition was observed. Long-term body weight measurements showed that the typical elevated weight gain in MPS IIIA mice corrected following scAAV9.U1a.hSGSH administration with mice treated at both 6 and 16 weeks showing no significant difference to their WT counterparts (Supplementary Fig. S2). In addition, both liver weights and bladder volumes in the treated MPS IIIA mice normalized, such that they were indistinguishable from WT mice at necropsy (Supplementary Fig. S3).
MPS IIIA mice displayed significantly reduced locomotor activity as measured by total distance travelled compared to WT mice at all three ages (4, 6, and 8 months) tested (Fig. 4A). The locomotor activity of the MPS IIIA mice treated at 6 weeks of age was comparable with WT mice at 4 months of age, whereas at 6 and 8 months of age, in MPS IIIA mice treated at both 6 and 16 weeks, the locomotor activity increased such that it was indistinguishable from WT (Fig. 4A). Anxiogenic behavior, measured by rearing activity, was impaired in the untreated MPS IIIA mice compared to WT, and those treated at both 6 and 16 weeks, although this did not reach significance (Supplementary Fig. S4). In the water cross-maze, MPS IIIA mice treated at 6 weeks had a similar number of correct entries to WT, whereas fewer correct entries were observed in mice treated at 16 weeks similar to MPS IIIA mice (Fig. 4B). MPS IIIA mice treated at 6 weeks produced normal gait patterns, and gait length, but not gait width, which normalized only in MPS IIIA mice treated at 16 weeks of age (Fig. 4C).
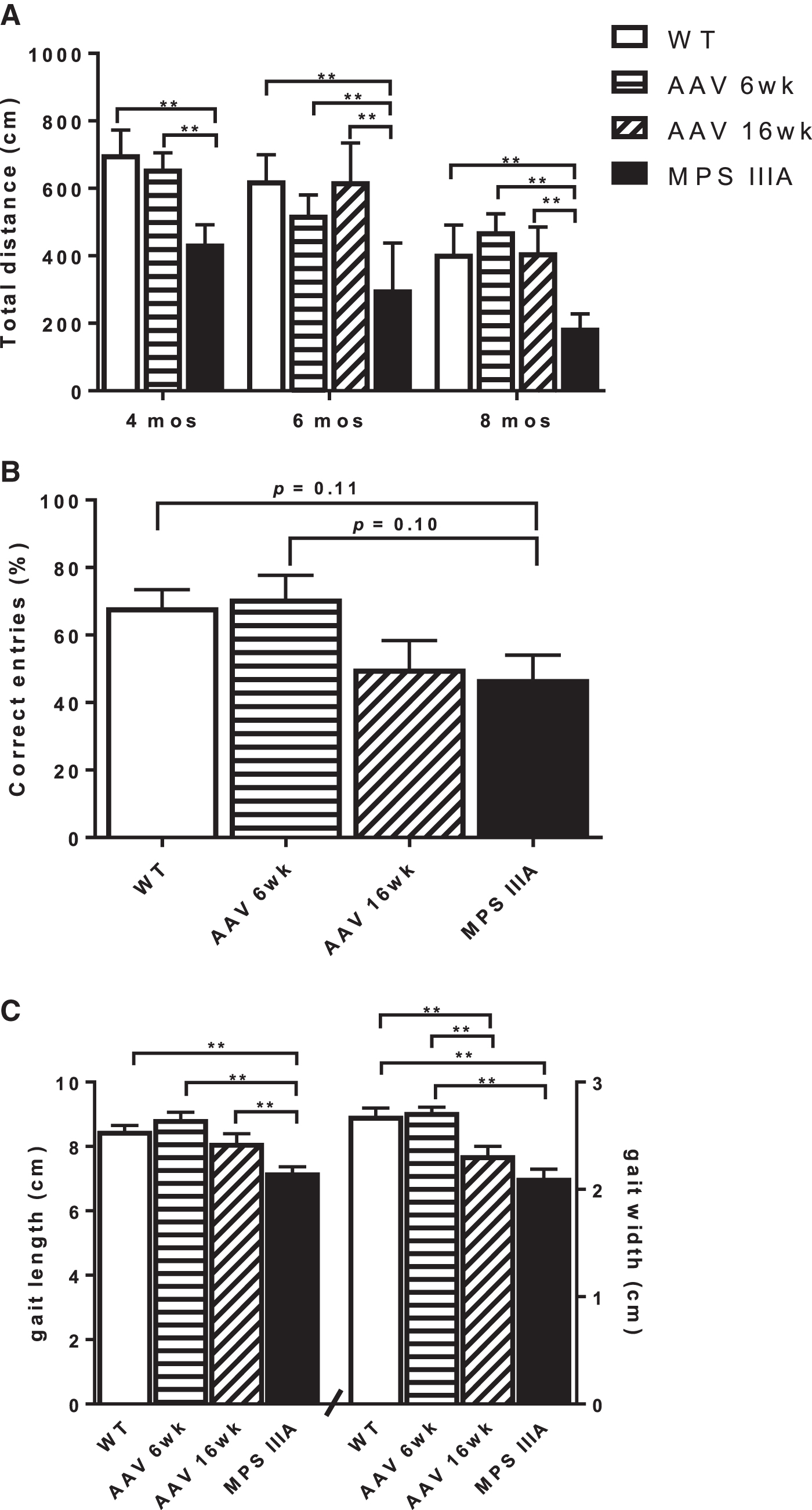
Behavioral assessment of MPS IIIA mice following treatment.
Cortical brain tissue was also assessed by immunohistochemical staining for GFAP to determine the impact of scAAV9.U1a.hSGSH administration at 6 weeks and 16 weeks on astrogliosis in MPS IIIA mice at 8 months of age. In the cortex of MPS IIIA mice, there was marked reactive astrocytic gliosis evident from increased immmunoexpression of the cytoplasmic intermediate filament protein marker, GFAP. This increased immunopositivity was due to enhanced GFAP content in individually enlarged astrocytic processes and cytoplasm (hypertrophy) and astrocytic proliferation (hyperplasia). Prominent astrogliosis was found not only in the parenchymal cortical neuropil but also in perivascular astrocytic endfeet and the subpial region (glia limitans). In both treated groups, there was, overall, a substantial decrease in cortical astrogliosis. Representative images are shown in Fig. 5A–D.
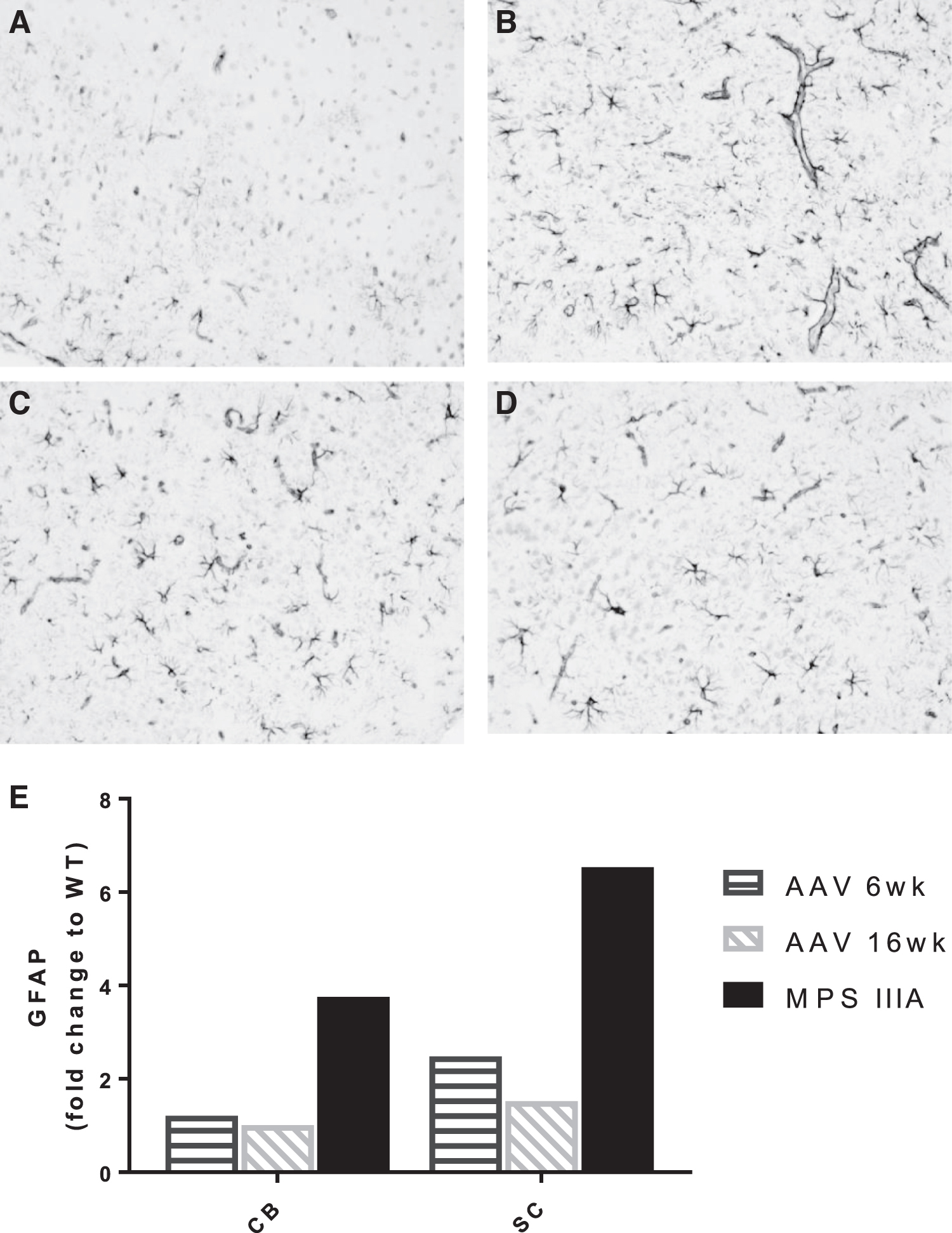
GFAP in the cortex at 8 months of age. Representative sections of positively stained astrocytes at magnification 100 × (GFAP; dark gray) counterstained with hematoxylin for nuclear staining (light gray).
Real-time PCR quantitation of gene expression of Gfap showed a 6.5-fold and 3.7-fold increase in the subcortex and cerebellum, respectively, compared to WT mice (Fig. 5E).
Discussion
Systemic scAAV9.U1a.hSGSH administration for MPS IIIA exploits the use of AAV9 to cross the blood–brain barrier, delivering SGSH to the CNS. Fu et al. 9 have shown that systemic delivery of 5 × 1012 vg/kg of scAAV9.U1a.hSGSH at 1, 2, or 3 months of age is effective at clearing HS storage and results in functional benefits. In this study, we report that 3 × 1013 vg/kg of scAAV9.U1a.hSGSH as late as 16 weeks of age also results in normalization of a HS-disaccharide biomarker in both the urine and CSF of treated mice (Fig. 1). This HS-disaccharide, consisting of an N-sulfated hexosamine and uronic acid [HN-UA(1S)], has previously been shown to be a diagnostic biomarker for MPS IIIA 15 and is currently being used to biochemically monitor the ABO-102 human gene therapy trial (NCT04360265). The measurement of this HS-disaccharide in the CSF reflects the normalization observed in the cerebellum, hippocampus, and subcortex, but does not yield the residual storage that remained in both the brain stem and cerebellum.
SGSH activity did not reflect clearance of substrate, remaining significantly reduced in all regions compared with WT. Of note is the higher residual SGSH activity of 34% reported in our study compared with 4% and 10% using the radiolabeled 21 and fluorescent 22 substrate, respectively. To address this discrepancy, we also measured SGSH activity in the cortex of the MPS IIIA mouse using the radiolabeled substrate, returning 5% of WT (Supplementary Table S1) activity similar to the residual SGSH activity previously reported. 21 Haurigot et al. 22 removed all blood from the tissues by perfusion with phosphate buffered saline before measuring enzyme activity, which likely accounts for the differences observed. This is because fluorescence measurements in tissue homogenates without substrate addition gave higher backgrounds, likely attributed to autofluorescence of lipofuscin in the brain and blood at the same excitation wavelength. 23,24 Subtraction of this background resulted in 4.5% of residual SGSH activity in the MPS IIIA cortex (Supplementary Table S1) similar to previous reports. 21,22 Given relative differences could still be observed, and due to the limited sample available, we were unable to perform a background subtraction for each sample.
Analysis of vector copy number demonstrated that scAAV9.U1a.hSGSH was indeed effectively distributed to the brain stem and cerebellum, analogous to the other regions, and therefore does not explain the remnant HS-disaccharide (Fig. 6). Furthermore, there was no difference in transgene expression (30–60% of normal) across the five brain regions and the secondary elevation in the lysosomal enzyme, β-hexosaminidase, normalized to WT levels inclusive of brain stem and cerebellum (Fig. 2). Ruzo et al. 8 treated MPS IIIA mice with a lower dose of AAV9 vector encoding SGSH (1 × 1012 vg/kg) and reported that the caudal domain of occipital cortex and rostral region of the brain stem only showed partial clearance of storage unlike other sections of the brain. No explanation was proffered for this finding. Given previous work has shown that both the brain stem and cerebellum, unlike the cortex and subcortex, do not continue to accumulate the HS-disaccharide with age, 16 there may be some particular intolerance for storage in these regions that warrants further investigation.
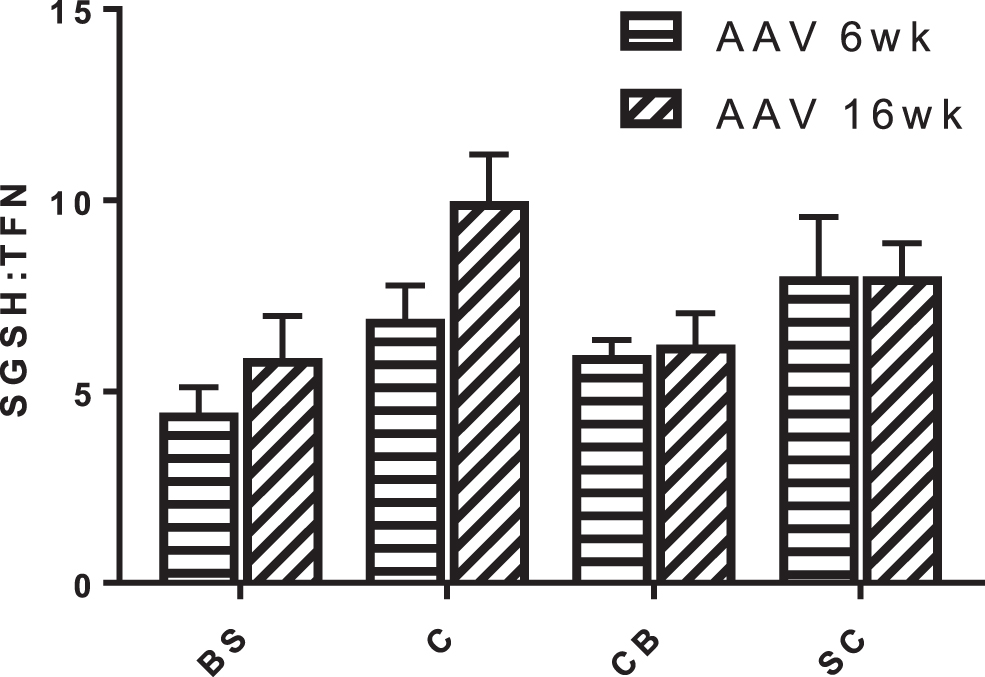
Distribution of vector genome across the brain regions of mice treated at 6 weeks (AAV 6wk; n = 4) and 16 weeks (AAV 16wk; n = 4). Data are expressed as vector genome (scAAV9.U1a.hSGSH) per diploid genome (transferrin [TFN]) and presented as mean ± SEM. BS, brain stem; C, cortex; CB, cerebellum; SC, subcortex.
Therefore, we sought to investigate whether residual ganglioside storage also remained in the brain stem and cerebellum following scAAV9.U1a.hSGSH administration. Secondary accumulation of gangliosides is well documented in the MPS IIIA brain, but the significance is not understood. 25 We have shown in this mouse model that the simple gangliosides GM2 and GM3 are elevated as early as 1 month of age, 16 and we show that these gangliosides normalized in the brain stem, cortex, hippocampus, and subcortex with treatment at both 6 and 16 weeks of age (Fig. 3). However, in the cerebellum, these gangliosides, although reduced compared with untreated mice, did not normalize and remained significantly higher than WT. This suggests that scAAV9.U1a.hSGSH administration also alleviates secondary ganglioside storage even as the disease progresses and the gangliosides continue to accumulate. We also showed that GFAP astrogliosis was reduced in MPS IIIA mice treated at both 6 and 16 weeks of age (Fig. 5), a finding supporting earlier reports of improvements in neuroinflammation. 9,22
From a biochemical standpoint, scAAV9.U1a.hSGSH has successfully cleared both primary and secondary ganglioside storage from the brain. This study also highlights the utility of CSF as an obtainable sample to measure the HS-disaccharide and gangliosides, as it does indeed reflect the biochemistry within the brain. As yet, no result has been published from the human clinical trial, but the study sponsor has reported a dose-dependent reduction in HS-disaccharide in the CSF of patients treated with low (5 × 1012 vg/kg), medium (1 × 1013 vg/kg), and high (3 × 1013 vg/kg) doses (NCT02716246). Nonetheless, our study shows that even at the high dose of 3 × 1013 vg/kg, storage was not completely cleared from the brain stem or the cerebellum, and although the aim of our study was to interrogate the brain biochemistry, we also performed a small range of functional tests to determine whether there was any association with this residual storage and behavior.
Similar to the data reported by Fu et al., 9 we reveal that treated mice improved neurocognitively in their ability to find a hidden platform in a swimming test, noting differences in methodology (Morris water maze was used by Fu et al. 9 ). Mice treated at 6 weeks of age performed better than mice treated at 16 weeks (Fig. 4B), highlighting the notion that treatment at an earlier age is preferential, and Fu et al. 9 also demonstrated that treatment before 3 months of age was superior to treatment at 6 months of age. To try and understand the functional consequences of the residual storage in the cerebellum, we performed additional behavior testing, including assessing locomotor activity and importantly gait, which is particularly sensitive to cerebellar function. Locomotor activity was indistinguishable from WT in treated mice regardless of age of treatment, suggesting that this parameter is easily corrected (Fig. 4A). Although mice treated at the early age of 6 weeks displayed normal gait, mice treated at 16 weeks displayed abnormal gait width (Fig. 4C). Thus, it is possible that residual storage in the cerebellum may be contributing to the abnormal gait noted in the MPS IIIA mice treated at the later stage of disease. No direct assessment was possible for the brain stem, a region of the brain involved in a number of basic functions through which all information relayed from the body to the cerebellum must traverse. It is not known whether there is a relationship between the storage in the brain stem and cerebellum, and what impact this has (if any) on the neurological improvement following scAAV9.U1a.hSGSH administration. Analysis of GFAP astrogliosis both by immunohistochemical methods in the cortex, and in the subcortex and cerebellum using gene expression analysis showed a reduction in reactive astrogliosis in both 6 and 16 weeks of age compared with the MPS IIIA-untreated counterparts. This is comparable to previous results reporting correction of GFAP astrogliosis at 9 months of age. 9 Further work is needed to interrogate the significance of residual storage and any possible downstream consequence.
Conclusions
We have shown that 3 × 1013 vg/kg of scAAV9.U1a.hSGSH—as used in the high dose cohort of the phase I/II clinical trial (NCT02716246) —clears storage within the brain when administered intravenously at both early (6 weeks) and later (16 weeks) stages of disease progression. Furthermore, we provide direct evidence that measuring the HS-disaccharide in both CSF and urine reflects concentrations within the brain per se. Quantification of the gangliosides, GM2 and GM3, in CSF may provide an additional biochemical assessment of therapeutic efficacy as both have been shown to be informative in a natural history study of MPS IIIA. 26 Although neurocognitive function improved at both treatment times, as expected, better outcomes were seen in mice treated at 6 weeks compared with 16 weeks. This study provides much needed preclinical data in the mouse model of MPS IIIA from which it is possible to collect brain tissue to assess the biochemical impact of systemic scAAV9.U1a.hSGSH administration.
Footnotes
Acknowledgments
The authors thank Wan Ling Ng for technical assistance, Sharon Chin for SGSH activity using the radiolabeled substrate, and Jim Manavis and John Finnie for GFAP immunohistochemistry, and are most grateful to Abeona Therapeutics for gifting the clinical grade scAAV9.U1a.hSGSH used in this study.
Authors' Contributions
M.F. designed the study. J.T.S., A.L.K.D.-R., and C.M. performed the experimentation and analyzed the data. J.T.S., A.L.K.D.-R., and M.F. wrote the article. All authors critically reviewed and approved the final published version.
Author Disclosure
M.F. declares that she has received travel support to attend educational meetings from Abeona Therapeutics.
Funding Information
This study was supported by a project grant from the Women's and Children's Hospital Research Foundation (South Australia).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.