
Abstract
Alphavirus M1 is a promising oncolytic virus for cancer therapy. Here, we constructed a fluorescent reporter virus for real-time visualization and quantification of M1 virus both in vitro and in vivo. The reporter-encoding M1 virus maintained the characteristics of parental virus in the aspects of structure, replication capacity, the feature to induce cytopathic cell death, and the property of tumor targeting. The fluorescence is positively correlated with virus replication both in vitro and in vivo. More importantly, the reporter can be stably expressed for at least 10 generations in a serial passage assay. In summary, we successfully constructed stable and authentic reporter viruses for studying M1 virus and provided a feasible technical route for gene modification of oncolytic virus M1.
INTRODUCTION
Oncolytic virus, which selectively kills cancer cells and stimulates the “cold tumor” into “hot tumor,” is emerging as a potential immunotherapy for cancer treatment. The first step of oncolytic virotherapy requires the virus to replicate in the tumor, then induces tumor cell death and release of tumor-antigen, which, in turn, stimulates the activation of dendritic cell and induces antitumor immunity. 1,2 During this process, the specific replication of oncolytic virus in tumor tissue rather than normal tissue is one of the most critical properties of an oncolytic virus to guarantee its safety. Therefore, tracking virus replication in vivo is extremely important for preclinical and clinical development of an oncolytic virus agent.
Alphavirus M1 is a natural single-stranded positive RNA virus, and it was first isolated from culicine in Hainan province of China. 3 According to the Togaviridae report of International Committee on Taxonomy of Viruses (ICTV), Alphavirus M1 belonged to the Semliki Forest complex among alphaviruses, and it was most closely related to getach virus and Sagiyama virus. The genome of M1 virus encodes four nonstructural proteins nsP1-nsP4, and five structural proteins, CP, E3, E2, 6K, and E1. 4 The nonstructural proteins are first translated after virus infection to promote the translation of structural proteins and synthesis of viral RNA. Structural proteins are translated into polyprotein by sub-genomic RNA, and they are cleaved by protease into five functional proteins. 5,6 Alphavirus M1 is characterized as a naturally existing novel systemic oncolytic virus, which selectively replicates in and kills tumor cells by intravenous injection in vivo. 7 Further, the virus was reported to be safe in cynomolgus monkeys after multiple rounds of repeated intravenous injections, 8 and to date, no M1 virus-related human diseases have been reported. For further preclinical and clinical development of oncolytic virus M1, a more convenient tool for real-time visualization and quantification of M1 virus in vitro and in vivo is in urgent need.
Optical imaging is a useful noninvasive method for molecular imaging study of oncolytic virus, such as vaccinia virus. 9,10 By continuous observation in the same animal, in vivo imaging can reduce the deviation induced by individual difference and minimize the animal use. Green fluorescent protein (GFP) and near infrared fluorescent protein (iRFP) are the most common reporter genes used because of their shorter acquisition time and high spatial resolution. 11 Further, the optical window of iRFP is from 650 to 900 nm, which can circumvent the spectral overlap of GFP and tissue autofluorescence and enables its utilization in small animal imaging; however, the application of iRFP in deep tissue imaging is still questionable due to autofluorescence. Compared with luciferase, the fluorescent proteins can also be detected without substrate and with no cytotoxicity. 12,13 To date, luciferase have been inserted into many alphaviruses for convenient virus research; however, the use of fluorescent proteins was limited.
The rationales for inserting the reporter gene at specific loci should also be considered. First, the recombinant virus should mimic the parental virus infection. Sun had compared three insertion sites of alphaviruses to express the transgenes, including a second viral subgenomic promoter at the 3′ end of the viral coding sequences, fusion of the reporter protein in frame within non-structural protein 3 (nsP3), or insertion of the reporter as a cleavable element between the CP and PE2 structural proteins. Results indicate that the CP-PE2 insertion and nsP3 fusion viruses exhibit the most authentic mimicking of parental virus infection. 1 Second, the infection of the reporter virus should lead to high-level expression of reporter proteins for tracking the replication of virus, especially in vivo. In Sun's study, the expression levels of reporter proteins of CP-PE2 insertion virus or 3′ double subgenomic promoter virus are much higher than that of nsP3 fusion virus. 1 Taking into account the two aspects cited earlier, we determined to insert the reporter gene between CP and PE2.
Here, we report a method to construct the reporter M1 virus, M1-GFP and M1-iRFP, and compared the property and stability of the new virus with its parental M1 virus. We demonstrated that the virus distribution in vivo by the fluorescence method was consistent with qPCR data reported. In summary, we have successfully constructed stable and authentic reporter viruses for real-time visualization and quantification of oncolytic virus M1 both in vitro and in vivo. These findings also provide a practical strategy for gene modification of M1 virus.
RESULTS
M1-GFP retains the characteristics of M1 virus
We used a wild-type alphavirus M1 as the backbone of the reporter virus, M1-GFP. Gene sequences encoding fluorescent protein as well as self-cleavage peptide P2A to cleave the fluorescent protein automatically were inserted between protein CP and PE2 of the M1 genome (Fig. 1A). In this way, the fluorescent protein can be simultaneously expressed with structural proteins of the M1 virus, thus representing the translational process of virus protein in the late stage of virus infection.
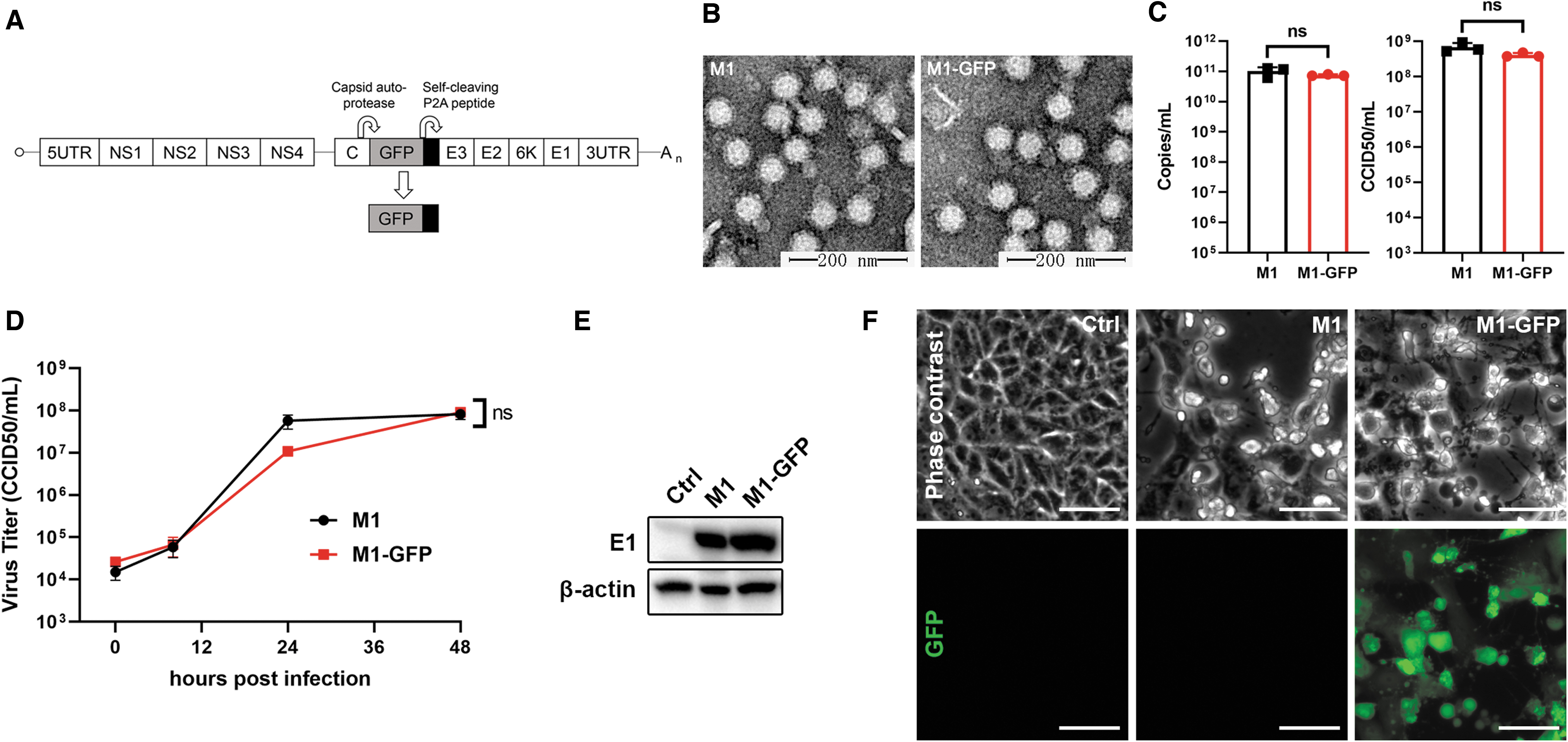
M1-GFP maintains the characteristics of alphavirus M1.
To confirm whether M1-GFP retains the characteristics of the wild-type M1 virus, we compared the two viruses in several aspects, including virus structure, replication curve, the expression of structural protein, and the cytopathic effect. Transmission electron microscope photos showed that particles of both viruses were enveloped and spherical and consist of a high electron density core and a halo-like structure surrounding (Fig. 1B). To compare the biological properties of M1-GFP with the parental M1 virus reliably, the virus stocks were purified by ultracentrifugation and then quantified via both qPCR method and CCID50 method. We found that both the physical and biological titers of M1 and M1-GFP were almost the same (Fig. 1C). Next, we demonstrated that the capacities of virus replication were the same, for the replication curve and the expression of virus envelope protein E1 in the infected Vero cells were similar for both viruses (Fig. 1D, E). We further compared the cytopathic effect (CPE) induced by both viruses, which is a critical feature of the oncolytic capacity of the virus. Phase-contrast photos showed that both viruses induced similar CPE in Vero cells, and fluorescent imaging confirmed the expression of GFP after M1-GFP infection (Fig. 1F). Our data proved that the insertion of GFP in the M1 virus maintained the virus structure, replication, and the CPE capacity of alphavirus M1.
M1-GFP is stable after 10 generations of serial passage
To further examine the stability of the M1-GFP, we repeatedly passaged the virus with an multiplicity of infection (MOI) of 0.1 for 10 generations in Vero cells. The culture supernatant collected from the M1-GFP RNA-transfected Vero cells was defined as P0, and the supernatant from each generation was defined as P1-P10. On passaging, we found that the virus-induced CPE was similar between P0 and P10 when the GPF protein was largely expressed (Fig. 2A). We further examined the locus where GFP was inserted in the virus genome by PCR and found that the transgene was stably inserted in the virus genome during the serial passages (Fig. 2B).

M1-GFP is stable after 10 generations of serial passage.
Visualization of tumor-specific replication of oncolytic virus M1 in vitro and in vivo by fluorescent imaging
Tumor selectivity is one of the most important properties of oncolytic virus M1, which has been previously illustrated. Here, we utilized GFP fluorescent imaging to directly visualize the selective replication of the M1 virus in cancer cells rather than normal cells. The phase-contrast photos and cell viability data showed that M1-GFP induced significant cell death in liver cancer cell line Hep3B and colorectal cancer cell line SW620, and no obvious cell death was observed in the respective normal cell line L02 and NCM460. Consistently, we found that the GFP was abundantly expressed in the sensitive cell lines, but not in the normal cell lines (Fig. 3A, B). Since GFP has a high background noise, and the short excitation wavelength limits its use in deep organ in vivo imaging, we choose iRFP702, 14 a near-iRFP, for in vivo visualization of the M1 virus. After intravenous injection, the fluorescence of iRFP was only observed in tumor tissues, and no signal was found in other normal tissues (Fig. 3C), which was consistent with our previous report. 7 Our data further proved the cancer selectivity of M1 virus by the fluorescence method both in vitro and in vivo.
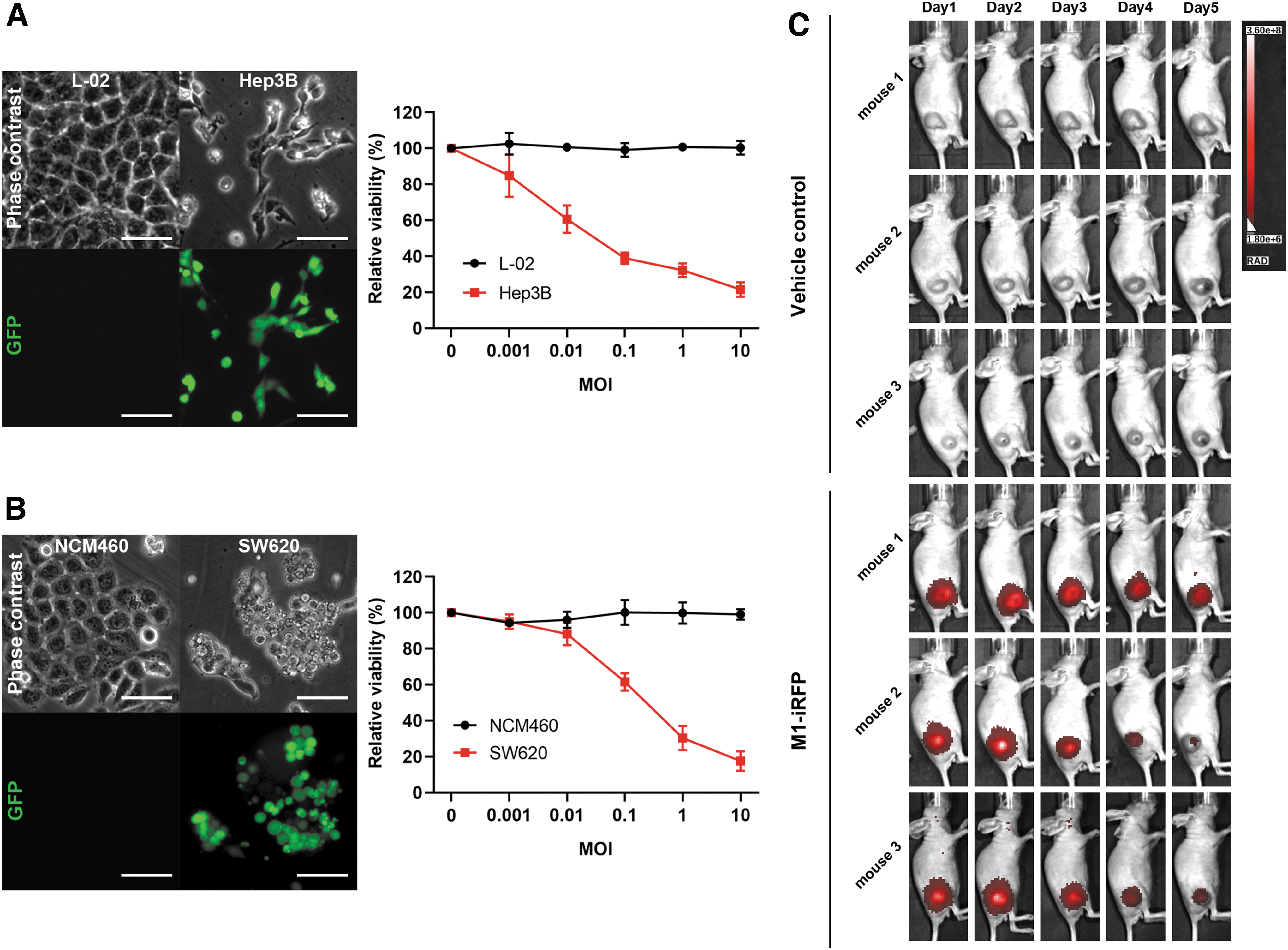
M1-GFP selectively replicates in tumor cells both in vitro and in vivo.
Quantification of M1 virus in vitro and in vivo by fluorescence
We further explored whether the reporter genes can precisely represent the replication of the M1 virus both in vitro and in vivo. Our data showed that the intensity of GFP was positively correlated to the virus titer after virus infection for 24 h, illustrating that GFP fluorescence can be used as an indicator of virus replication (Fig. 4A). By utilizing this convenient tool, we closely monitored the replication curve of the M1 virus under five different infection MOI in Vero cells. We found that high infection of MOI resulted in fast virus growth, whereas low infection of MOI led to slow but a high level of viral replication (Fig. 4B). In our previous study, we demonstrated that dibutyryl-cAMP (dbcAMP) significantly promoted the replication of M1 virus by directly detecting virus titer, 15 and here we further validated that dbcAMP treatment obviously elevated the ratio of GFP-positive cells, suggesting that dbcAMP increased M1 virus replication by promoting viral spread (Fig. 4C).
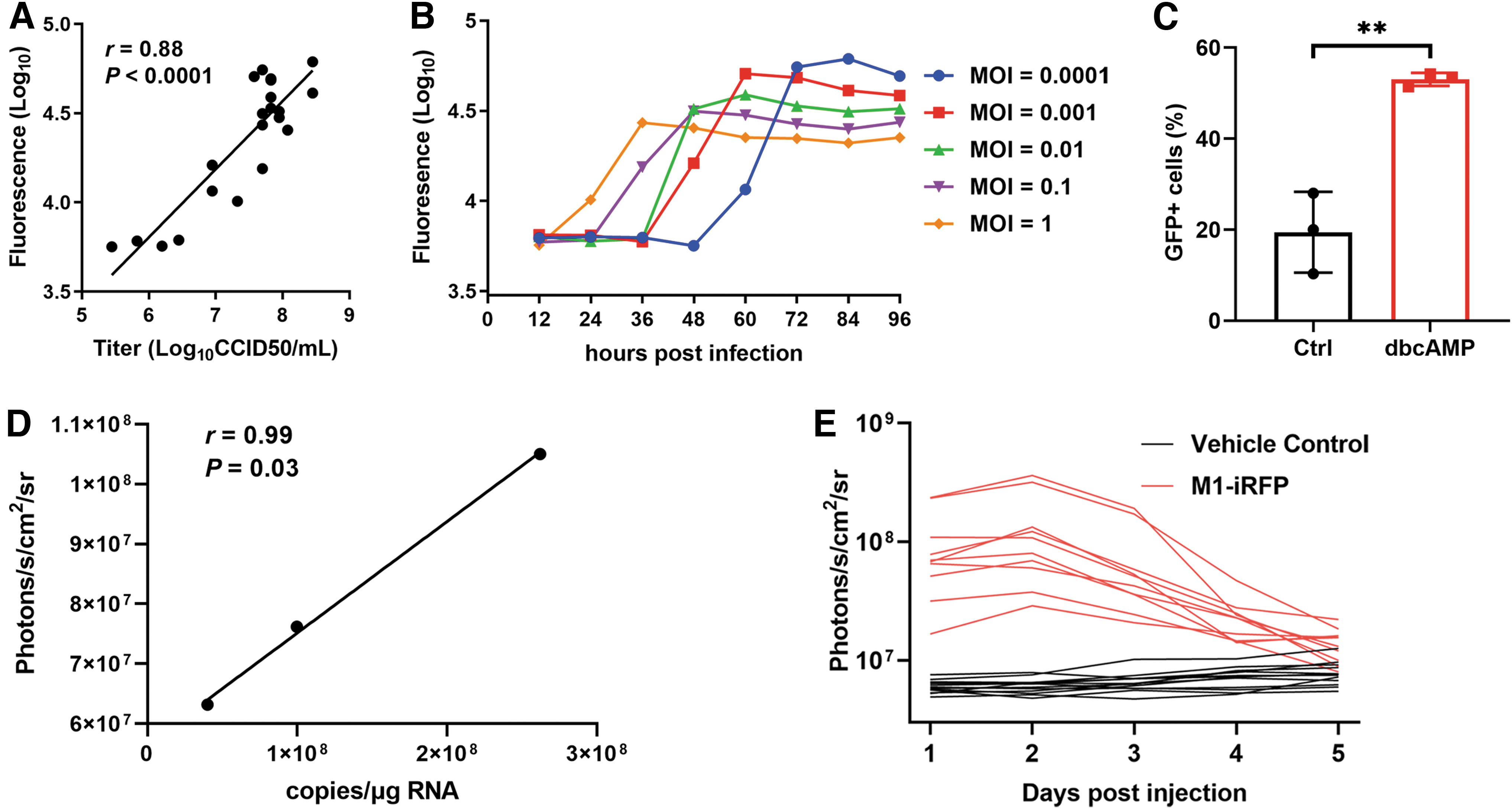
Quantification of M1 virus both in vitro and in vivo by fluorescence.
Precise quantification of virus in tumor-bearing animal model is important for the preclinical pharmacokinetic study of an oncolytic virus. Although qPCR detection of viral nucleic acid is the gold standard for quantification, it is labor-consuming and requires a lot of animals when data of many time points are needed. In vivo imaging, on the other hand, is much more convenient and can function as a complementary method to qPCR. We hereby compared the qPCR data and iRFP photons in the Hep3B subcutaneous tumor model with one single intravenous injection of M1 or M1-iRFP virus, respectively, and found that iRFP photons were significantly and positively correlated with qPCR data, suggesting that iRFP fluorescence can precisely quantify the replication of the M1 virus in vivo (Fig. 4D). We next monitored the replication of the M1 virus within tumor tissue for five consecutive days by in vivo imaging. The viral replication reached maximum at 48 h post-injection and lasted for about 5 days (Fig. 4E).
These data proved that replication of the oncolytic virus M1 can be quantified by using fluorescent reporter viruses both in vitro and in vivo.
DISCUSSION
Our data provided a feasible method to construct a stable reporter virus based on the fluorescent protein-P2A auto-cleaving system. M1-GFP was similar to the M1 virus in the structure, replication capacity, and the feature to induce CPE, and the expression of GFP precisely represented the replication of the M1 virus. M1-iRFP resembled the characteristics of cancer selectivity of M1 virus both in vitro and in vivo with a convenient continuous measurement fluorescence assay.
Reporter virus is a common method to study the alphavirus infection. Molecularly, fluorescent proteins are exploited to study the protein interaction between virus and host. Sindbis virus was constructed to tag nonstructural proteins with GFP to study the localization and interacting proteins. 16 For the most common use, GFP was inserted into the virus genome to monitor the replication of the virus both in vitro and in vivo. Chikungunya virus (CHIKV) was also constructed to insert the iRFP gene into nsP4 to study the virus-induced toxicity. However, the virus titer of CHIKV-iRFP was 10 times less than the wild-type CHIKV, and its genome was stable for less than 5 generations. 17 Compared with CHIKV-iRFP, M1-GFP possessed an inserted GFP gene between capsid protein and PE2 and showed better virus production capacity and stability. One possible cause of the difference in virus replication between CHIKV-iRFP and M1-GFP is the location of fluorescent protein gene, for previous research also demonstrated that Capsid-PE2 insertion and nsP3 fusion of transgene mostly mimicked the parental virus infection. 18
The utility of the reporter virus for the nonclinical research of oncolytic virus can highly boost the efficiency of virus biodistribution detection. In vivo imaging makes it possible to monitor the replication of virus in real time, which tremendously reduces the operational procedures of traditional qPCR methods and improves the reproducibility of the biodistribution data. The reporter virus based on vaccinia virus GLV-1h68 had already been exploited for the study of biodistribution, tumor therapy, and synergistic interaction with another virus. 19,20 However, one of the most important obstacles for the widely use of reporter virus in nonclinical research is the validation of the method. QPCR method had been extensively validated by many oncolytic viruses, so it is broadly used in Good Laboratory Practice labs to detect virus distribution. Nevertheless, some validation research of the reporter virus has been reported. GLV-1h68-expressing GFP was used to study the oncolytic effect of lymphatic metastasis, and GFP was successfully demonstrated to specifically and sensitively detect the virus in the lymph nodes. 21 However, systematic method validation is still needed to further support the nonclinical use of reporter viruses based on the oncolytic virus M1.
Alphavirus M1 is a natural oncolytic virus, and we have discovered many combination strategies to enhance the oncolytic effect of M1. Here, we further identified a feasible site in M1 for directly enhancing the antitumor effect by inserting genes into the backbone of the virus. Oncolytic viruses take advantage of gene therapy to construct antitumor genes in their genome, whereas the viruses selectively replicate in the tumor tissue; the inserted genes may be simultaneously expressed to boost the antitumor effect. 22 Alphavirus replicates fast in sensitive cells, which is good for the expression of transgene. Moreover, the alphavirus-transducing systems are important and mature tools for expressing genes of interest, and sindbis virus had been modified by this method successfully. 23 More importantly, M1-GFP has provided a successful example for the further genetic modification of the oncolytic virus M1.
In summary, we have successfully constructed a stable and authentic reporter virus for real-time visualization and quantification of the oncolytic virus M1 and provided a feasible strategy to arm the M1 virus with therapeutic genes.
MATERIALS AND METHODS
Cell culture and viruses
All the cell lines were purchased from the American Type Culture Collection (MD) and cultured in Dulbecco's modified Eagle's medium (DMEM) medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and 1% penicillin/streptomycin (Thermo Fisher Scientific). The M1 virus, M1-GFP, and M1-iRFP were cultured in Vero cells, with DMEM medium supplemented with 10% fetal bovine serum. The M1 virus titer was determined by the CCID50 assay.
Construction of M1-GFP and M1-iRFP
The infectious clone of pBR322-M1 plasmid was used as the backbone to construct M1-GFP and M1-iRFP plasmid. The cDNA sequences of GFP or iRFP702 were chemically synthesized (GENEWIZ, Suzhou, China) and introduced into the backbone by restriction digestion and ligation. Briefly, the GFP or iRFP702 reporter genes were inserted into the site between capsid (C) and PE2 genes in the full-length M1 genome. All the constructs were verified by DNA sequencing. The plasmids of WT M1, M1-GFP, and M1-iRFP reporter viruses were linearized by XbaI followed by in vitro transcription using an SP6 RiboMAX Large-Scale RNA Production System (Promega) according to the manufacturer's protocols. The transcribed RNAs were transfected into Vero by Lipofectamine MessengerMAX Transfection Reagent (Thermofisher). When ∼80% of Vero cells showed CPE, the supernatants were collected and aliquoted at −80°C.
Transmission electron microscope
Vero cells were infected with M1 or M1-GFP (0.01 CCID50/cell) for 36 h. Supernatants were collected by centrifugation at 1,000 g for 10 min, and they were further centrifuged at 32,000 g for 1 h at 4°C. Samples were then transferred to the Zhongshan School of Medicine (Sun Yat-sen University) Electron Microscopy Facility for standard transmission electron microscopy ultrastructural analysis.
CCID50 assay
The samples were diluted with MEM and added to BHK-21 cells 24 h after inoculation. The CPE of the M1 virus was recorded 72 h after infection, and the viral titer was calculated with Spearman-Karber assay.
PCR assay
RNA was extracted from each frozen sample by using TRIzol (Life Technologies), and reverse transcription was performed by using oligo(dt) and GoScript reverse transcription system (Promega) according to the manufacturer's instructions. PCR was performed with 2 × Taq PCR Mastermix (TIANGEN), and the PCR sample was detected by agarose gel electrophoresis. Amplification primers (Thermo Fisher) were as follows:
F: AGGTTCACAATCCCGACAGGC
R: CATCGGAAGCCTCATCCCTAAT
Western blotting
Vero cells were infected with M1 or M1-GFP (0.01 CCID50/cell) for 24 h; cell samples were collected and prepared by using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) and then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The membranes were inoculated with antibody to E1 (Beijing Protein Innovation company, China) and β-actin (AP0063; Bioworld). The proteins in the membrane were visualized by Immobilon Western Chemiluminescent HRP Substrate (Millipore, Darmstadt, Germany) with a ChemiDoc XRS+ System (Bio-Rad, Hercules, CA).
Cell viability assay
Cells were seeded in 96-well plates and infected with the M1 virus with different MOI for 72 h. Then, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added with the concentration of 1 mg/mL and incubated at 37°C for 3 h. The MTT precipitate was dissolved in 100 μL of dimethyl sulfoxide (DMSO). The optical absorbance was determined at 570 nm by a microplate reader (synergy H1, Gene Company, Hong Kong, China).
Virus distribution detection
This study was approved by the Ethics Committee of ZSSOM on Laboratory Animal Care, China. Nude mice were subcutaneously inoculated with Hep3B human hepatoma cells in the right limb, 10 mice for each group. When the tumors of Balb/c nude mice reached a volume of 100 mm3, M1-iRFP was intravenously injected via tail vein at the dosage of 2 × 106 CCID50, and the expression of iRFP was detected by the in vivo imaging system (IVIS) daily for 5 days. The M1 was intravenously injected via the tail vein at the dosage of 1 × 108 CCID50. Then, animals were sacrificed at 24, 48, and 72 h after administration, three mice for each time point, and tumor tissues were collected and detected by the RT-qPCR method.
RT-qPCR
RNA was extracted by the Eastep Super Total RNA Extraction Kit (QIAGEN) according to the manufacturer's instructions. The RNA was diluted to 0.25 μg/μL with distilled water for QPCR. FastKing One Step RT-qPCR Kit (TIANGEN) was used for reverse transcription and quantitative amplification according to the manufacturer's instructions. The concentration of M1 RNA in each sample is plotted as the copy number per microgram of RNA.
Statistical analysis
Statistical analysis was performed by using GraphPad 6 software. Comparisons between different groups were performed with analysis of variance (ANOVA). The correlation between fluorescence and virus RNA was performed with Pearson correlation. All error bars indicate the standard deviation. Differences were considered significant if the p-value was <0.05.
Footnotes
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Guangzhou people's Livelihood Science and technology tackling key project (No. 201803010113), the Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011564), the National Major Scientific and Technological Special Project for Significant New Drugs Development (No. 2018ZX09733002), the Leading team for entrepreneurship in Guangzhou, Guangdong Province (No. 201809020004), the Pioneering talents project of Guangzhou Development Zone, Guangdong Province (No. CY2018-012), and the Fundamental Research Funds for the Central Universities (No. 19ykpy166).