
Abstract
Epstein-Barr virus (EBV) infections in healthy individuals are usually cleared by immune cells, wherein CD8+ T lymphocytes play the most important role. However, in some immunocompromised individuals, EBV infections can lead to the development of cancer in B, T, natural killer (NK) cells and epithelial cells. Most EBV-associated cancers express a limited number of virus-specific antigens such as latent membrane proteins (LMP1 and LMP2) and nuclear proteins (EBNA1, -2, EBNA3A, -B, -C, and EBNA-LP). These antigens represent true tumor-specific antigens and can be considered useful targets for T cell receptor (TCR) gene therapy to treat EBV-associated diseases. We used a TCR isolation platform based on a single major histocompatibility complex class I (MHC I) K562 cell library for the detection, isolation, and re-expression of TCRs targeting immunodominant peptide MHC (pMHC). Mature dendritic cells (mDCs) were pulsed with in vitro-transcribed (ivt) RNA encoding for the selected antigen to stimulate autologous T cells. The procedure allowed the mDCs to select an immunogenic epitope of the antigen for processing and presentation on the cell surface in combination with the most suitable MHC I molecule. We isolated eight EBV-specific TCRs. They recognize various pMHCs of EBV antigens LMP1, LMP2A, and EBNA3C, some of them described previously and some newly identified in this study. The TCR genes were molecularly cloned into retroviral vectors and the resultant TCR-engineered T cells secreted interferon-γ after antigen contact and were able to lyse tumor cells. The EBV-specific TCRs can be used as a basis for the generation of a TCR library, which provides a valuable source of TCRs for the production of EBV-specific T cells to treat EBV-associated diseases in patients with different MHC I types.
Introduction
Adoptive T cell therapy (ATT) using T cell receptor (TCR) gene-engineered T cells (TCR gene therapy) has moved from an experimental approach to clinical application. However, TCR gene therapy has shown success only in a limited number of cancer patients. This is because of the fact that most clinical trials of TCR gene therapy target tumor-associated antigens (TAAs), which are self-antigens and therefore are also expressed on normal cells. Targeting of such antigens accepts the lack of cancer specificity and is often accompanied by severe side effects. 1 Such on-target/off-tumor side effects can be prevented by targeting tumor-specific antigens (TSAs; mutated antigens, and neoantigens). 2 Mutated antigens can be considered as genuinely tumor specific and their use as targets should substantially improve the benefit-to-risk ratio of ATT.
At present, it is not entirely known how many of these mutations encode truly tumor-specific epitopes. Data indicate that this number can vary between ∼50 mutations in most adult solid tumors and 1,000 mutations in cancers with genomic instability. 3 The majority of these mutations are individuum specific, whereas some may occur repeatedly in different cancers. Taking these facts into account, TCR gene therapy that targets mutated antigens is rather challenging. For each presented mutation, the specific TCR genes have to be identified, isolated, and molecularly cloned into gene transfer vectors. TCR gene-engineered T cells have to be generated and thoroughly characterized before application in ATT.
In addition to mutated TSAs, viral antigens associated with cancer represent another group of TSAs, which are also not expressed on normal cells but are not individuum specific. Thereby, viral antigens represent ideal targets for TCR gene therapy.
Epstein-Barr virus (EBV), a human herpes virus, infects ∼90% of the human population. In healthy individuals, diseases caused by EBV are usually cleared by immune cells, wherein cytotoxic CD8+ T lymphocytes (CTLs) play the most important role. 4 EBV is associated with different nonmalignant diseases such as infectious mononucleosis or multiple sclerosis. 5,6
Most of the EBV-associated malignancies derive from B cells, whereas others are derived from T cells, natural killer (NK) cells, as well as nasopharyngeal and gastric epithelial cells. The B cell lymphomas include post-transplant lymphoproliferative disorders (PTLD), Hodgkin lymphoma (HL), Burkitt lymphoma, and diffuse large B cell lymphoma. In accordance with the above-mentioned types of infected cells, EBV contributes to the development of T/NK lymphomas/leukemias, nasopharyngeal carcinomas (NPC), and gastric carcinoma. 7 –11 Most of the EBV-associated cancers express only a limited number of EBV-specific antigens such as the two latent membrane proteins (LMP1 and LMP2) and the six nuclear proteins, EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP. 10,11 All these antigens represent TSAs and can be considered as attractive targets for TCR gene therapy of EBV-associated diseases, including cancer. Hence, several groups have isolated EBV antigen-specific TCRs and evaluated their potential to be utilized in TCR gene therapy. 12 –19 In most of these reports, T cells isolated from EBV+ donors were expanded by means of EBV-transformed B-lymphoblastoid cell lines (LCLs) or EBV peptide pools. However, in some situations, only peptide-loaded cells were used as targets to demonstrate the cytotoxic capability of TCR-engineered T cells instead of tumor cells where the TSA is naturally processed and presented on the cell surface. 12,13
Recently, we established a TCR isolation approach for the efficient detection, isolation, and re-expression of TCRs targeting immunologically relevant peptide-major histocompatibility complexes (pMHCs). 20 In this approach, dendritic cells (DCs), which are professional antigen-presenting cells, were pulsed with in vitro-transcribed (ivt) RNA encoding the full-length sequence of the selected antigen to stimulate autologous T cells. This procedure allows the DCs to freely select one epitope of the antigen for processing and presentation on the cell surface in conjunction with the most suitable MHC I complex. Thus, the TCR isolation procedure does not discriminate against epitopes presented by less frequent MHC molecules or depend on a wealth of pre-existing experimental data for a reliable epitope prediction.
In this study, we report on the isolation and characterization of eight TCRs, which recognize epitopes of EBV antigens LMP1, LMP2A, and EBNA3C presented by various MHC I molecules.
Materials and Methods
Cell culture
RPMI 6666 21 (Hodgkin's lymphoma-derived cells, ATCC CCL-113), K562 (chronic myelogenous leukemia cells, ATCC CCL-243), L591 22 (LCLs of a Hodgkin's lymphoma patient, kindly provided by Dr. U. Höpken, MDC, Berlin), EBV-transformed LCLs BM14, BSM, DEM, EK, HO104, KASO11, SA and WIN (courtesy of Prof. D. Schendel, Medigene AG, Martinsried), peripheral blood lymphocytes (PBLs), and CD8-enriched T cells were grown in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal calf serum (FCS; Biochrom), 1 mM sodium pyruvate, 1 × Minimum Essential Medium Eagle (MEM) nonessential amino acids solution, 1 × penicillin–streptomycin (10,000 U/mL) (all Gibco; lymphocyte medium [LM]) at 38°C and 6.5% CO2. HG820-GALV packaging cells 23 were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% heat-inactivated FCS and 1 × penicillin/streptomycin at 37°C and 5% CO2. An ethic committee approval to recruit voluntary blood donors was obtained from the ethics committee of the Charitè Universitätsmedizin Berlin, Germany (EA1/141/18).
Maturation of DCs
Autologous mature DCs (mDCs) were generated from plate-adherent monocytes as follows: 7.5 × 107 peripheral blood mononuclear cells were seeded into a 75 cm2 cell culture flask in 15 mL of DC medium [RPMI 1640 VLE (Biochrom) supplemented with 1.5% human serum (Biochrom)] and incubated for 30 min at 38°C and 6.5% CO2. The flasks were tapped two times, and the suspended cells were collected with the supernatant and used as source for T cell isolation. Plate-adherent cells were washed three times in phosphate-buffered saline and incubated in 15 mL DC medium supplemented with interleukin (IL)-4 (20 ng/mL) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (100 ng/mL) (both PeproTech) for 48 h. Then, 3 mL of DC medium supplemented with a DC maturation cocktail [GM-CSF (100 ng/mL), IL-4 (20 ng/mL), IL-1β (10 ng/mL), tumor necrosis factor alpha (10 ng/mL), interferon gamma (IFN-γ) (250 ng/mL), PGE2 (250 ng/mL), poly (I:C) (20 ng/mL), R848 (20 ng/mL) (all PeproTech)] was added for 24 h. The detached cells were harvested and used directly for staining with monoclonal antibodies (mAbs) specific for MHC class II molecules, CD80, CD86 (Cat. No.: 555558, 557227, and 555660; all BD Bioscience), and CD83 (BioLegend).
Generation of EBV antigen-expressing mDCs
EBV antigen-encoding genes were molecularly cloned into the expression plasmid pcDNA3.1(-) (Invitrogen) under the control of the T7 promoter. Plasmids were linearized by restriction enzyme digestion at the 3′-end of the transgene. Ivt RNA was generated using in vitro synthesis of capped RNA followed by poly-A tailing (Ambion) and electroporated into mDCs (GenePulser, 250 μF, 0.35 kV; Bio-Rad). Half of the resulting antigen-expressing mDCs were used directly and the other half was stored at −80°C for later restimulation.
T cell isolation and antigenic stimulation
T cells were isolated from the peripheral blood of healthy EBV+ donors with predetermined MHC I haplotypes after informed consent by Biocoll gradient centrifugation (Biochrom) 24 (Supplementary Table S1). CTLs were obtained by negative selection using the human CD8+ T Cell Isolation Kit (Miltenyi Biotec) and cocultured with mDCs expressing EBV full-length antigen LMP1, LMP2A, and EBNA3C, respectively, at an effector:target (E:T) cell ratio of 10:1 in a 24-well plate containing 2 mL/well of LM. Starting at 72 h after stimulation, CTLs were fed every second day with IL-2 (25 U/mL; Proleukin) and IL-7 (5 ng/mL; PromoCell) and split in a ratio of 1:2, if cell density was too high. At day 13, CTLs were restimulated with thawed mDC-presenting EBV antigens, and at day 18, CTLs were analyzed for responses to EBV antigens.
Identification of antigen-MHC I-specific T cell responses
To identify EBV antigen-specific T cell responses, a K562 single-MHC I cell library was used. 20 In brief, 48 h before the coculture, K562 cells stably expressing the MHC I alleles of the T cell donor as single gene were transiently transfected by electroporation with 10 μg of the pcDNA3.1(−) vector encoding for one of the full-length EBV antigens LMP1, LMP2A, and EBNA3C, respectively, in combination with 10 μg of the pcDNA3.1(−) vector encoding for CD80. After coculturing of CTLs with antigen-presenting K562 cells at an E:T ratio of 5:1 in 96-well plates in 200 μL/well LM for 20 h, two assays were applied for the identification of antigen-MHC I-specific T cells. First, IFN-γ release into the supernatant was determined using a human IFN-γ enzyme-linked immunosorbent assay (ELISA) kit according to the manufacture`s instruction (BD Biosciences). Second, the expression of the T cell activation marker CD137 was measured using a CD137-specific mAb (BioLegend). Only those T cells that responded in both assays were regarded as antigen MHC I responsive.
Isolation and identification of antigen-specific TCRs
Total RNA was isolated from the fraction of sorted CD8+/CD137+ EBV-specific T cells using the RNeasy Micro Kit (Qiagen) and transcribed into cDNA using the SMARTer RACE 5′/3′ kit and the 5′-CDS primer A included in the kit (Takara Bio). TCR-α variable (TRAV)- and TCR-β variable (TRBV)-specific chain amplification was performed using 5 μL of 5′-RACE cDNA reaction mix, 5 μL universal primer mix (long for a first PCR and short for a “nested” PCR) (all Takara Bio), 10 μL of 5 × Phusion GC buffer, 1 μL dNTP (dATP, dCTP, dGTP, dTTP, each 10 mM), 0.5 μL Phusion DNA polymerases (all Invitrogen, Thermo Fisher Scientific), and 0.5 μL of TCR RACE primers: TRAC-RACE, TRBC-RACE (each 50 pmol/μL) and/or “nested” PCR-specific primers: TRAC-RACE-deep sequencing, TRBC-RACE-deep sequencing (each 50 pmol/μL) (primer sequences will be provided on request), and up to 50 μL H2O. RACE-PCR conditions were provided in the SMARTer RACE 5′/3′ kit user manual.
TCR-α and TCR-β chains were isolated and sequenced as described. 20 TRAV and TRBV sequences of TCR chains, which were dominantly expressed in antigen-MHC I-specific T cells (above 10% of total reads) were combined with codon-optimized murine TRAC and TRBC regions. 25,26 The individual chimeric TCR-α and TCR-β chains were then molecularly cloned into the retroviral vector MP71. 27 Combined expression of the most abundant TCR-α and TCR-β chains was used for the identification of functional TCR heterodimers. For this, individual TCR-α and TCR-β chain vectors were used to co-transduce primary human T cells. After the coculture of TCR-engineered T cells with the respective antigen-MHC I-expressing K562 cells, the amount of IFN-γ release was measured by ELISA and used to judge the functionality of the TCR. Functional TCR-α and TCR-β chain pairs were then molecularly cloned using a P2A peptide linker in the configuration 5′-TCRβ-P2A-TCRα-3′ into the MP71 vector. 27 All transgene constructs were completely codon optimized (GeneArt). 25
Production of retroviruses and transduction of cells
γ-retrovirus particles were generated by transient calcium phosphate transfection of HG820-GALV packaging cells in 6-well plates with 18 μg of MP71 plasmid carrying the different transgenes. 27 Forty-eight hours after transfection, virus-containing supernatant was harvested, filtrated through a 0.45-μm pore size filter to remove cells and debris, and directly utilized for the transduction of K562, L591, and RPMI 6666 cells. PBLs and CTLs (1 × 106 per well and 1 mL) were transduced in 24-well nontissue culture plates, precoated with 12.5 μg/mL of RetroNectin (TaKaRa Bio) by spinoculation in RPMI 1640 GlutaMAX medium. 27
Flow cytometry
Surface expression of transgenic TCRs was analyzed utilizing a phycoerythrin (PE)- or allophycocyanin (APC)-labeled mAb (Biolegend) directed against the murine constant region of the TCR-β chain. For further T cell characterization, both PE- and APC-labeled anti-CD8 and anti-CD137 mAbs (clone: RPA-T8, 4B4-1, all BioLegend), were used. For the characterization of mDCs, the following mAbs were used: PE-Cy7-labeled anti-CD83 (BioLegend), PE-labeled anti-CD80, APC-labeled anti-CD86, and fluorescein-5-isothiocyanate labeled anti-MHC II (HLA-DR) (all BD Biosciences). Fluorescence intensity was measured using a MACS Quant flow cytometer (Miltenyi Biotec). Data were analyzed using FlowJo software (Tree Star).
IFN-γ release assay
TCR-engineered T cells and target cells were cocultured at an E:T ratio of 1:1. After 24 h at 38°C and 6.5% CO2, culture supernatants were harvested. Secretion of IFN-γ by CTLs and PBLs was measured using the OptEIA Human IFN-γ ELISA Kit (BD Biosciences) according to the manufacturer's instructions. The reagents of the kit were diluted 1:1,000 before use. Standard curves were generated using the lyophilized recombinant human IFN-γ (included in the kit) at a starting concentration of 4,000 or 8,000 pg/mL.
Peptide titration assay
For exogenous peptide pulsing, K562 cells carrying the respective MHC I complex, were loaded with titrating amounts of the corresponding peptide, ranging from 10−6 to 10−13 M. After 2 h of incubation at 38°C and 5% CO2, K562 cell were pelleted and resuspended in LM. K562 cells were cocultured with TCR-engineered T cells at an E:T ratio of 1:1. Untransduced and TCR-transduced T cells without stimulation were used as a negative control (MIN), whereas untransduced and TCR-transduced T cells unspecifically stimulated with PMA (Sigma, 5 ng/mL) and ionomycin (Calbiochem, 1 μm) for IFN-γ release served as a positive control (MAX). After 20 h of cultivation at 38°C and 5% CO2, IFN-γ release was determined using ELISA and flow cytometry. All peptides were synthesized by Custom Peptide Synthesis service (JPT Peptide Technologies), providing peptides with a purity >95%.
Cytotoxicity assay
L591 tumor cells expressing both the respective MHC I complex and EBV antigen were seeded into 96-well round bottom plates at a concentration of 5 × 105/mL. TCR-engineered T cells (CD8+/TCR+) or an equivalent number of untransduced T cells were added at indicated E:T ratios in triplicates and incubated at 38°C for 6 h. The number of surviving target cells was determined by flow cytometry using a MACS Quant flow cytometer. Target cells incubated without effector cells were used to measure spontaneous cell death. Triplicate wells were averaged, and the percentage of surviving cells was calculated in relation to the values obtained from the samples treated with untransduced T cells: % specific survival = 100 × (test value)/(average background).
Prediction of immunogenic epitope sequences recognized by EBV-specific TCRs
Sequences of immunogenic epitopes for EBV antigen-specific TCRs were determined using the NetMHCpan4.0 Server (
Results
Generation and identification of EBV-specific T cells
mDCs were prepared from plate-adherent monocytes and characterized by the expression of MHC class II molecules, CD80, CD83, and CD86 (Supplementary Fig. S1A). mDCs were transfected with ivt RNA encoding the full-length sequence of the selected EBV antigens LMP1, LMP2A, and EBNA3C, respectively, to ensure unbiased endogenous antigen processing and MHC I expression. To judge the transfection efficiency, a GFP-encoding ivt RNA was electroporated in parallel in all experiments in a separate sample of mDCs. Because GFP expression in all experiments was >80%, we concluded that the expression of EBV antigens was in a similar range as well (Supplementary Fig. S1B).
After stimulation of autologous CTLs of different EBV+ donors (Supplementary Table S1) for 14 days with antigen-presenting mDCs, an MHC cell library was used to identify antigen-specific T cell responses. To do so, K562 cell lines that expressed the six MHC I complexes of the donor, each as a single allele, were selected from the library. K562 cells were then transfected with the selected EBV antigen-encoding gene and cocultured with expanded EBV-specific T cells. Responding T cells were identified by upregulation of the T cell activation marker CD137 and by the release of IFN-γ, as given in Fig. 1A, B. Here, mDCs were simultaneously transfected with full-length genes encoding LMP1 and LMP2A. Upregulation of CD137 and release of IFN-γ indicated that EBV-specific antigens were presented to donor T cells by the MHC I molecules A*02:01 and B*57:01, respectively. Therefore, reactive CTLs were enriched by fluorescence-activated cell sorting (FACS), total RNA was isolated, and RACE-PCR amplification of TCR-α (TRAV)- and TCR-β (TRBV) chain-specific sequences was performed.
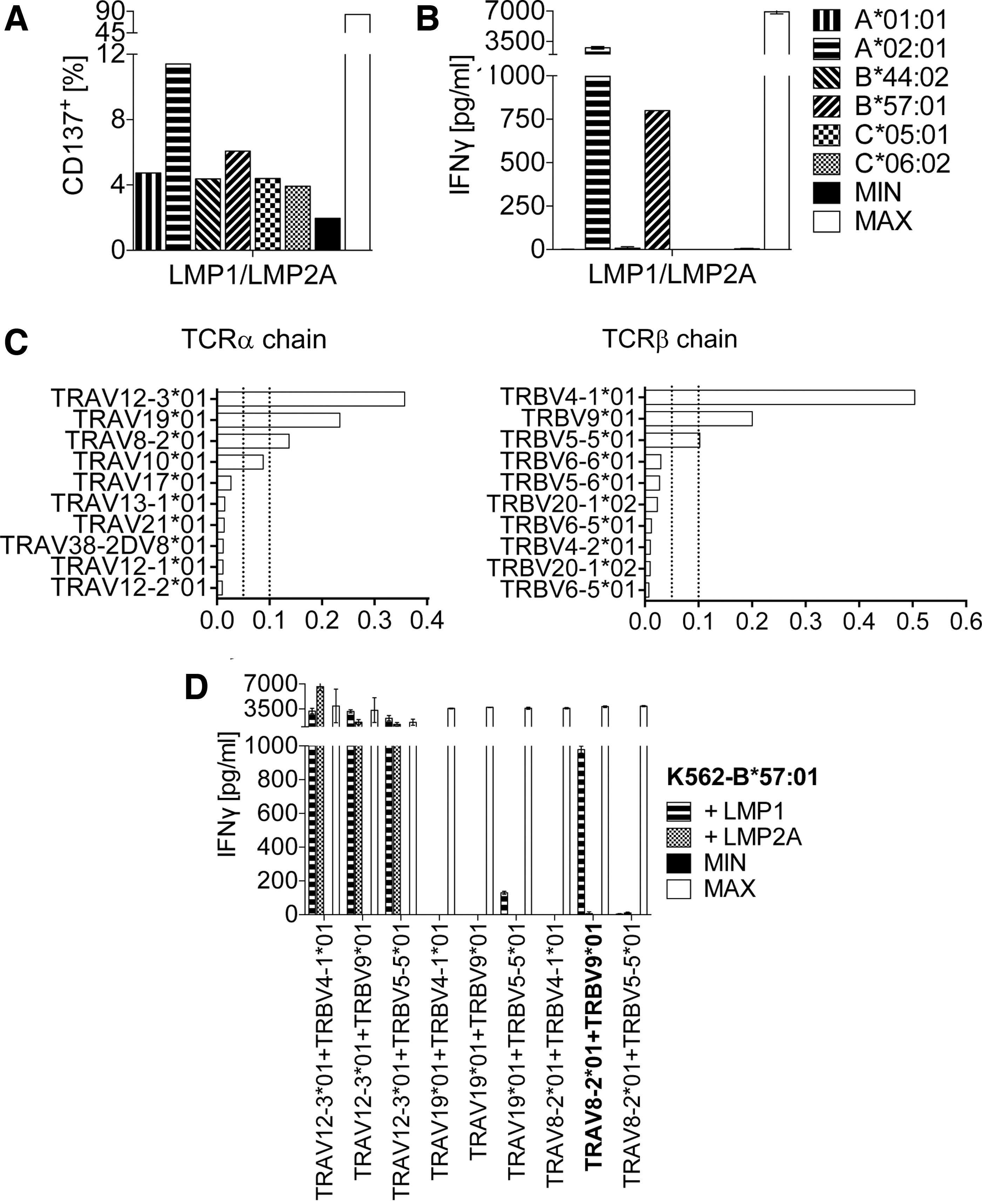
Detection of T cell response and identification of functional TCRαβ chain combination. T cells expanded on EBV antigen-presenting DCs were cocultured with K562 cells of an MHC I cell library harboring the respective MHC alleles of the donor. Screening for immunogenic EBV antigen–MHC combinations was performed by
Exemplified for the TCR that recognized the EBV antigen presented by MHC I B*57:01, next-generation sequencing revealed three dominant TRAV- and TRBV sequences, each covering >10% of all reads (Fig. 1C). To identify the functional TCR-α/TCR-β chain combination, single-chain TCR-α and TCR-β retroviruses were constructed and used to perform combinatorial chain expression in primary human T cells. Figure 1D shows, as indicated by the high amount of IFN-γ secretion, that the combination of the third prominent TCR-α chain (TRAV8-2*01) and the second prominent TCR-β chain (TRBV9*01) (highlighted in Fig. 1D) yielded a functional TCR, subsequently designated as TCR50.
Only this combination recognized the full-length LMP1 antigen presented by K562 cells, which carried the relevant MHC I molecule B*57:01. The combination of the most abundant TRAV12-3*01 TCR-α chain with the three most prominent TCR-β chains did not result in the formation of an LMP1-specific TCR, as LMP2A was unspecifically recognized as well. The combination of the second dominant TCR-α chain TRAV19*01 with the three prominent TCR-β chains did not reveal the formation of a functional TCR (Fig. 1D).
The functional TCR-α/TCR-β chain combination of another EBV LMP1-specific TCR, termed TCR83, was identified by the same process (Table 1).
Summary of T cell receptor features
CDR3 sequences of TRAV and TRBV will be provided on request.
LMP, latent membrane protein; MHC I, major histocompatibility complex class I; TCR, T cell receptor; TRAV, TCR-α chain variable region; TRBV, TCR-β chain variable region.
Identification of antigenic peptides recognized by LMP1-specific TCRs
For an in-depth characterization of the LMP1-specific TCR50 and TCR83, the functional chains of each TCR were re-cloned as a single transgene cassette into the γ-retrovirus vector MP71 using a 2A-peptide linker in the gene configuration TCRβ-P2A-TCRα. TCR retroviruses were used to generate TCR-engineered T cells for the identification of the immunogenic epitopes recognized by the TCRs. For this, full-length LMP1 was C-terminally truncated to generate different gene fragments (LMP1/2, LMP1/1) (Fig. 2A). These fragments were cloned into plasmid vectors, expressed in K562 cells carrying the relevant MHC I molecule (B*57:01 for TCR50, C*15:02 for TCR83), and tested for their ability to present the epitope recognized by the TCRs. TCR-engineered T cells were cocultured with antigen-expressing K562 cells and IFN-γ secretion was determined as a parameter for epitope recognition. Both TCR50 and TCR83 recognized epitopes translated from nucleotides (nt) 316–624 of the coding region of LMP1, locating the epitope in this part of the gene (Fig. 2B, C).

Mapping of the antigenic peptide region recognized by LMP1-specific TCR50 and TCR83.
Next, candidate peptides of the corresponding protein region were identified using the epitope prediction algorithm NetMHCpan4.0 (Fig. 3A, B). Predicted peptides were generated and loaded onto K562 cells carrying the relevant MHC I molecules B*57:01 for TCR50 and C*15:02 for TCR83, respectively, and screened in coculture experiments for their ability to be recognized by TCR-engineered T cells. Figure 3C shows that 4 of 13 predicted peptides (highlighted in Fig. 3A) were recognized by TCR50, whereas 1 of 4 peptides was identified for TCR83 (Fig. 3D, highlighted in Fig. 3B). The peptides, which were capable of stimulating the highest amount of IFN-γ secretion, were considered as cognate epitopes of TCR50 and TCR83, respectively. Because the four peptides recognized by TCR50-engineered T cells contained the same core amino acid sequence ALYLQQNW, it is difficult to predict the true epitope of this TCR. However, the nonamer peptide IALYLQQNW was already described as an epitope in the context of MHC I B*57 and B*58 antigen presentation. 28 In contrast, the nonamer peptide QQNWWTLLV presented by MHC I C*15:02 and recognized by TCR83-engineered T cells was not known previously (Table 1).

Identification of the immunogenic epitope of LMP1-specific TCR50 and TCR83. The protein region identified as epitope-positive sequence (LMP1/2 nt 316–624) was used to select for candidate peptides by applying the epitope prediction algorithm NetMHCpan4.0.
The peptide sensitivity (half maximum IFN-γ release; 1/2 IFN-γ) of TCR50 and TCR83 was analyzed in peptide titration experiments. For this purpose, peptides representing the cognate epitopes were loaded in titrating amounts onto the appropriate K562 target cells, and the ability of TCR-engineered T cells to recognize their target epitope was assessed by IFN-γ ELISA. TCR50 recognized all three peptides predicted to have a strong binding affinity (SB), although, as expected, with different sensitivity, whereas the peptide ALYLQQNWW predicted to have a weak binding affinity (WB) was recognized to a much lesser extent (Fig. 4A). Untransduced T cells were not able to react with peptide-loaded K562 cells. Based on the peptide sensitivity, the 1/2 IFN-γ was calculated for the most efficiently recognized peptide, IALYLQQNW, as 3 × 10−8 M (Table 1).

Peptide titration of LMP1-specific TCR50- and TCR83-engineered T cells. Untransduced, TCR50-transduced T cells
Thus, our results indicate that the peptide IALYLQQNW is probably the cognate epitope of TCR50. Of interest, the experimentally measured sensitivity of the different peptides did not precisely correspond to their predicted position in the epitope prediction algorithm NetMHCpan4.0. Peptides IALYLQQNW and IIALYLQQNW, which the algorithm placed at the third and fifth position, respectively (Fig. 3A), were more efficiently recognized by TCR50-engineered T cells than peptide IALYLQQNWW, which was placed at the second position. Peptide ALYLQQNWW, based on its relatively low binding affinity of 853.78 nM placed at position 11 of the algorithm and classified as a weak binder, was hardly recognized by TCR50-engineered T cells even at a high peptide concentration of 10−7 M.
For TCR83, the peptide QQNWWTLLV was the only epitope, which was recognized by TCR-engineered T cells (1/2 IFN-γ of 2 × 10−9 M) after coculture with peptide-loaded K562-C*15:02 cells (Fig. 4B and Table 1). TCR83 recognized the respective epitope with ∼10-fold higher sensitivity than TCR50 (2 × 10−9 vs. 3 × 10−8 M), although it is classified as a weak binder.
Identification, isolation, and functional characterization of an EBV LMP2A-specific TCR
The same strategy was applied to isolate an EBV LMP2A-specific TCR designated as TCR06. After stimulation of autologous CTLs from an EBV+ donor (Supplementary Table S1) for 14 days with full-length LMP2A-presenting mDCs, T cells were cocultured with LMP2A-presenting K562 cells carrying the six MHC I complexes of the T cell donor. Thereby, A*02:01 was identified as the antigen-presenting MHC I complex (Fig. 1A, B). After identification of the functional TCR-α/TCR-β chain combination of TCR06 (Table 1) and the generation of TCR retroviruses, truncations of the full-length LMP2A gene were utilized to determine the antigenic peptide recognized by the TCR (Fig. 5A). The truncated gene fragments were expressed in K562-A*02:01 cells, which were subsequently cocultured with TCR06-engineered T cells. Indicated by the secretion of IFN-γ, it is evident that the antigenic peptide is located between nt 1006 and 1494 (Fig. 5B). The epitope prediction algorithm NetMHCpan4.0 revealed 14 peptides that could be considered as a cognate epitope of TCR06 (Fig. 5C). When TCR06-engineered T cells were cocultured with individually peptide-loaded K562-A*02:01 cells, the two overlapping peptides MCLGGLLTMV (tenmer, classified as SB) and the N-terminal truncated nonamer CLGGLLTMV (classified as WB) induced an IFN-γ response (Fig. 5D). Functional analysis revealed a similar peptide sensitivity (1/2 IFN-γ 6 × 10−9 M) of TCR06-engineered T cells to MCLGGLLTMV and CLGGLLTMV, respectively (Fig. 5E). Thus, TCR06 recognized the described nonamer CLGGLLTMV and the N-terminal extension MCLGGLLTMV when presented by MHC I A*02:01. 29 However, we did not analyze whether the N-terminal methionine is cleaved off before loading the peptide into the binding grove.
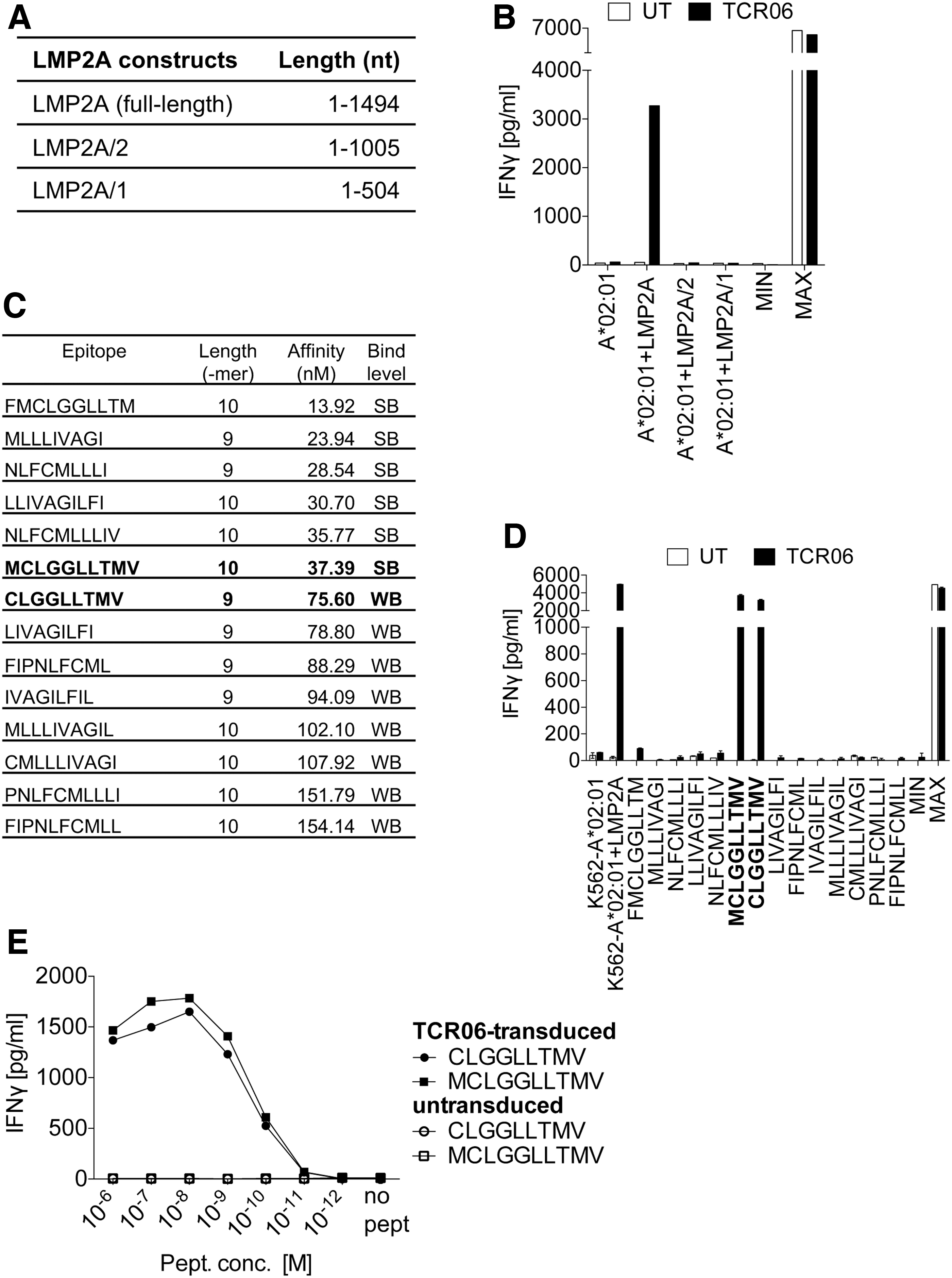
Epitope mapping to identify the antigenic peptide of the LMP2A-specific TCR06 and functional analysis. For TCR06, the restriction element MHC-A*02:01 was identified by using the K562 MHC I cell library (Fig. 1A, B).
Identification, isolation, and functional characterization of EBV EBNA3C-specific TCRs
Next, we generated EBNA3C-specific TCRs. Again, mDCs were transfected with full-length EBNA3C antigen-encoding ivt RNA and cocultured with autologous CTLs of three EBV+ donors (Supplementary Table S1). After the coculture of T cells with EBNA3C-presenting K562 cells carrying the MHC I complexes of the different T cell donors, we identified B*07:02, B*44:02, and C*06:02 (Supplementary Fig. S2) as antigen-presenting MHC I complexes. Functional TCR-α/TCR-β chain combinations were determined for B*07:02-restricted TCR01 and TCR27, B*44:02-restricted TCR25 and TCR58 as well as for the C*06:02-restricted TCR64 (Table 1). The two B*07:02-restricted TCR01 and TCR27 have identical TRAV- and TRBV gene segments but differ in their CDR3 amino acid composition. The two B*44:02-restricted TCRs 25 and 58 share the same V gene segment TRBV2*01 but exhibit different TRAV segments. Whereas the TCR25 α-chain carries the V segment TRAV8-2*01, the TCR58 α-chain possesses TRAV12-1*01 (Table 1). In addition, both TCRs are different in the amino acid composition of their CDR3 regions.
Truncated versions of the EBNA3C antigen were generated (Fig. 6A) to identify the gene segment recognized by the different TCRs in coculture experiments of TCR-engineered T cells and K562 cells carrying the relevant MHC I complex (Fig. 6B–F). The antigenic peptides are encoded between nt 2071 and 2979 for TCR01 and TCR27, between nt 1 and 567 for TCR25 and TCR58, and between nt 568 and 1559 for TCR64. Using the epitope prediction algorithm NetMHCpan4.0, we identified 27 potential epitopes each for TCR01 and TCR27, both restricted to MHC I B*07:02 (Supplementary Fig. S3A), whereas four potential epitopes were identified for TCR25 and TCR58, which were restricted to MHC I B*44:02 (Supplementary Fig. S3B). In addition, 27 potential epitopes were predicted for TCR64 restricted to MHC I C*06:02 (Supplementary Fig. S3C).
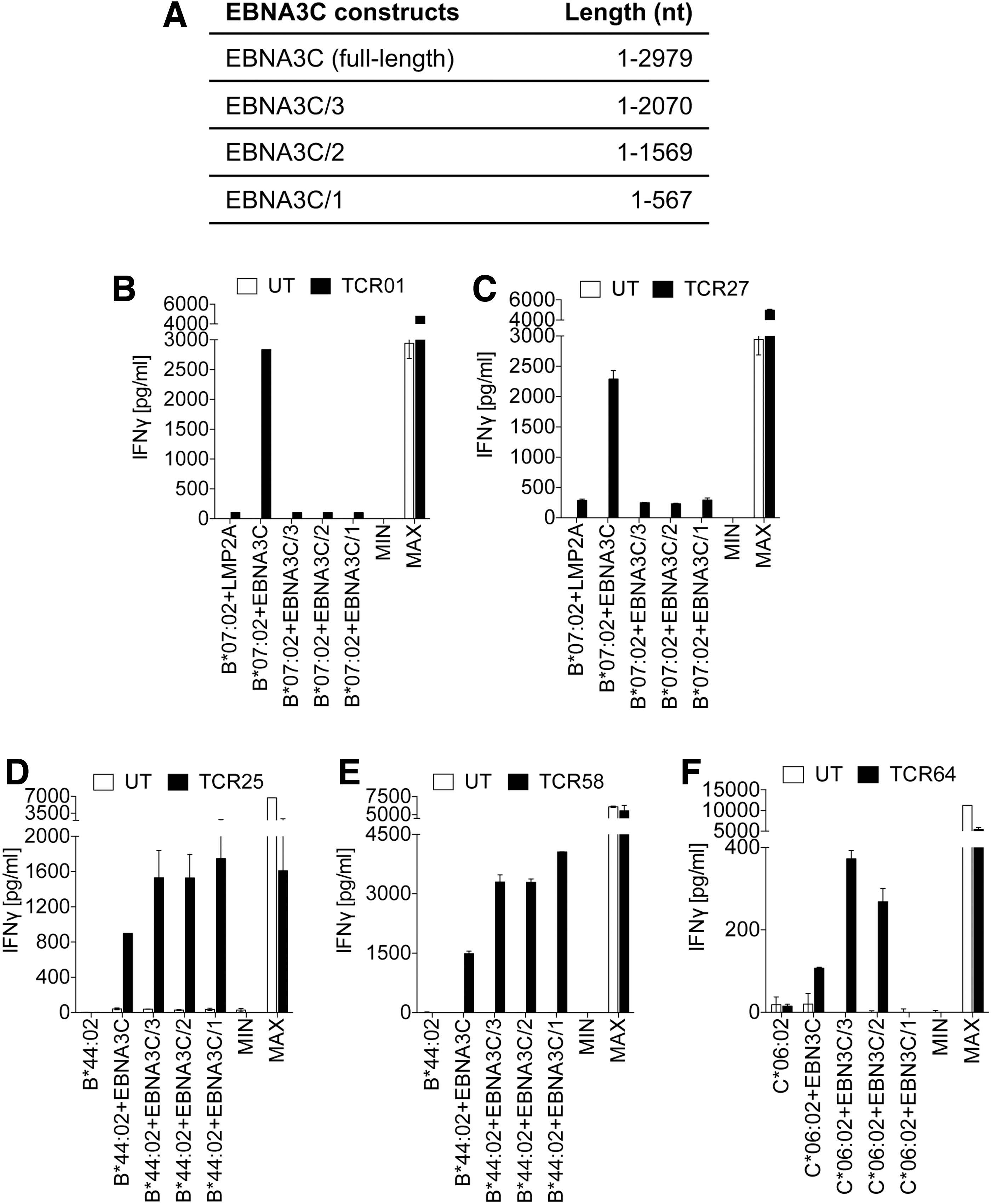
Mapping of the antigenic peptide regions of EBNA3C-specific TCRs.
Subsequently, TCR-engineered T cells were cocultured with the corresponding peptide-loaded K562 cells expressing the relevant MHC I complex. IFN-γ secretion of EBNA3C TCR-engineered T cells revealed peptides SPQPRAPIRPIP and QPRAPIRPIP (both SB), and QPRAPIRPIPT (WB) as cognate epitopes of TCR01 (Fig. 7A). Similarly, peptides QPRAPIRPI, SPQPRAPIRPI, SPQPRAPIRPIP, PQPRAPIRPI, and QPRAPIRPIP (all SB), and QPRAPIRPIPT, as well as QPRAPIRP (both WB), can be considered potential epitopes of TCR27 (Fig. 7B). Peptides of the potential EBNA3C epitopes were loaded in serial dilutions onto K562 target cells and the ability of TCR-engineered T cells to recognize their target epitope was determined by IFN-γ secretion. Figure 7C shows that TCR01-engineered T cells were able to recognize the NetMHCpan4.0-categorized SB peptide SPQPRAPIRPIP and the WB peptide QPRAPIRPIPT with a similar 1/2 IFN-γ of 6 × 10−8 M and that the titration curves between 10−8 and 10−11 M were identical. Another SB peptide QPRAPIRPIP was recognized to a considerably lower extent.

Identification of the immunogenic epitopes of EBNA3C-specific TCR01 and TCR27 and determination of peptide sensitivity. Twenty-seven peptides were predicted by the epitope prediction algorithm NetMHCpan4.0 27 as potential binder for
Exactly the same three peptides, although with different affinity compared with TCR01, were recognized by TCR27-engineered T cells (Fig. 7D). Here, the two peptides QPRAPIRPIP and QPRAPIRPIPT induced a distinct T cell response (1/2 IFN-γ 6 × 10−8 M), which resulted in nearly identical titration curves. The peptide SPQPRAPIRPIP was recognized to a lesser extent and the remaining peptides were hardly recognized. Because all epitopes share the same core amino acid sequence, QPRAPIRPIP, it is difficult to specify the accurate sequence of the cognate epitope. We did not investigate whether the N-terminal serine and proline are cleaved off before loading the peptide SPQPRAPIRPIP into the binding grove. However, proline as the C-terminal amino acid of an epitope is rather unusual. Therefore, we suggest the tenmer QPRAPIRPIPT as potential epitope of TCR01 and TCR27.
TCR25 and TCR58, which were able to react with peptides presented by B*44:02, recognized the peptide AEGGVGWRHW classified as SB (Fig. 8A, B and Supplementary Fig. S3B). Peptide titration experiments revealed a 1/2 IFN-γ of 6 × 10−7 M for TCR25 and 3 × 10−7 M for TCR58, respectively (Fig. 8C, D). Of interest, it was reported that EBNA3C-specific T cell clones lysed more efficiently LCLs loaded with the N-terminal truncated peptide EGGVGWRHW in comparison with LCLs loaded with the peptide AEGGVGWRHW and other N- and C-terminal amino acid extensions. Unfortunately, a peptide titration of all peptides was not performed. 30 We show that a titration experiment of both peptides clearly revealed a substantial higher peptide sensitivity of TCR25-engineered T cells to AEGGVGWRHW (1/2 IFN-γ of 6 × 10−7 M) than to EGGVGWRHW (1/2 IFN-γ of 5 × 10−6 M) (Supplementary Fig. S4). The similar result was obtained using TCR58-engineered T cells cocultured with either EGGVGWRHW- or AEGGVGWRHW-loaded K562-B*44:02 cells. Thus, further analyses have to provide evidence which peptide represents the cognate epitope of TCR25 and TCR58.
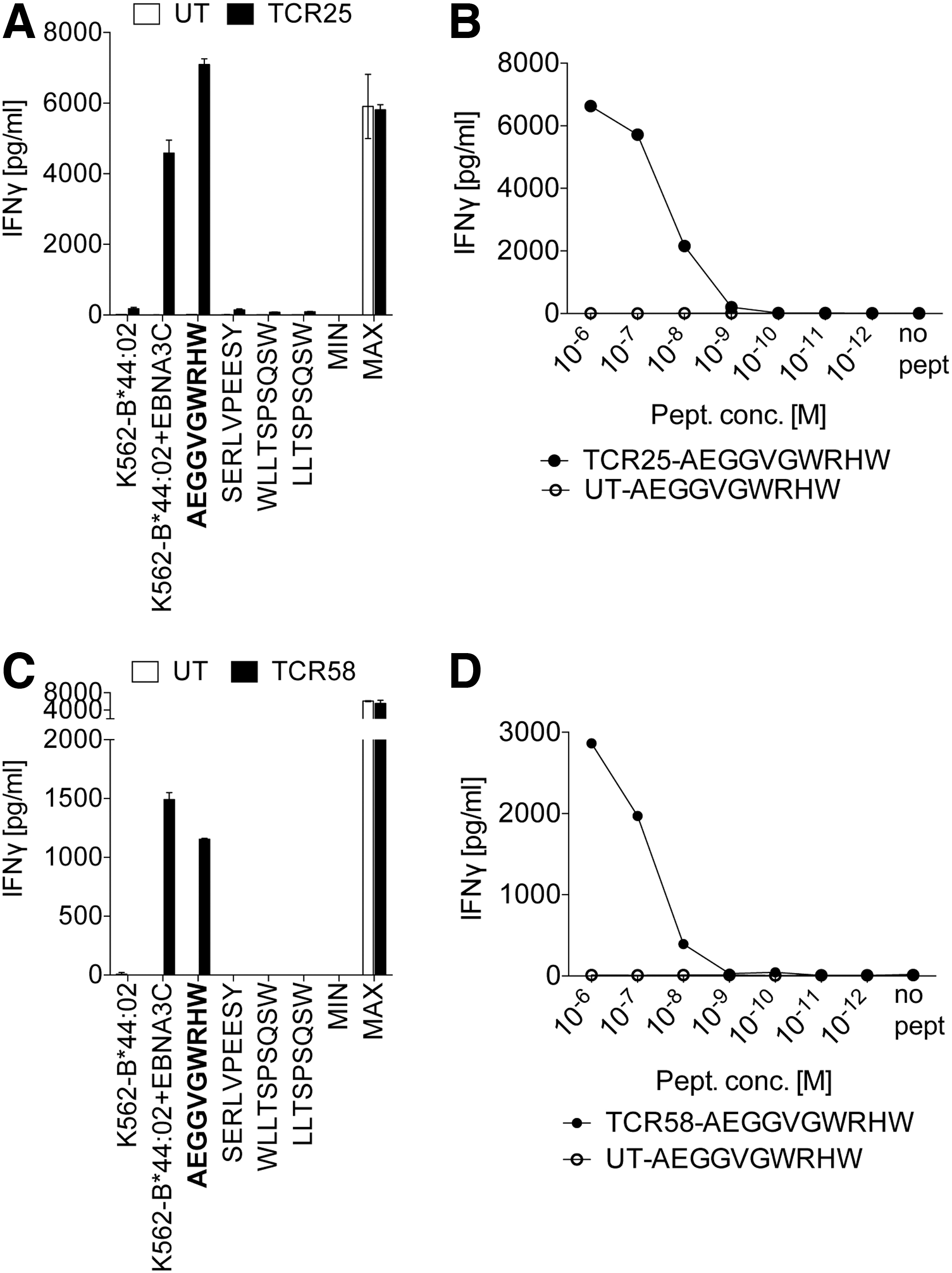
Identification of the immunogenic epitopes of EBNA3C-specific TCR25 and TCR58 and determination of peptide sensitivity.
Finally, TCR64 was able to recognize only the peptide FRKAQIQGL (classified as SB) presented by the MHC I complex C*06:02 (Fig. 9A, B). Compared with all other EBNA3C-specific TCRs, the 1/2 IFN-γ of TCR64 (7 × 10−6 M) was relatively low (Table 1).

Identification of the immunogenic epitope of EBNA3C-specific TCR64 and determination of peptide sensitivity.
TCR-engineered T cells recognize and kill tumor cells in vitro, which naturally present EBV antigens
Next, the capability of the different TCRs to recognize their relevant epitopes was analyzed. For this, a panel of tumor cells and LCLs was utilized, which either were naturally equipped with EBV antigens and the relevant MHC I molecule or had been transfected with the antigen and/or the proper MHC I allele. Exemplified by one TCR of each antigenic specificity (LMP1/TCR50, LMP2A/TCR06, and EBNA3C/TCR64), Fig. 10A–C shows that TCR-engineered T cells secreted IFN-γ after coculture with naturally antigen-expressing cells. However, it is obvious that cells that were transfected with the antigen, the MHC I allele, or both, mostly secreted higher amounts of IFN-γ in comparison with those cells that expressed both the antigen and the MHC I complex at physiological levels. The same result holds true for the other EBV-specific TCRs. Again, both antigen- and MHC I-transfected cells, as well as naturally antigen+/MHC I+ cells, were recognized by TCR-engineered T cells, but T cells secreted different amounts of IFN-γ (Supplementary Fig. S5A–E).
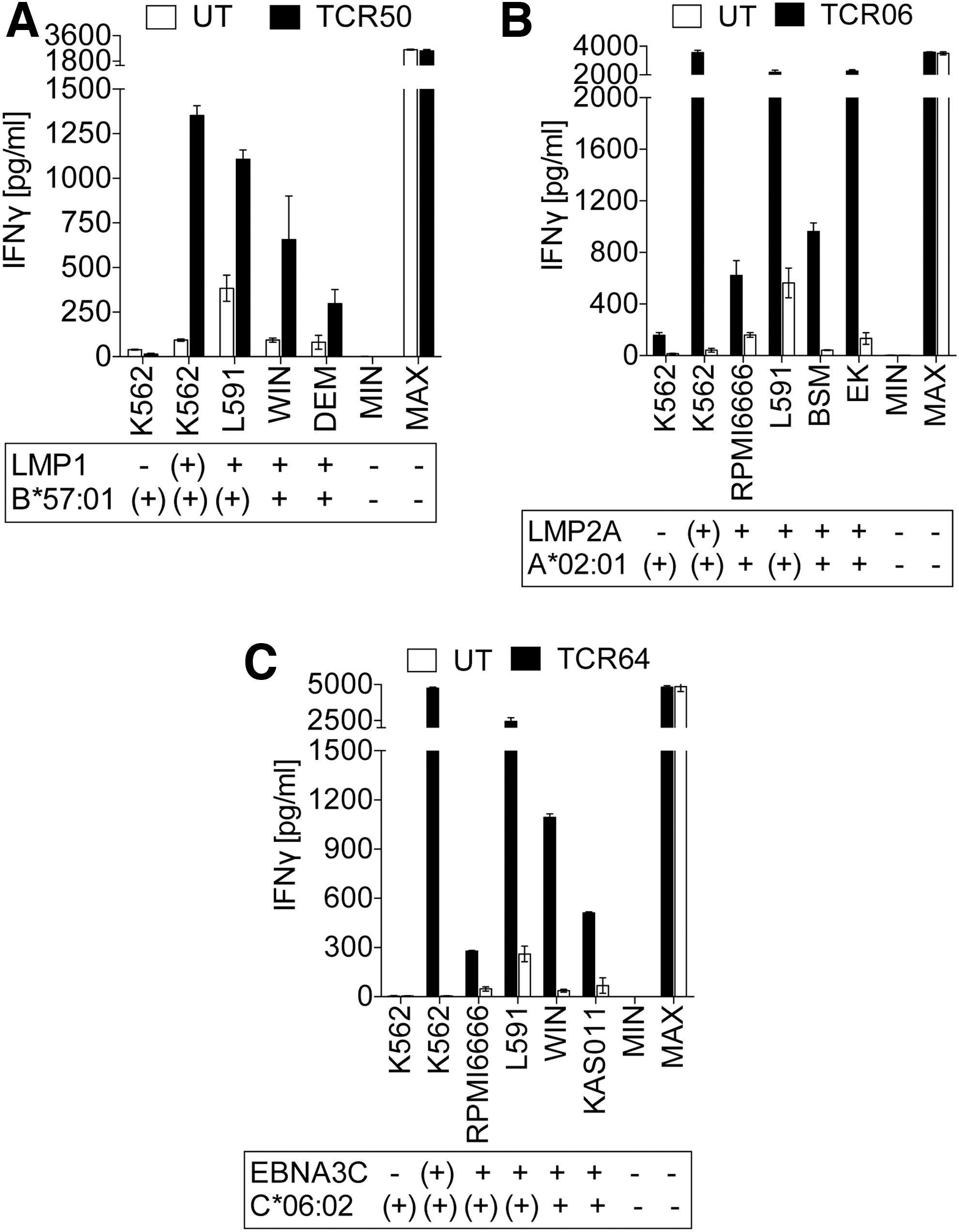
EBV antigen-specific TCRs recognize cancer cells.
To further characterize EBV TCR-engineered T cells for their potential to kill tumor cells, we selected TCR50 (LMP1-specific), TCR06 (LMP2A-specific), and TCR64 (EBNA3C-specific) from our TCR panel and cocultured TCR-engineered T cell with L591 tumor cells in different E:T ratios. Figure 11 shows that each TCR-engineered T cell population was able to kill tumor cells. At an E:T ratio of 5:1, TCR50- and TCR06-engineered T cells killed ∼25% of L591 cells, whereas no killing of tumor cells was observed using TCR64-engineered T cells. At an E:T ratio of 10:1, most effective cell killing was achieved with the LMP2-specific TCR06-engineered T cells (∼50%) followed by the LMP1-specific TCR50-engineered T cells (30%) and the EBNA3C-specific TCR64-engineered T cells (∼15%). Of interest, even at an E:T ratio of 1:10, TCR06-engineered T cells showed a slight killing of L591 tumor cells. This result seems to correlate with the different affinity of the three TCRs (Table 1).

EBV-specific TCR-engineered T cells kill cancer cells. LMP1-specific TCR50-, LMP2A-specific TCR06-, and EBNA3C-specific TCR64-engineered T cells were co-cultured with L591 EBV-positive tumor cells, which naturally express LMP1, LMP2A, and EBNA3C but were transfected with the respective MHC I allele. At indicated E:T cell ratios triplicate wells were averaged and the percentage of surviving cells was calculated in relation to the values obtained from the samples treated with untransduced T cells: % specific survival = 100 × (test value)/(average background). Data are means of triplicates ± mean deviation of one out of two independent experiments. UT: untransduced T cells.
Discussion
For several decades, T cells have been equipped with new antigen specificities by TCR gene transfer. Initial problems, such as nonefficient transgene expression or formation of mispaired TCRs, were solved and TCR-engineered T cells became a valuable tool for TCR gene therapy in clinical studies. 31 However, an issue that still hampers the success of TCR gene therapy is the identification of potent TCRs that efficiently target pMHC complexes exclusively expressed by cancer cells. Several approaches have been used to generate peptide-specific CTLs as a source for the isolation of TCR-encoding genes. These include (1) priming of autologous or allogeneic T cells with selected peptides, 32 –34 (2) generation of T cells in HLA-A*02:01 transgenic mice that recognize human target antigens, 35,36 and (3) utilization of transgenic mice carrying the human TCR repertoire restricted to HLA molecules. 37,38 All these approaches, on the one hand, are successfully used for the generation of antigen-specific T cells clones and the isolation of TCR genes but, on the other hand, are also accompanied by various limitations.
We used our recently established approach for the identification, isolation, and characterization of TCRs. 20 This enabled us to isolate EBV antigen-specific TCRs in an unbiased manner, meaning in the absence of preselected target epitopes and MHC binding complexes. The antigen-presenting DCs themselves select for the processing and presentation of the most suited epitope of a given antigen and the appropriate MHC I complex. Therefore, the pMHC combination should be immunological relevant and recognized by T cells with proper affinity for TCR gene therapy. Because we used exclusively EBV+ donors, the T cells are most probably reactivated from the T cell memory pool. By using this approach, we eliminated two possible disadvantages concerning the isolation of potent TCRs for immunotherapy. First, currently available software algorithms do not precisely predict which pMHC combination would be able to induce a strong T cell response and allow the isolation of high-affinity TCRs. In this study, most of our EBV antigen-specific TCRs (except TCR25 and TCR58) recognized epitopes, which were not ranked at the highest positions in the corresponding NetMHCpan4.0 prediction algorithm. This was not only true for the rather rare MHC I proteins B*07:02, B*57:01, C*06:02, and C*15:02 but was also evident for the broadly distributed MHC I protein A*02:01. Therefore, peptide prediction algorithms like NetMHCpan4.0 or others can be considered only as helpful tools to select and restrict for a number of possible candidate epitopes, which subsequently have to be thoroughly analyzed. Second, we excluded the possibility that a database predicted peptide is not properly endogenously processed, and therefore cannot serve as a suitable target antigen. This was previously shown for an epitope of the fusion protein TEL-AML1, an important transforming factor in leukemogenesis. Here, a nonamer peptide of the fusion region of TEL-AML1 was described as immunogenic in an MHC I A*02:01-restricted manner. However, although a specific T cell response against this peptide was induced, the defined epitope was not endogenously processed and hence not applicable as a target for TCR gene therapy. 39
However, a system based on the antigenic stimulation of autologous T cells is also not without limitations, which have to be considered for the isolation of TCRs to achieve a therapeutic success. T cells equipped with high-affinity TCRs recognizing TAAs are eliminated by thymic selection to prevent autoimmunity. Thus, usually only T cells with low-affinity TCRs are usually released into the periphery, which do not possess the optimal pMHC affinity to be used as candidates for successful TCR gene therapy. 34,38 It may also be difficult to use our approach for the isolation of TCRs that recognize epitopes of mutated TSAs, because the frequency of such T cells may be too low for identification.
To circumvent these disadvantages, we selected viral antigens as targets that are associated with diseases and represent TSAs. Thereby, we avoid the potential risk of on-target/off-tumor toxicity as a potential side effect of TCR gene therapy. 40
Among viruses that are associated with different diseases, including the induction of cancer or the maintenance of the malignant cell phenotype, we selected EBV. 7 –11 EBV-associated malignancies are categorized according to the latency gene expression profile. 8 In HL, GC, NPC, and some NK cell and T cell lymphomas LMP1, LMP2, and EBNA1 are expressed (type II EBV latency), whereas in PTLD (type III EBV latency) nearly the complete number of EBV latency antigens including LMP1, LMP2, and the EBNA3 family is expressed. In previous studies, it has been shown that diseases, which are caused by EBV, are usually cleared by immune cells, wherein CTLs play the most critical role. 4 Subsequently, EBV-specific T cells were adoptively transferred to treat virus-associated diseases or to control the reactivation of EBV after allogenic bone marrow transplantation. 41 –44 However, the application of EBV-specific TCR-engineered T cells for adoptive transfer was until now not reported.
In our study, we isolated and characterized eight EBV antigen-specific TCRs, which recognized epitopes of LMP1, LMP2A, and EBNA3C, respectively. Among these TCRs, TCR83 recognized a not yet described LMP1 epitope (QQNWWTLLV) restricted by MHC I C*15:02. In some cases, the TCRs we isolated and characterized were novel, but their recognized epitopes had previously been identified in response to CTL clones (e.g., TCR01, TCR25, TCR27, TCR50, and TCR58). In another case, we identified a TCR (TCR06) that utilizes a different TRAV/TRBV composition including CDR3 region compared with the published TCRs recognizing the LMP2A-specific MHC I A*02:01-restricted epitopes CLGGLLTMV or SLGGLLTMV, 12 –16,19 and shows a very high pMHC affinity (Table 1).
Of interest, the EBNA3C-specific MHC I C*06:02-restricted epitope FRKAQIQGL, which we identified as the ligand for TCR64, was previously shown to be also presented by other MHC I proteins like B*08:01, B*14:02, B*27:02, B*27:04, B*27:05, and B*39:01. 45 This is not unusual as it was shown for >240 well-defined EBV and HIV epitopes. These viral epitopes were analyzed in 100 individuals regardless of their MHC I type. Approximately 50% of all detected responses were observed in the absence of the originally reported restricting MHC I allele, whereas only 3% of epitopes were recognized exclusively in the presence of their original MHC I allele. 46
Based on our approach for TCR isolation, the EBV LMP1, LMP2A, and EBNA3C epitopes were presented by a variety of different MHC I alleles, indicating that mDCs transfected with the full-length antigen indeed selected for the most suitable MHC I to present the best suited epitope to T cells. All TCRs and the resultant TCR-engineered T cells recognized naturally processed and presented epitopes and were able to lyse antigen-expressing LCLs and tumor cells.
In addition, stimulation of T cells using one mDC sample primed with more than one antigen allowed the isolation of TCRs specific for different pMHC combinations in one experiment, as we have shown for TCR06 (LMP2A/A*02:01) and TCR50 (LMP1/B*57:01). Thus, our method allows the identification of different TCRs that recognize different antigens in the context of different MHC I proteins in a relatively short time. This is a requirement for the generation of an EBV-specific TCR library, which would provide a valuable source for the rapid identification of EBV-specific T cells for TCR gene therapy. Moreover, the availability of TCRs targeting either different epitopes of one EBV-specific antigen or different epitopes on several EBV antigens, respectively, is a prerequisite for a combinatorial TCR gene therapy. Such kind of therapy could increase the efficacy of TCR gene therapy or could overcome problems arising from the occurrence of antigen-loss variants, which make tumor cells resistant to antigen-based immunotherapies.
In summary, we described a panel of novel TCRs that recognize EBV epitopes of LMP1, LMP2A, and EBNA3C. TCR-engineered T cells efficiently recognized antigen-expressing LCLs and tumor cells and displayed potent cytotoxicity. Because all TCRs were generated in an unbiased setting concerning epitope selection and MHC I presentation, the resultant TCR-engineered T cells should be valuable tools for immunotherapy of EBV-associated diseases.
Footnotes
Acknowledgments
The authors thank F. Lorenz (MDC) for his initial support in the project, A. Grybowski, J. Hauchwitz, C. Hesse, M. Naschke (all MDC), K. Hummel (Humboldt-University Berlin) for their excellent technical assistance, and H.-P. Rahn (MDC) for support with FACS. The authors are thankful to T. Blankenstein (MDC) and A. Moosmann (Helmholtz Zentrum Munich) for valuable suggestions and critical discussions on the article.
Author Disclosure
W.U. is an inventor on a patent covering the TCR isolation platform approach used in this study. The MDC applied for a patent on the TCRs described in this study where K.D. and W.U. are named as inventors. No competing financial interests exist for W.U. K.D., K.W., and E.N.
Funding Information
This work was supported by grants of the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich TR36), the Berlin Institute of Health and the Wilhelm Sander-Stiftung to W.U.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.