
Abstract
Oncolytic virotherapy represents an ideal therapeutic platform for cancer in which natural or engineered viruses selectively replicate in and destroy tumor cells, whereas sparing normal cells. Oncolytic virotherapy is considered as a key contributor in modern immunotherapy. However, several challenges remain with regard to exploiting the potential of oncolytic viruses, such as the lack of biomarkers for precise treatment, the difficulty of systemic administration because of pre-existing neutralizing antibodies to popular oncolytic viral vectors in human serum and the lack of mature lyophilization technology for convenient transport and preservation of viral preparations. The M1 strain, which was isolated on Hainan Island of China in the 1960s, is a member of the alphavirus genus Togaviridae family. It was identified as a novel oncolytic virus in 2014. During the development of M1 virus, many challenges have been overcome: several biomarkers have been identified for precise treatment; systematic administration of M1 is suitable and feasible because of the extremely low percentage of pre-existing neutralizing antibodies in the general population, and a lyophilized powder that maintains high biological stability has been developed. This review provides an encyclopedia of studies supporting M1 as an oncolytic virus, including the biological characteristics, tumor selectivity and its mechanism, tumor killing mechanism, combination therapy, and nonclinical pharmacokinetics of M1 virus. The future development direction of oncolytic virus M1 is also discussed at the end of the review.
INTRODUCTION
Oncolytic viruses are naturally existing or genetically engineered organisms, due to their abilities to selectively self-amplify in and kill tumor cells but leave normal cells intact, oncolytic virotherapy represents an ideal therapeutic platform to treat tumor patients.
1,2
The ability of viruses to kill cancer cells has been recognized for nearly a century, but only for the past decade have clinical trials documented a benefit in patients with cancer.
1,3
In fact, interests in oncolytic viruses have been skyrocketed based on a better understanding of viral biology, tumor immunology, and molecular genetics. Direct cell lysis has long been identified as the mechanism by which oncolytic viruses kill tumor cells. In recent years, oncolytic virotherapy has been considered as a key member of immunotherapy due to its capacities in modulating the tumor microenvironment (TME) and activating both innate and adaptive antitumor immune responses.
4,5
Talimogene laherparepvec (IMLYGIC®; Amgen), which is derived from herpes simplex virus type 1 (HSV-1) and encodes granulocyte-macrophage colony-stimulating factor (GM-CSF), was the first oncolytic virus approved in the United States and Europe for unresected melanoma.
6
Since then, incredible interests aroused in the scientific community and pharmaceutical industries to develop oncolytic viruses. To date, hundreds of oncolytic viruses derived from >10 types of viral vectors are in different stages of clinical trials (see
The exciting outcome of clinical trials from several oncolytic viruses brings renewed hope for cancer patients. However, several challenges remain in the exploitation of oncolytic viruses. (1) Few biomarkers have been identified for precise treatment with oncolytic viruses. Biomarkers mean measurable and quantifiable biological parameters, such as the level of a certain messenger RNA (mRNA) or protein, the mutation of a certain gene, the mutation burden of DNA, the amount of certain type of cells, which can be evaluated to determine whether a patient is suitable for oncolytic virotherapy. Without biomarkers, oncolytic viruses are applied in a trial-and-error principle that similar to cytotoxic anticancer chemicals, failing to precisely identify a therapeutic window at the cost of patients who receive ineffective treatment. In 2017, the U.S. Food And Drug Administration (FDA) approved Keytruda (Keytruda®; Merck & Co) for cancer patients with the biomarker referred to as microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR). This is the first time that the FDA has approved a cancer treatment based on a common biomarker rather than the location in the body where the tumor originated. Since then, identifying biomarkers has become a new paradigm in the development of anticancer drugs. The search for predictive biomarkers of the oncolytic virus pelareorep (Reolysin®; Oncolytics Biotech) derived from reovirus is also ongoing. Although several candidates, such as the mutation status of K-RAS and expression of hypoxic inducible factor 1α (HIF-1α), appeared to predict sensitivity to pelareorep in nonclinical studies, no certain biomarkers were identified during the clinical study. 7
(2) Pre-existing neutralizing antibodies to popular oncolytic viral vectors in human serum make them suitable only for administration by local injection to elicit sufficient therapeutic efficacy. Although intratumoral injection concentrates oncolytic viruses in target foci, it largely prevents them from reaching distal metastatic foci and sites deep inside the body. Systematic administration represents the most effective way of drug delivery for oncolytic viruses, and a growing number of pharmaceutical agencies are targeting it as the next stage for the development of oncolytic viruses. (3) The preparation of most oncolytic viruses is still liquid injection, which requires a temperature of −80°C to maintain their biological activity, hampering transport and preservation. Accordingly, there is an urgent need to develop a method to preserve oncolytic viruses as lyophilized powder, which can be transported and preserved at 2–8°C.
Alphavirus M1 is a novel natural oncolytic virus identified by our team, and during the development of M1 virus, we have been attempting to overcome the challenges described earlier. In brief, several biomarkers enabling precise treatment have been identified, and systematic administration has been found to be suitable for M1 because of the extremely low percentage of pre-existing neutralizing antibodies in the general population. A lyophilized powder formulation that maintains high biological stability has also been developed. In this review, we will introduce an encyclopedia of the researches about alphavirus M1, which includes the biological characteristics, tumor selectivity and its mechanism, tumor killing mechanism, combination therapy, and nonclinical pharmacokinetics of M1 virus.
THE BIOLOGICAL CHARACTERISTICS OF M1 VIRUS
During investigation on arboviral diseases in Mao'an Village (Long 109°07′ E, Lat 18°07′ N), Baoting Country, Hainan Island, China, in 1964, by the former National Institute for the Control of Pharmaceutical and Biological Products, a virus (strain M1) isolate was obtained from a pool of Culex mosquitos captured in the wild. Cross-hemagglutination-inhibition tests indicated strain M1 to be an alphavirus 8,9 (called alphavirus M1 or M1 virus in this review). Alphavirus M1-related antibodies were detected in humans, swine, equine, goats, and chickens native to Hainan (with positivity rates of 3.6–26.4%, 17.7–27.3%, 25%, 37.5%, and 30.8%, respectively), suggesting that these species may have been infected by alphavirus M1. 8 M1 virus is not related to any arboviral diseases, and it had not been further investigated for nearly 50 years since it was isolated, until our group identified its potential to kill tumor cells in 2009. 10
The genome of alphavirus M1 is a positive-sense single-stranded RNA (+ ssRNA) ∼12 kb in length that is organized with nonstructural proteins (nsPs) at the 5′ end and structural proteins at the 3′ end. The complete sequence information for alphavirus M1 is available. 11,12 nsPs are translated from genomic RNA and structural proteins from subgenomic RNA. The genome of alphavirus M1 encodes a total of five structural proteins, including three envelope proteins (E1, E2, and E3) as well as capsid protein (C) and 6K protein (6K), and four nonstructural proteins (NS1, NS2, NS3, and NS4). As the replication of alphavirus M1 depends on viral encoded RNA-dependent RNA polymerase (NS4), without a DNA stage in its life cycle, it is impossible for viral nucleic acid to recombine with host DNA; thus, there is no risk of long-term latency or carcinogenic mutations caused by chromosome integration.
The cytopathic morphology induced by M1 virus and its life cycle have been studied by transmission electron microscopy in a M1-sensitive HepG2 liver carcinoma cell line. 13 First, M1 virus enters HepG2 cells through endocytosis, and the endocytosed vesicle contains a plurality of virus particles. Second, assembly of newly synthesized nucleocapsids and the packaging of RNA occur in the cytoplasmic and endoplasmic reticulum (ER). Third, the new nucleocapsids acquire envelopes and mature not only at the plasma membrane but also at vacuole membranes inside the cell. Another kind of particle, 59 nm in diameter, can also be detected in the ER cavity. Fourth, M1 virus is released in two ways: nucleocapsids budding through the plasma membrane and mature virus-like particles moving through vacuole transportation. 13 The detailed process of viral entry, assembly, maturation, and release provides the structural basis for the study of M1 virus–host interactions.
Although the biological characteristics of M1 virus have been well reported, several details of the biophysical events remain to be explored, as follows: How does the virus facilitate endocytosis into the cell? What is the exact antigen motif in the viral particle that is recognized by the receptor or neutralizing antibodies? How do viral genes or proteins interplay with the host genome or proteins to facilitate viral replication? How much potential exists for modifying the genome of M1 virus? Addressing these questions will help us better understand and further develop M1 virus.
PROOF OF CONCEPT: THE IDENTIFICATION OF M1 AS AN ONCOLYTIC VIRUS
Since alphavirus M1 was isolated in the 1960s, no research on M1 was reported for nearly 50 years, until we first verified that M1 virus could be used to kill cancer cells in 2009. 10 Before 2009, alphaviruses had already emerged as preferred gene vectors to specifically induce apoptosis in cancer cells, namely, Sindbis virus 14 and Semliki forest virus. 15 We first assessed whether alphavirus M1 has oncolytic ability in glioma. M1 induced apoptosis rather than necrosis in the rat malignant glioma cell line C6, a result that might correlate with cell cycle arrest in S phase. Detailed examination identified that an intrinsic mitochondrial pathway represented by an increased proapoptotic protein Bax/antiapoptotic protein B cell lymphoma-2 (Bcl-2) ratio and the release of cytochrome c contributed to the apoptosis induced by M1 viral infection. Furthermore, downregulation and nucleolar translocation of the p21WAFI/CIPI protein contributed to M1-induced C6 apoptosis. 10 It was the first study to prove that M1 virus could be used to kill cancer cells, it also revealed a novel mechanism of alphavirus-induced apoptosis by illustrating the relationship between cell cycle redistribution and apoptosis.
The concept that alphavirus M1 represents a novel oncolytic virus was achieved in 2014. 16 In addition to murine malignant cells, M1 markedly induces cell death in a large panel of human cancer cell lines, including >10 types of high incidence malignancy, in a dose-related and viral replication-dependent manner. In contrast, normal human primary cells derived from heart, muscle, bladder, colon, and liver tissues are highly resistant to M1 virus. M1 also shows in vivo antitumor activity in various types of xenograft mouse models by intravenous or intratumoral injection. Moreover, M1 effectively inhibits the viability of primary human hepatic and colorectal tumor explants. M1 does not cause mortality in mice or rats, and all examined animals remained asymptomatic throughout the treatments. Detection of viral RNA showed that M1 was >1,000 times enriched in tumor tissues compared with any other normal tissues, including brain, heart, kidney, liver, lung, muscle, and spleen, in the tumor-bearing mouse model, suggesting that the antitumor activity and safety of M1 are based on high tumor tropism. 16
The definition of oncolytic viruses requires selective tropism for cancerous cells and tissues without targeting normal cells and tissues. This research first identified M1 virus as a promising member of the oncolytic virus family with the advantage of being suitable for systemic infusion. This initial proof-of-concept study has become the milestone of oncolytic virus M1 development.
After the initial proof-of-concept study in 2014, the efficacy of oncolytic virus M1 by intravenous injection was also thoroughly studied in bladder carcinoma. 17 Bladder cancer is the most common malignancy of the urinary system, 18 and approximately one-quarter of bladder cancers are muscle-invasive bladder cancers (MIBCs), with elevated incidence and mortality in China. 19 Oncolytic virus M1 selectively kills bladder cancer cells but not normal cells, and transcriptome analysis revealed that interferon (IFN)-stimulated genes (ISGs) are expressed at low levels in sensitive bladder cancer cells and at high levels in resistant cells. In orthotopic MIBC mice, tail vein injections of M1 significantly inhibit tumor growth and prolong survival, and the antitumor effects of M1 are stronger than those of the first-line chemotherapy agent cisplatin (CDDP). 17 This research indicates that M1 virus might be a good alternative treatment for bladder cancer and further affirms the proof of concept that M1 is a promising oncolytic virus.
Although the safety of M1 virus was evaluated in mice, 16 differences between rodent and human anatomy, physiology, and pathology are too great to accurately predict the safety of M1 virus in humans. Cynomolgus macaques (Macaca fascicularis), which share similar physiology and infection patterns with humans, have been used to study the safety of M1 virus. 20 Three rounds of repeated intravenous injections of a high dose of M1 virus were performed, with six injections in each round and a 50- or 130-day interval between each round. Body weight, temperature, complete blood count, clinical biochemistry, cytokine profiles, lymphocyte subsets, neutralizing antibodies, and clinical symptoms were closely monitored at different time points. Magnetic resonance imaging was also performed to assess the possibility of encephalitis or arthritis, which can be induced by other type of alphavirus. 21,22 All the extensive analysis yielded no evidence of toxicity. 20 Conclusively, we identified M1 as a nonpathogenic virus in nonhuman primates.
MECHANISM OF ACTION: BIOMARKERS FOR PRECISE TREATMENT OF M1 VIRUS
It is of great importance to identify biomarkers that can predict the antitumor efficacy of oncolytic viruses. 1 It is well known that the specificity of oncolytic viruses to replicate in tumor cells stems mainly from the deficiency of antiviral activity in tumor cells. In contrast to normal cells, tumor cells often eliminated or inactivated key gene products that control antiviral signaling, such as type I IFNs. 1 Research into the tumor selectivity mechanism offers the opportunity to identify biomarkers for prediction of oncolytic efficiency. The search for predictive biomarkers for various oncolytic viruses is ongoing; to date, a few conclusive biomarkers have been identified, including reovirus requiring activated Ras signaling 23 and vesicular stomatitis virus (VSV) requiring defects in the IFN pathway. 24 Four biomarkers for M1 virus have been identified: zinc-finger antiviral protein (ZAP), 16 inositol-requiring kinase 1α (IRE1α), 25 ras homolog family member Q (RHOQ), 26 and K-RAS mutation 27 (Fig. 1).
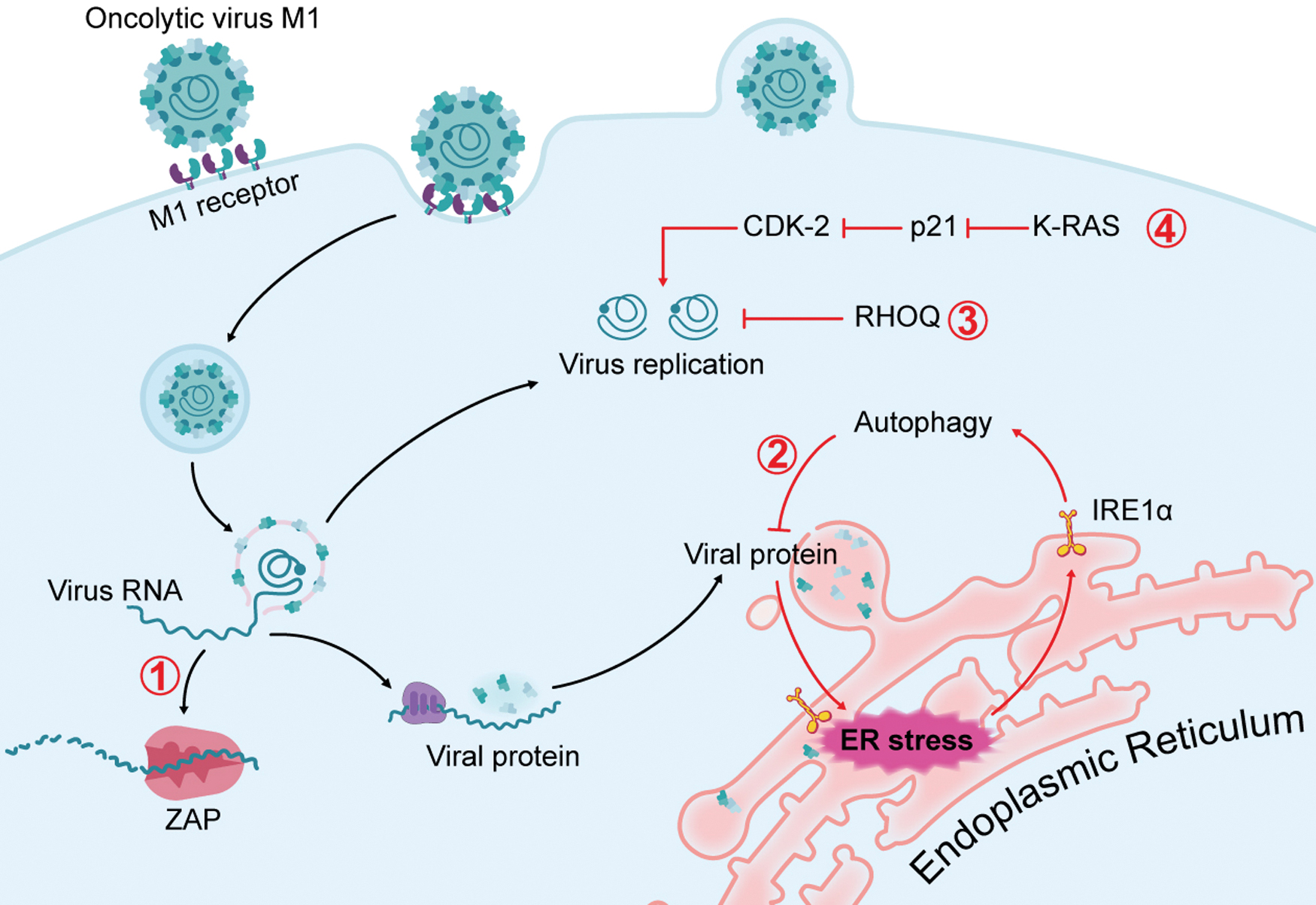
Mechanism of action: biomarkers for precise treatment of M1 virus. Four biomarkers for M1 virus have been identified: ① ZAP; ② IRE1α; ③ RHOQ; ④ K-RAS mutation. After the binding of M1 virus with receptor on the membrane and membrane fusion, the viral RNA is released to the cytoplasm to replicate progeny viral genome or translate to viral proteins. ZAP can bind with the viral RNA and recruit degrading machines to degrade viral RNA, the deficiency of ZAP mediates sensitivity of cancer to M1 virus. The RHOQ axis, which is a side branch of the cholesterol biosynthesis pathway, has an antiviral role during M1 replication; the deficiency of RHOQ also mediates the sensitivity of cancer to M1 virus. The activation mutation of K-RAS promotes the replication of M1 virus by inhibiting p21. The expression of viral proteins leads to the accumulation of misfolded proteins in the ER lumen, which can activate IRE1α, one of the UPR pathway. The activation of IRE1α in turn triggers autophagy, which counteracts the viral protein load and subsequent oncolysis, so the lower expression of IRE1α, the higher sensitivity of tumor to M1 virus. ER, endoplasmic reticulum; IRE1α, inositol-requiring kinase 1α; RHOQ, ras homolog family member Q; UPR, unfolded protein response; ZAP, zinc-finger antiviral protein. Color images are available online.
Zinc-finger antiviral protein
During the research that identifying M1 as a novel oncolytic virus, the molecular mechanism of M1 tumor selectivity was also probed. 16 Transcriptional profile and arrayed small interfering RNA (siRNA) screening identified ZAP as the highest ranked host factor against M1 virus. ZAP is an ISG that inhibits the replication of multiple viruses by inducing viral RNA degradation and translational inhibition. 28 Expression of ZAP protein in sensitive tumor cell lines is significantly lower than that in resistant tumor and normal cell lines. In addition, depletion of ZAP expression overcomes resistance to M1, as it leads to increased viral replication, viral RNA and protein expression and M1-induced cell death. Consistently, susceptible tumor cells transfected with vectors expressing ZAP show decreased viral yield, decreased viral RNA and protein expression, and finally suppressed viral oncolysis. 16 Such gain- and loss-of-function data suggest that ZAP deficiency marks the potential for personalized therapy with M1 in cancers. Indeed, large-scale tumor tissue microarrays containing paired tumor and adjacent non-neoplastic specimens from patients indicate high percentages of ZAP deficiency in liver, bladder, and colon cancer, suggesting high potential of M1 virus in these cancer types. 16 In addition to M1 virus, ZAP was also found to be an antiviral factor of a wide range of viruses, 29 highlighting its potential as a predictive biomarker for other types of oncolytic viruses.
The high percentages of ZAP-deficient tumors encouraged us to explore its potential tumor-suppressive role. Pancancer analyses demonstrate that ZAP is downregulated in a wide range of tumor types and associated with poor patient survival. In vitro and in vivo studies show that ectopic expression of ZAP inhibits tumor growth. Furthermore, loss of function of ZAP co-operates with Apc deficiency to promote colorectal cancer in mice, demonstrating that ZAP is a genuine tumor suppressor in colorectal carcinoma. 30 This research provides an example of an antiviral factor acting as a tumor suppressor. 30 Canonical tumor suppressor genes, including TP53 and PTEN, have also been identified as positive regulators of innate immunity, such as the antiviral response. 31,32 The tight relationship between tumor suppression and intrinsic antiviral response, if not pure coincidence, suggests that a broad range of tumor suppressors have the potential to serve as antiviral factors and, therefore, biomarkers of oncolytic viruses.
Inositol-requiring kinase 1α
Replication of oncolytic virus leads to the accumulation of misfolded viral proteins in the ER lumen, which activate three major unfolded protein response (UPR) pathways: the IRE1α-XBP1 pathway, the protein kinase-like endoplasmic reticulum kinase (PERK)-eIF2α pathway, and the ATF6 pathway. The IRE1 pathway is the most ancient branch of the UPR and is conserved from yeast to mammals. 33 Infection of glioma cells with M1 virus primarily activates IRE1α, which in turn counteracts the viral protein load and subsequent oncolysis in glioma cells. 25 Furthermore, inhibition of IRE1α increases the oncolytic effects of M1 virus by overcoming this limitation. Consequently, IRE1α might constitute a second predictive biomarker of M1 virus. Moreover, IRE1α expression levels in glioma patients decrease gradually during malignant progression and correlate negatively with glioma progression. 25 This suggests a potential for M1 treatment in high-grade glioma, and the expression level of IRE1α might predict the efficacy of M1 in the future clinical study.
Ras homolog family member Q
Lipids are important bioactive molecules that are extremely relevant to virus infection, either by providing structural ingredients or by regulating intracellular signaling pathways. Regarding the relationship between lipid metabolism and M1 replication, cholesterol biosynthesis genes are decreased after M1 infection, and the mevalonate/protein farnesylation/RHOQ axis, which is a side branch of the cholesterol biosynthesis pathway, has an antiviral role during M1 replication in refractory tumor cells. 26 Loss of RHOQ renders colorectal cancer and pancreatic cancer cells permissive and sensitive to M1 virus. Consistently, tumor cell lines with lower levels of RHOQ are more sensitive to M1 virus. 26 This discovery suggests great potential for personized cancer therapy through prescreening for RHOQ expression in tumor biopsies.
K-RAS mutation
The tumor selectivity of oncolytic viruses is largely conferred by tumor-specific aberrations in signaling pathways that normally sense and block viral replication. It is now well established that cancer-specific aberrations in BCL-2, WNT, EGFR, RAS, TP53, RB1, PTEN, and other cancer-related genes predispose cancer cells toward viral infection. 34 –36 The relationship between M1 viral infection and oncogenic mutations in a large panel of tumor cell lines has been analyzed, and among oncogenic mutations, K-RAS mutation correlated with the oncolytic efficiency of M1 virus. 27
Experimental elimination of the K-RAS mutation renders tumor cells resistant to M1 virus. The oncogenic RAS/RAF/MEK pathway promotes the replication and oncolytic effect of M1 virus by inhibiting expression of the key antiviral factor CDKN1A, also known as p21. 27 Despite more than three decades of intensive effort, no effective pharmacological inhibitors of RAS oncoproteins are available in the clinic, promoting the widely held perception that RAS proteins are “undruggable.” 37,38 Nonetheless, M1 virus might offer renewed hope for the treatment of cancer with abnormal RAS activation and transform RAS-driven tumors to be “druggable.” Among all cancer-driving genes, RAS is the most frequently mutated oncogene family in human cancers. The top four cancer types harboring RAS mutations are pancreatic ductal adenocarcinoma (97.7%), colorectal adenocarcinoma (52.2%), multiple myeloma (42.6%), and lung adenocarcinoma (32.2%). 37 The high percentage of RAS mutations in tumors indicates that M1 virus might be effective in a large proportion of cancer patients.
Precision medicine for cancer represents the most efficient method and is becoming the ultimate goal of the pharmaceutical industries. 39,40 Indeed, prescreening of biomarker levels greatly helps to decrease costs and expedite treatment of cancer patients. 39,40 Although multiple biomarkers have been identified for M1 virus, different biomarkers might function heterogeneously in distinct cancer types. Overall, there remains an urgent demand to develop effective biomarker or biomarker combinations for each cancer type. Most importantly, the correlation of biomarker levels and oncolytic efficiency needs verification through large-scale clinical trials.
THE TUMOR KILLING MECHANISM OF M1 VIRUS
M1 virus destroys tumors through multiple mechanisms, including (1) infection and replication within cancer cells, resulting in direct cell lysis and ER stress-mediated apoptosis or autophagy; (2) release of cytokines by infected cells, which promote death in surrounding cancer cells, the so-called bystander killing effect; and (3) release of antigens by killed tumor cells, which trigger natural and acquired immune responses, enhancing both local and systemic antitumor effects. The tumor killing mechanisms of M1 virus resemble those of other oncolytic viruses, 1,41,42 establishing them as promising anticancer bioagents (Fig. 2).
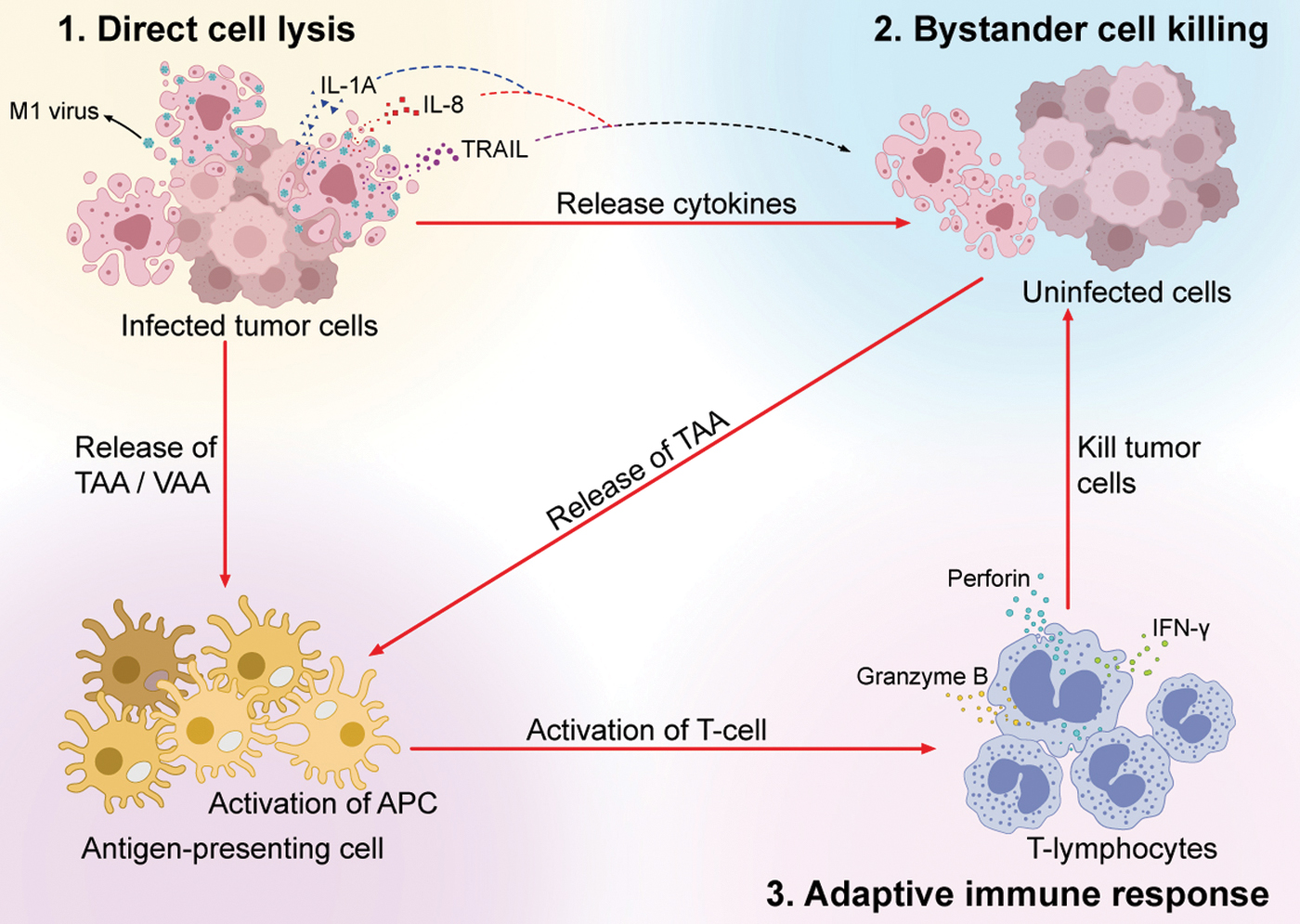
The tumor killing mechanism of M1 virus. M1 virus destroys tumors through multiple mechanisms. The infection and replication of M1 within cancer cells result in direct cell lysis mediated by ER stress-induced apoptosis. The infected tumor cells release several cytokines such as IL-1A, IL-8, and TRAIL to destroy neighboring uninfected tumor cells, which represents the bystander killing effect. The direct cell lysis and bystander killing of tumor cells release the TAAs and VAAs, which can activate the antigen-presenting cells and subsequently prime T-lymphocytes (the activation of adaptive immune response). The primed T-lymphocytes elicit perforin, IFN-γ and granzyme B to kill uninfected tumor cells. IFN, interferon; TAAs, tumor-associated antigens; VAAs, virus-associated antigens. Color images are available online.
Direct cell lysis
Replication of oncolytic viruses results in the accumulation of large amounts of viral protein in the ER of host cells, which are recognized as heterologous proteins and activate the UPR to handle unfolded foreign proteins and restore cellular homeostasis. When the accumulation of viral proteins is insurmountable after UPR activation, downstream pathways are initiated, leading to irreversible ER stress and apoptosis. Scanning electron microscopy revealed a progressive distension of the ER lumen as early as 6 h post-M1 infection in sensitive cells, catastrophic destruction and condensation of chromatin were observed over time, indicating prolonged and severe ER stress that induces apoptosis. 16 Two symbolic events of ER stress include the induction of chaperone BIP and phosphorylation of eIF-2α accompanying the change in ER morphology. Of three specific ER stress-mediated apoptosis pathways (CHOP, c-Jun N-terminal kinase [JNK] and caspase-12), 43 JNK and caspase-12 are activated by M1. 16 Understanding the molecular biology of M1-induced ER stress and subsequent cell death has facilitated the discovery of synergistic compounds for combination therapy and enabled the identification of multiple ER stress inducers to enhance the oncolytic effect of M1 virus.
Autophagy is an evolutionarily conserved process involving the formation of double-membrane vesicles called autophagosomes. 44 Autophagy plays key roles in maintaining cellular homeostasis by eliminating unwanted proteins and damaged organelles through self-digestion in cell lysosomes, thereby promoting cell survival. Moreover, autophagy activation protects cells against certain pathogens. 45 Nevertheless, uncontrollable foreign pathogen replication induces unbalanced autophagy, which results in cell death. In glioma, M1 virus can induce autophagy in host cells, which might function to counteract the replication and expression of viral proteins in the cells. 25 Recent reports have revealed that the ER serves as a subcellular platform for autophagy initiation, 46,47 indicating that ER stress and autophagy might work together to combat the infection and replication of M1 virus.
Bystander killing
Viral infection causes the release of viral pathogen-associated molecular patterns (PAMP) and cellular danger-associated molecular patterns (DAMP), which activate the host innate immune system to secrete cytokines, such as TNF-α, TRAIL, IFN-γ, and IL-12, as well as dozens of other cytokines and chemokines. 48 –50 Some of these cytokines might directly destroy neighboring cancer cells. Upon M1 virus treatment, a large proportion of dying cancer cells labeled by caspase 3/7 are M1-uninfected, suggesting that M1 virus elicits a bystander killing effect, which is characterized by cell death in neighboring uninfected cells. 51 However, the biological significance of bystander killing induced by M1 virus might be more related to the cancer cell host than the virus. Death of both virus-infected and virus-uninfected cells forms a barrier that can help the host limit the infection. In contrast, the cytokines and chemokines induced by oncolytic viruses might recruit and activate multiple immune cells to the TME, leading to the warm-up of the tumor and pushing the immune response toward an antitumor one. 5
Activation of the antitumor immune response
Recently, oncolytic virotherapy has been considered as a novel type of immunotherapy due to its capacities to activate both innate and adaptive antitumor immune responses. 52 The detailed mechanisms include promotion of immunogenic cell death, which leads to the release of DAMP and attraction of innate immune cells, particularly dendritic cells, modulation of the TME, recruitment of tumor infiltrating lymphocytes, priming of immune responses mediated by CD8+ T cells and innate immune cells, and vascular modifications (such as inhibition of tumor angiogenesis and neovascularization). 41 Activation of the antitumor immune response of M1 virus has been extensively investigated in the context of intravenous injection. 53 M1 induces immunogenic tumor cell death and subsequently resumes priming of antitumor T cells by dendritic cells. Intravenous injection of M1 disrupts immune tolerance in the privileged TME, reprogramming immune silent (cold) tumors into immune inflamed (hot) ones. The antitumor response of M1 is totally dependent on CD8+ T cells. Moreover, intravenous injection of M1 virus establishes long-term antitumor immune memory in poorly immunogenic tumor models, especially in an orthotopic glioma model. 53 M1 virotherapy represents a versatile immunotherapy due to its therapeutic activity by combining direct in situ tumor killing with the ability to immunologically warm up tumors and provoke a powerful and long-lasting antitumor immune response.
Among the three tumor killing mechanisms of M1 virus, activation of both local and systemic antitumor immune responses represents the ultimate strategy to eliminate cancer, direct and bystander killing provides the biomaterial basis such as tumor antigens to prime the immune response. Investigation into the molecular details of the M1-induced antitumor immune response may provide the foundation for rational-design combination immunotherapy regimens to eliminate cancer.
COMBINATION THERAPY WITH ONCOLYTIC VIRUS M1
The antitumor activity of M1 mono-virotherapy differs substantially among cancer cell lines, with some cancer cells showing a poor response to M1 virus. 16 Clearly, there are great opportunities to potentiate oncolytic efficiency by using combinations. Various combination regimens have been designed to potentiate the oncolytic effect of M1 virus in different cancer types, and some of the chemicals function through multiple mechanisms. The combination regimens are summarized in Table 1.
Summary of combination regimens of M1 virus
cAMP, cyclic adenosine monophosphate; Epac1, exchange protein directly activated by cAMP1; ER, endoplasmic reticulum; IFN, interferon; IRE1α, inositol-requiring kinase 1α; JNK, c-Jun N-terminal kinase; PKA, protein kinase A; RHOQ, ras homolog family member Q; SMC, second mitochondria-derived activator of caspases (Smac) mimetic compound; VCP, valosin-containing protein.
Chemicals enhancing direct lysis
As oncolytic virus M1 directly lyses tumor cells to induce apoptosis, chemicals that induce apoptosis might be an appropriate approach to potentiate the efficiency of M1 virus. Bcl-2 family members are the major regulators of the apoptotic pathway in the decision phase. In fact, Bcl-2-related antiapoptotic proteins (Bcl-2, Bcl-XL, and Bcl-w) are frequently expressed at high levels in various cancers. 54 Pan Bcl-2 antiapoptotic family inhibitors ABT-263 and ABT-737 and the selective Bcl-XL inhibitor WEHI-539 sensitize cancer cells to M1 oncolysis but spare normal cells, whereas selective Bcl-2 inhibitors ABT-199, GX15-070, and HA14-1 display no synergistic effect with M1 virus. siRNAs specifically targeting Bcl-2, Bcl-XL, and Bcl-w confirm the selectivity of Bcl-XL in the combination treatment. 55 Moreover, the combination of M1 and Bcl-XL selective inhibitors exerts strong mitochondrial apoptosis-induced cell death but does not affect viral replication. 55
Another attractive approach to inducing apoptosis is the expression of second mitochondria-derived activator of caspases (Smac). Smac is an endogenous proapoptotic protein that upon releasing from mitochondria, binds to and antagonizes several members of the inhibitors of apoptosis (IAP) family (mainly X-IAP, c-IAP1, and c-IAP2), which regulate programmed cell death in tumor cells. Smac mimetic compounds (SMCs) have been rationally designed based on the properties of Smac. 56 By inhibiting c-IAP1 and c-IAP2 but not X-IAP, SMCs LCL161 and birinapant potentiate M1-induced apoptosis in tumor cells but not normal cells. Subsequent scanning electron microscope and protein assays have revealed irreversible ER stress and apoptosis through JNK signaling contribute to the enhanced oncolytic effect. 51
To identify molecular targets to improve the efficacy of M1 virus in hepatocellular carcinoma (HCC), a combinatorial drug screening of 350 anticancer small molecules that inhibit pathways involved in growth, metabolism, and apoptosis was performed. The valosin-containing protein (VCP, also known as P97) inhibitor Eeyarestatin I (EerI) is identified as the strongest sensitizer for M1 virus in HCC cells. 57 VCP is a ubiquitous, abundant, and essential adenosine triphosphatase that controls protein homeostasis by acting as a molecular segregase that extracts specific ubiquitin-modified client proteins from the ER and delivers them to the proteasome for degradation. 58 Inhibition of VCP induces ER stress and ER stress-associated apoptosis. 59 The VCP inhibitors NMS-873 and CB-5083 also sensitize HCC cells to M1 virus, and this combination treatment has been verified in subcutaneous or orthotopic HCC xenograft models in both immunodeficient and immunocompetent mouse models. 57 Further exploration indicated that inhibition of VCP does not enhance viral production or promote M1-induced bystander killing. The IRE1α-XBP1 pathway (part of the UPR) and subsequent JNK- and caspase-12-mediated ER stress-induced apoptosis are key to the enhanced efficacy. 57 The toxicity of intravenously injected M1 virus and VCP inhibitor EerI was evaluated in the nonhuman primate M. fascicularis. Hematology and serum chemistry support the conclusion that EerI combined with M1 virus is safe for nonhuman primates, thus providing important safety evidence for the clinical use of VCPI/M1 combination therapy in the future. 57
A comment about this research was published in Nature Reviews Gastroenterology and Hepatology, 60 in which the author highlights that “as preclinical evidence of the efficacy of oncolytic virus therapies mounts, the optimization of the therapy through the application of myriad combination strategies to synergize with viral oncolysis is coming to the forefront as a rational means to bring these therapies closer to the clinic. The study of Zhang et al. is, therefore, extremely timely and clinically relevant.” 57 In addition, high VCP expression is associated with increased sensitivity to the combination treatment, 57 suggesting that VCP expression might be a useful biomarker to identify subsets of HCC that are sensitive or resistant to this combination treatment.
DNA-PK is a serine/threonine protein kinase complex composed of a Ku heterodimer (Ku70/Ku80) and a catalytic subunit (DNA-PKcs) and plays important roles in DNA damage repair. As DNA-PK was recently identified as a candidate driver of tumor progression, it has also become a potential anticancer target. 61 Similar to HSV and KSHV, infection of M1 virus causes DNA damage that is associated with the phosphorylation of H2AX (p-H2AX), 62 a sensitive marker of DNA double-strand breaks (DSBs). 63 Most DSBs in human cells are repaired by nonhomologous end-joining (NHEJ), which directly ligates broken DNA ends through the critical regulator DNA-PK. Inhibition of DNA-PK by chemicals hinders DSB repair in the context of M1 infection, leading to DNA damage response-mediated cell death. 62 In addition, the combination of DNA-PK inhibitors and M1 virus induces irresolvable ER stress. 62
Cyclic adenosine monophosphate (cAMP) signaling is integral to a variety of cellular activities, including immunosuppression, apoptosis, generation of reactive oxygen intermediates, and phagocytosis. In eukaryotic cells, protein kinase A (PKA) is the primary effector of cAMP, controlling many cellular mechanisms such as gene transcription, ion transport, and protein phosphorylation. 64 In addition, the guanine nucleotide exchange factor exchange protein directly activated by cAMP1 (Epac1) has also been identified as a sensor for cAMP. 65 Activation of cAMP signaling by small molecules, such as db-cAMP (an analog of cAMP), forskolin (a ubiquitous activator of eukaryotic adenylyl cyclase that increases intracellular cAMP levels), and 8-CPT-cAMP (another analog of cAMP), enhanced M1 virus oncolysis by prolonging and increasing ER stress. Furthermore, Epac1 but not PKA is necessary for cAMP-enhanced oncolysis. 66 The classical PKA inhibitor H89 was also found to synergize with oncolytic virus M1 in various cancers through ER stress-mediated apoptosis. Interestingly, H89 was verified to enhance the oncolytic effect of M1 virus by activating Epac1. 67 Both studies illustrate the necessity of activating Epac1 to potentiate the oncolytic effect of M1 virus.
Chemicals inhibiting autophagy
In glioma, M1 induces autophagy in host cells, which might counteract replication and viral protein expression in host cells. In detail, IRE1α mediates M1 viral protein degradation by autophagy induction, 25 which suggests that autophagic inhibitors can enhance the oncolytic effects of M1 virus. Inhibition of IRE1α by STF083010 increases the viral protein load and subsequent oncolytic effect of M1 virus by abrogating M1 virus infection-induced autophagy. 25 The increase in viral load is not due to suppression of the innate antiviral response because IRE1α inhibition does not compromise the antiviral immunity induced by the virus. 25 Consistently, classical autophagy inhibitors, such as rapamycin and chloroquine, have been reported to increase the potency of several oncolytic viruses, such as HSV, 68 vaccinia virus 69 and Newcastle disease (NDV). 70 In recent years, autophagy has been found to promote the immune evasion of pancreatic cancer by targeting MHC-1 for lysosomal degradation. 71 Moreover, inhibition of autophagy either with chloroquine or chemical inhibitors of vacuolar protein sorting 34 (Vps34) reprograms cold into hot tumors. 71,72 The tight connection between autophagy and antitumor immunity suggests a rationale for the combination of autophagic inhibitors and oncolytic viruses.
Chemicals potentiating the bystander killing effect
Various studies have reported that by inhibiting the function of IAPs, SMCs sensitize tumor cells to the bystander killing effect induced by cytokines, such as TNF-α, 73 TRAIL, 73 and IL-1β. 74 SMCs might also potentiate the bystander killing effect mediated by virus-induced cytokines. In hepatic and colorectal carcinoma, SMCs LCL161 and birinapant greatly enhance the bystander killing triggered by M1 virus. When combined, only 30% of the dead cells were infected by M1 virus; 70% of the dead cells were uninfected by M1 virus and bystander-killed. 51 Transcriptomics revealed that the cytokines IL-8, IL-1A, and TRAIL are elicited by M1-infected cancer cells and co-operate with SMCs to induce a stronger bystander killing effect. 51
Chemicals increasing M1 viral replication
In addition to enhance ER stress-mediated direct cell oncolysis, some of the aforementioned chemicals function by promoting M1 virus replication in tumor cells, namely, SMCs, DNA-PK inhibitors, cAMP signaling activator inhibitors, and the classical PKA inhibitor H89. 51,62,66,67 Except for SMCs, with an undetermined mechanism by which M1 viral replication is increased, the other three types of chemicals enhance M1 viral replication by inhibiting the innate antiviral immune response. The key antiviral factors involved differ substantially; for example, IRF9, IFNL1, OASL, IFIT1, IFIT2, and IFIT3 are the key antiviral factors inhibited by DNA-PK inhibitors, IFNA, IFNB, IRF3, IRF7, MDA5, and IFIT1 are the key antiviral factors inhibited by cAMP signaling activators, and NF-κB is involved in H89-mediated type I IFN response inhibition.
Innate immune pathways might drive the adaptive immune response toward tumors, 75 mounting evidence highlights the importance of the cGAS-STING pathway in dictating antitumor immunity through potentiating type I IFN production, 76 so inhibition of the innate antiviral immune response, especially type I IFN, might result in poor outcomes in immunocompetent systems, despite enhanced M1 virus replication. Nonetheless, more studies are needed to balance the role of the innate antiviral response in antiviral effects and priming antitumor immune responses.
RHOQ has been identified as a biomarker of oncolytic virus M1, 26 whereby the cholesterol biosynthesis pathway was downregulated by M1 virus and the mevalonate/protein farnesylation/RHOQ axis was shown to have an antiviral role against it. By abrogating the antiviral response, the farnesyl transferase inhibitor tipifarnib promotes replication of M1 virus and potentiates its oncolytic efficacy in vitro and in vivo. 26 Regarding the antitumor immune response, high concentrations of cholesterol in the TME function as an immunosuppressive factor that dampens the antitumor efficacy of T lymphocytes by upregulating levels of immune checkpoints such as PD-1, 2B4, TIM3, and LAG3. 77,78 The ability of M1 virus to downregulate the cholesterol biosynthesis pathway demonstrates the potential of M1 virus to strengthen antitumor immunity by relieving the immune inhibition of cholesterol.
Combinations with immune checkpoint inhibitors
Cancer immunotherapy with immune checkpoint blockade (ICB) constitutes a breakthrough strategy for the past decade. 79 –82 However, despite great efforts, the majority of patients fail to respond to this novel therapeutic regimen. 83,84 Effective cancer immunotherapy depends on the status of the immune response in the TME. In general, the benefits of ICB are greatest in tumors with a high level of tumor-infiltrating lymphocytes, a high mutational burden and increased PD-L1 expression, which are referred to as immunologically “hot.” In addition, a lack of therapeutic benefit has been observed in immunologically “cold” tumors. These facts focused research on next-generation therapeutic solutions to ICB resistance with the aim of creating immunologically “hot” tumors. 41 Oncolytic viruses are, therefore, ideal candidates for combination with ICBs. For oncolytic virus M1, in addition to immunologically warming up tumors, expression of PD-L1 in the TME is upregulated. 53 The efficacy of M1 virotherapy in melanoma and prostatic cancer can be augmented by PD-L1 antibodies and overcome PD-L1 blockage resistance. 53 Furthermore, the ability of M1 to induce antitumor immunity may also synergize with other antitumor immunotherapies, which is being investigated in ongoing researches.
The combination of M1 with synergistic compounds to elicit intensive antitumor immunity represents a promising strategy to fight cancer, as the direct lysis, bystander killing, and autophagy induced by M1 virus can produce the antigens needed to stimulate the antitumor immune system. Among all synergistic drugs, SMCs might have the highest potential to enhance the oncolytic effect of M1 virus by various mechanisms; in addition to promoting both direct lysis and bystander killing of M1 virus, SMCs are also reported to enhance CD8+ T cell-mediated antitumor response of other oncolytic viruses, such as VSV. 85 Importantly, all combination regimens need further validation in the clinical research.
THE NONCLINICAL PHARMACOKINETICS OF ONCOLYTIC VIRUS M1
Most oncolytic viruses are developed for intratumoral administration, of which adenoviruses, herpesviruses, and vaccinia virus have been the most widely evaluated DNA virus vectors. A small number of clinical trials have focused on evaluating the safety and efficacy of traditional virus vectors administered intravenously, but few have been successful. For M1 virus, toxicokinetic (TK), biodistribution (BD), and pharmacodynamic (PD) after intravenous injection were studied. 86 The TK pattern of M1 virus in the blood was evaluated using healthy SD rats and cynomolgus monkeys. After the first injection, the concentration of the virus in the blood reached a peak at 2 min after intravenous injection in both animal models and then rapidly decreased. After 24 h, the virus levels in the blood decreased more than 104 times, but was still detectable. By day 44, the virus was distributed throughout different tissues, especially in the lymph nodes and spleen. The amounts of virus significantly decreased to undetectable levels in most tissues by day 72, but small amounts of M1 virus were still detectable in the lymph nodes and spleen. Further experiments verified that the viruses in the immune tissues were unreplicative, indicating nonspecific uptake and subsequent clearance by these organs.
In tumor-bearing immunocompromised mice, M1 virus was rapidly distributed to all tissues by 2 h after intravenous injection and then eliminated from normal tissues 28 days postinjection. However, this pattern was very different in tumor tissues, where the virus rapidly accumulated, peaked at 48 h postinjection, and then slowly decreased. Viruses in tumor tissue were viable, and importantly, intravenously injected M1 virus is capable of infiltrating the blood–brain barrier and replicating in malignant glioma. These findings emphasize the tumor selectivity and safety of M1 virus, supporting further clinical development potential. 86 Regardless, taking the immune system into account, experiments in immunocompetent tumor-bearing models need to be conducted to further reveal the pharmacokinetics of oncolytic virus M1.
Conclusions and Perspectives
Mounting evidences in recent years have identified M1 virus as a promising oncolytic virus with several advantages, such as various predictive biomarkers, clear mechanism of action, multiple combination regimens, capable of intravenous injection, and the development of lyophilized powder. To transform these nonclinical data into clinical practice, the first human study is eagerly awaited. Nonetheless, further investigations are required to achieve the maximum clinical benefit of oncolytic virus M1. For biomarkers, a rational biomarker prescreen strategy is needed to efficiently screen potential patients. Whether neutralizing antibodies impact the efficacy of M1 virus will drive development of the optimal administration schedule. Oncolytic virus essentially constitutes an immunotherapy that can affect almost all aspects of the cancer-immunity cycle. However, the communication between M1 virus with each component in the TME requires further study to devise more tailored combination approaches that restore effective host antitumor immunity. Based on the various combination strategies with other anticancer drugs, the development of genetically modified M1 viruses armed with specific gene fragments that express gene products along with M1 viral replication is currently under investigation. This approach may potentiate the efficacy of M1 virus and eliminate the toxicity of anticancer drugs administered systematically.
Footnotes
Author Disclosure
The authors have no conflicts of interest to declare.
Funding Information
This study was supported by the National Major Scientific and Technological Special Project for Significant New Drugs Development (No. 2018ZXO9733002), the Pioneering talents project of Guangzhou Development Zone, Guangdong Province (CY-2018-012) and the leading team for entrepreneurship in Guangzhou, Guangdong Province (No. 201809020004).