
Abstract
This phase II clinical trial investigated the efficacy and safety of intramuscular injection of plasmid pUDK-HGF, which encodes the human hepatocyte growth factor gene in patients with critical limb ischemia. Resting pain patients (n = 119) and patients with leg ulcers (n = 121) were enrolled as two cohorts and randomized to receive pUDK-HGF treatment on days 0, 14, and 28. In the resting pain cohort, the proportion of patients with complete pain relief on day 180 after receiving pUDK-HGF injection, as the primary outcome, was significantly higher than that of the placebo group on the same day (p = 0.0148). More responders with >50% pain reduction were also observed in the pUDK-HGF groups than in the placebo groups (p = 0.0168). In the ulcer cohort of patients, pUDK-HGF treatment tended to be superior to the placebo in the percentage of patients with both complete ulcer healing and >50% ulcer healing. No significant differences in the incidence of adverse events (AEs) or serious AEs were observed among the groups. The mid-dose pUDK-HGF (6 mg) was the most efficacious, and is therefore an appropriate dose for use in a phase III clinical trial.
This study was approved by the China Food and Drug Administration (2013L00637), China Clinical Trial Registry URL:
Introduction
Peripheral arterial disease (PAD), a local manifestation of atherosclerosis, has become a global problem. A recent survey reported that 6.6% (estimated 45.3 million) of the adult (>35 years of age) Chinese population have PAD, the prevalence of which increases significantly in those aged ≥75 years (11.8%). 1 Older age, Han ethnicity, smoking, education level, hypertension, coronary artery disease, diabetes, dyslipidemia, and rural residences are associated with a higher risk for PAD. China still faces a prevention and management challenge for PAD. Critical limb ischemia (CLI), the most severe limb manifestation of PAD, is associated with chronic ischemic pain at rest and/or the presence of ischemic skin lesions. Although open bypass surgery and endovascular revascularization are currently considered the first treatment choices for CLI, ∼50% of patients with CLI are poor candidates for these standard therapies. 2 Therefore, a less invasive therapy is needed for improving limb perfusion in these patients and angiogenesis offers hope for patients who cannot undergo standard revascularization treatment. Hepatocyte growth factor (HGF) is one of the most potent angiogenic proteins. Indeed, human HGF gene-encoding plasmids are currently considered to be the most promising type of gene therapy for CLI, and several clinical trials using this type of CLI therapy have been performed in the United States, Japan, and China. 3 –12 However, some results from these clinical trials are controversial, and a large-sample-sized clinical study is needed to verify the potential beneficial efficacy of HGF plasmid therapy for CLI.
pUDK-HGF is a naked plasmid DNA encoding the human HGF gene inserted into pUDK vector. 13 Based on our rat and dog ischemic hind limb models, our preclinical studies have indicated that pUDK-HGF has therapeutic potential in both cases. 13 –17 Intramuscular injection of pUDK-HGF into the rat hind limb resulted in strong HGF expression and significantly increased capillary density in the ischemic tissue. It also significantly increased the blood flow in collateral vessels in either a completely or partly femoral artery ligation hind limb dog model. Furthermore, no obvious toxic effects were observed in the rat and dog toxicology studies. In fact, the results of a phase I clinical study 3 showed that intramuscular injection of pUDK-HGF is safe, and no serious adverse events (SAEs) were observed over the 3-month follow-up. Notably, pUDK-HGF therapy significantly decreased the pain severity and improved ulcer healing in CLI patients. The mean visual analog scale (VAS) for all the patients decreased significantly from a baseline of 4.52 to 0.30 (p < 0.01 vs. baseline) on day 90 posttreatment with pUDK-HGF. Fourteen patients reported complete pain relief and a 100% VAS score reduction on day 90. Of the four patients with ulcers, two ulcers showed complete closure, while the other two patients showed ulcer size reductions >25% in the long axis diameter after pUDK-HGF treatment. Of the five patients with gangrene, one gangrenous wound was completely closured on day 90 and two gangrenous wounds showed marked size reductions. These results provided the rationale for conducting a larger sized clinical study to evaluate the safety and efficacy of pUDK-HGF for CLI gene therapy.
In this phase II study, we performed a multicenter, randomized, double-blinded, placebo-controlled trial of pUDK-HGF injection in two cohorts of patients with CLI, including 119 resting pain patients and 121 patients with ulcers, to evaluate the safety and efficacy of pUDK-HGF, and to determine an appropriate dose for a phase III clinical trial.
Materials and Methods
Plasmid pUDK-HGF
The human HGF gene, which is driven by the human cytomegalovirus promoter (EcoRI/NotI, 2.2 kbp), was inserted into the pUDK vector to construct recombinant plasmid pUDK-HGF. 13 pUDK-HGF was provided by Humanwell Healthcare (Group) Co. Ltd., Wuhan, China. The product was supplied in a sterile glass vial at a concentration of 2 mg/mL in the formulation solution, and stored at 4–8°C. pUDK-HGF solution and saline are visually indistinguishable.
Patient population
Inclusion criteria
Patients aged 20–80 years with ischemic lower limb arterial disease (Rutherford Categories 4–6), which included those with arteriosclerosis obliterans, diabetic arteriosclerosis obliterans, and thromboangiitis obliterans, were enrolled. Patients were required to have significant stenosis (75%) of one or more of superficial femoral and popliteal arteries as verified by digital subtraction angiography, computerized tomography angiography, or magnetic resonance angiography within 12 months. Patients were also required to have VAS scores of 3. The resting pain patients were required to have baseline ankle pressures below 40 mmHg or toe pressures below <30 mmHg. Patients with ischemic ulcers or local gangrene were required to have baseline ankle pressures below 60 mmHg or toe pressures below 40 mmHg. Patients were also required to have normal laboratory examinations for tumors, including chest X-ray, tumor marker detection (including alpha-fetoprotein), chorioembryonic antigen, prostate-specific antigen, carbohydrate antigen 19-9, cancer antigen 125, Pap smear, breast X-ray mammography, and fecal occult blood. The female patients must have undergone sterilization or have experienced amenorrhea for more than 1 year, and the pregnancy tests must be negative.
Exclusion criteria
The major exclusion criterion included patients with a vascular disease prognosis who would require amputation within 3 months of the study initiation. It also included patients who had undergone a successful revascularization procedure within the previous 12 weeks, or experienced vascular reconstruction failure 4 weeks before the study initiation. Patients with stage 3 or higher retinopathy, malignant tumors or had suspected malignant tumors (by screening tests), or had resistant hypertension (systolic blood pressure >200 mmHg or diastolic blood pressure >115 mmHg), or had any type infection (e.g., hepatitis B and/or C, AIDS, or syphilis) were also excluded, as were patients with the following laboratory results: hemoglobin <80 g/L, white blood cell count <3.0 × 109/L, platelets <75 × 109/L, aspartate transaminase or alanine transaminase >2 × upper limits of normal (ULN), creatine or blood urea nitrogen (Bun) >1.2 × ULN, and hemoglobin A1c >11.0%.
Withdrawal criteria
Cilostazol tablets were provided as a basic therapeutic drug in this trial, and the patients were withdrawn from using any other antiplatelet or vasodilation agent. For ethical consideration, patients could choose acetaminophen tablets (Tylenol), tramadol (sustained-release tablets), or controlled-release morphine tablets according to the degree of pain. Patients were also withdrawn from the study for poor compliance, unwillingness to continue, for other severe concomitant diseases, or after missing a visit.
Study design
Randomization
This double-blind, randomized, placebo-controlled, 14-center trial was designed to assess the safety and efficacy of intramuscular injection of pUDK-HGF in CLI patients. Two cohorts of patients were enrolled, and 120 patients with resting pain were evaluated for pain relief, whereas 120 patients with ulcers were evaluated for ulcer healing. The patients in each cohort were randomized 1:1:1:1 to receive intramuscular injection of placebo (0.9% saline), or 1 of 3 doses of pUDK-HGF (low-dose, 4.0; middle-dose, 6.0; high-dose, 8.0 mg) on days 0, 14, and 28.
Intramuscular injection of plasmid pUDK-HGF
The patients received 32-site injections in the calf and/or thigh muscles of the affected limb on days 0, 14, and 28. The high-dose pUDK-HGF (8 mg) group of patients received 32- site injections of 0.25 mg/0.5 mL pUDK-HGF, the mid-dose pUDK-HGF (6 mg dose) group of patients received 24 injections of 0.25 mg/0.5 mL pUDK-HGF, or 8 injections of 0.5 mL of the placebo. The low-dose pUDK-HGF (4 mg) group of patients received 16 injections of 0.25 mg/0.5 mL pUDK-HGF, or 16 injections of 0.5 mL of the placebo. Patients in the placebo group received 32 injections of 0.5 mL of 0.9% saline. The first injection site was chosen to be 2 cm above the starting point of the stenosis based on the vascular disease anatomy, whereas the others were regularly distributed following the blood vessel direction at 2 cm apart.
Efficacy assessments
Efficacy assessments included pain scale, ulcer size, toe brachial index (TBI), ankle brachial index (ABI), and transcutaneous oxygen pressure (TcPO2). They were evaluated on screening, and on days 0, 14, 28, 60, 90, and 180 after treatment. Pain intensity was measured using the VAS score on screening and on days 0 (preinjection), 14 (preinjection), and 28 (preinjection), and on days 30, 60, 90, and 180. The ulcer size was measured using Anshutuo sticking strips at baseline and at every visit throughout the 6-month follow-up period.
The TcPO2 values for the chest, thigh, calf, and foot were measured. The minimum ratio of limb to chest TcPO2 on each visit was used to evaluate changes in transcutaneous oxygen tension.
The primary treatment endpoint was the proportion of patients with complete pain relief (for resting pain patients) or with complete ulcer closure (for ulcer patients) on day 180. The secondary outcome included the percentage of patients with >50% reduction in the pain VAS score or a >50% reduction in ulcer size, as well as a change in TcPO2, ABI, and TBI, amputation rate, and mortality from baseline to the 6-month follow-up.
Safety assessments
Safety analysis was based on the occurrence of adverse events (AEs) according to the NCI Common Terminology Criteria for Adverse Events v3.0, and included physical examination, concomitant medication, laboratory examinations such as electrocardiogram, blood chemistry, hematology, coagulation, urinalysis, retinopathy screening, and tumor screening.
The protocol was approved by the Research Ethics Boards for all sites and all the patients provided written informed consent before participating in this study. The trial was carefully monitored by the members of an independent data monitoring committee who reviewed the accruing and unblinded data.
Statistical analysis
Statistical analysis was performed using SAS, version 9.2, with p < 0.05 considered to be statistically significant. A full analysis set and a per-protocol set were performed to evaluate the comprehensive efficacy. Safety analysis was evaluated using the safety analysis set. All continuous data (ABI, VAS) were expressed as the median ± standard deviation and interquartile ranges. Comparisons between the baseline data and posttreatment data were performed by a χ 2 test. Categorical variables were expressed using frequency or percentage (n, %) and analyzed by Fisher's exact test.
Results
Baseline demographics and medical histories
Between May 10, 2013, and August 16, 2014, the 288 patients who consented to participate in this study were screened in accordance with the inclusion and exclusion criteria. Resting pain patients and ulcer patients were enrolled as two cohorts, with each cohort randomized 1:1:1:1 to receive intramuscular injection of placebo or 1 of 3 doses of pUDK-HGF (4.0, 6.0, or 8.0 mg) on days 0, 14, and 28, as shown in Fig. 1.
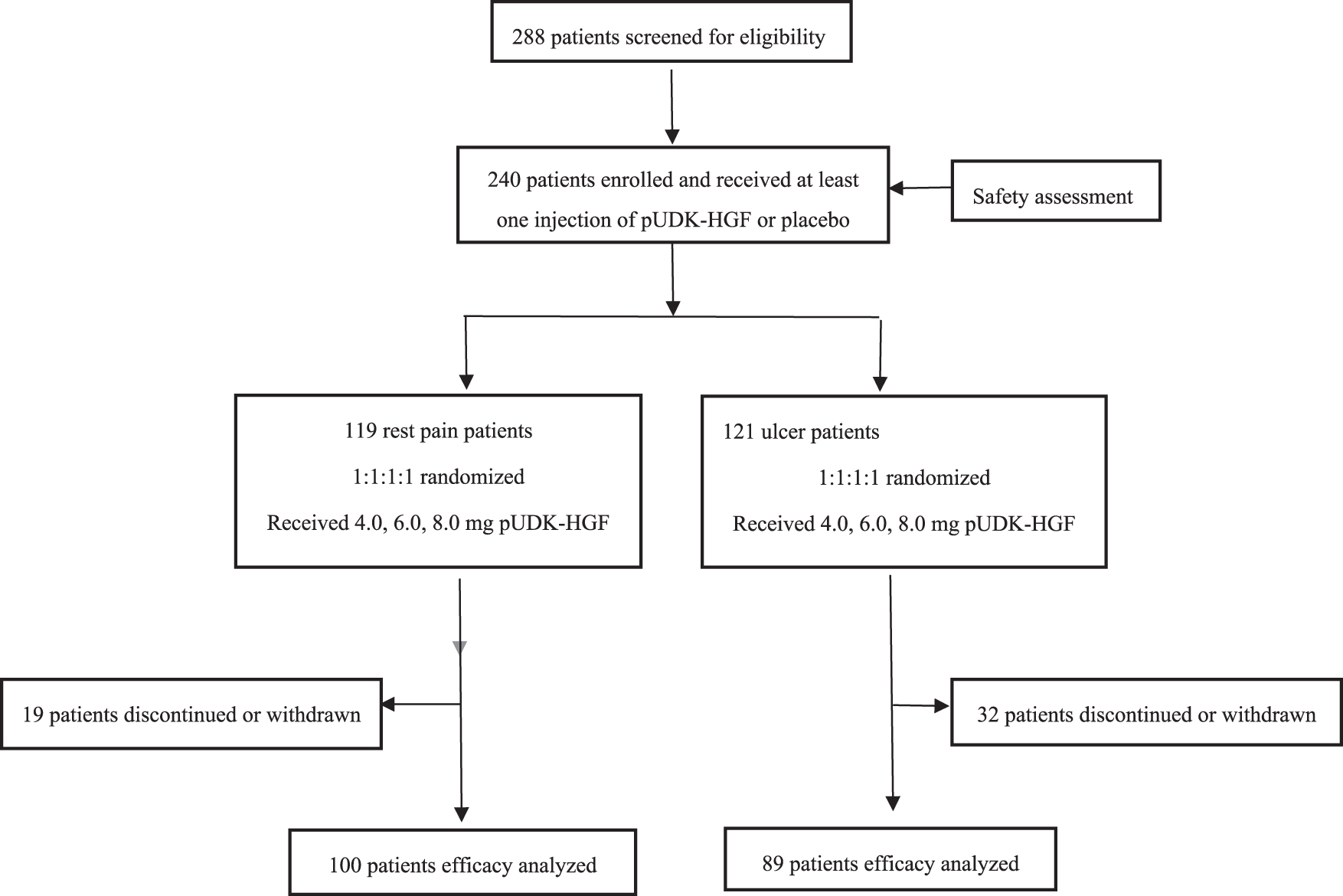
pUDK-HGF phase II clinical trial flowchart. Two cohorts of patients (119 pain-at-resting patients and 121 ulcer patients) were enrolled and randomized 1:1:1:1 to receive intramuscular injection of placebo (0.9% saline), or 1 of 3 doses of pUDK-HGF (low-dose, 4.0; mid-dose, 6.0; high-dose, 8.0 mg) on days 0, 14, and 28. In the pain-at-resting cohort, 19 patients discontinued or were withdrawn and 100 patients were available for the efficacy analysis. In the ulcer patient cohort, 89 patients completed the study and were available for the efficacy analysis. Two hundred forty patients in two cohorts who received at least one injection of pUDK-HGF or placebo were available for safety assessment.
The resting pain cohort comprised 119 patients and randomized into the following three pUDK-HGF treatment groups: high-dose (n = 28), mid-dose (n = 30), low-dose (n = 27), and placebo (n = 34). Five patients who experienced SAEs or were lost to follow-up did not complete the study, and 14 patients were withdrawn for using nonpermitted antiplatelet agents, or for violating the enrollment criteria. Finally, 100 patients (high-dose, n = 22; mid-dose, n = 25; low-dose, n = 24; placebo, n = 29) completed the study and were available for the efficacy analysis. The baseline characteristics of the four groups were homogeneous and comparable with no significant differences in age, sex, demographic origin, or pain characteristics (Table 1).
Resting pain patient characteristics at baseline
ASO, arteriosclerosis obliterans; SD, standard deviation; TAO, thromboangitis obliterans.
The cohort of ulcer patients (n = 121) was randomized into the following four groups: pUDK-HGF high-dose (n = 30), mid-dose (n = 30), low-dose (n = 31), or placebo (n = 30). Nineteen patients who were lost to follow-up, underwent bypass, or died did not complete the study. Thirteen patients who violated the inclusion criteria were withdraw. Eighty-nine patients (high-dose, n = 22; mid-dose, n = 25; low-dose, n = 19; placebo, n = 23) completed the study and were available for the efficacy analysis. The baseline characteristics of the patients are summarized in Table 2.
Ulcer patient characteristics at baseline
Efficacy endpoints in resting pain patient cohort
Resting pain was evaluated at each in-clinic visit (screening, and on days 0, 14, 28, 60, 90, 180) using VAS. The primary endpoint was the proportion of patients with complete pain relief and 100% reduction in their VAS scores on day 180. As shown in Fig. 2, a significantly higher proportion of patients in the pUDK-HGF groups (62.50%, 72.00%, or 63.64% in the high-dose, mid-dose, or low-dose group, respectively) experienced complete pain relief and achieved the primary endpoint on day 180 when compared with patients in the placebo group (31.03%, p = 0.0148).
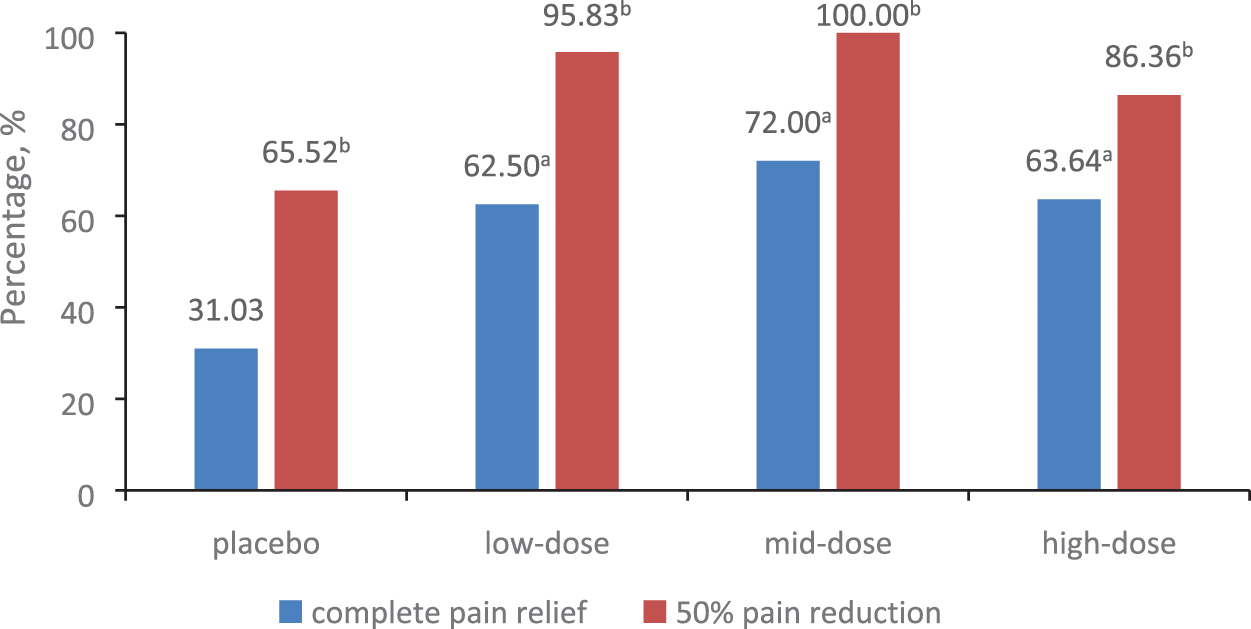
The percentages of complete pain relief and >50% pain reduction in the resting pain patient cohort on day 180. A significantly higher proportion of patients in the pUDK-HGF groups (62.50%, 72.00%, or 63.64% in the high-dose, mid-dose, or low-dose group, respectively) achieved complete pain relief at the primary endpoint on day 180, compared with patients in the placebo group (31.03%, p = 0.0148). The number of responding patients with >50% pain relief in all the pUDK-HGF groups was also significantly higher than the number in the placebo group (65.52% for the placebo vs. 86.36%, 100.00%, or 95.83% for the high-dose, mid-dose, or low-dose groups, respectively; p = 0.0168). a,b p < 0.05 by Fisher's exact test, placebo versus low-, mid-, or high-dose pUDK-HGF, respectively. Color images are available online.
The secondary efficacy measures included the VAS change from baseline and the number of responding patients. The VAS score was reduced in all of the pUDK-HGF patients and showed statistically significant differences when compared with the placebo group on days 60, 90, and 180 (Table 3 and Fig. 3). A much greater reduction was observed in the group that received the mid-dose pUDK-HGF (compared with the other groups), with a 5.42-point reduction seen in the baseline VAS score on day 180 (95% confidence interval, −5.98 to −4.86, p = 0.0041).

The VAS score change from baseline over the 6-month follow-up in the resting pain patient cohort. After the first injection of pUDK-HGF on day 0, the VAS score reduced and showed statistically significant differences on days 60, 90, and 180 among the placebo and pUDK-HGF groups. pUDK-HGF treatment experienced long-term pain relief. a,b,c p < 0.05, placebo versus low-, mid-, or high-dose pUDK-HGF, respectively. VAS, visual analog scale. Color images are available online.
Visual analog scale score changes in resting pain patients available for efficacy
Patients experiencing a >50% VAS reduction from baseline were defined as responding patients. As shown in Fig. 2, the number of responding patients in all the pUDK-HGF groups was significantly higher than the number in the placebo group (65.52% for the placebo vs. 86.36%, 100.00%, or 95.83% for the high-dose, mid-dose, or low-dose groups, respectively; p = 0.0168).
Of the 100 patients, 6 used analgesics during the trial (4 in the placebo group, 1 in the high-dose group, and 1 in the low-dose group), and the VAS changes in the 94 remaining patients not taking analgesics showed a similar trend to that seen in all the available patients (Supplementary Table S1).
There were no statistically significant differences in the TcPO2 ratio among the four groups throughout the study period, but the TcPO2 ratio in each group significantly increased from baseline onward (as shown in Fig. 4). There was no difference in the ABI or the TBI among the groups at any of the time points. Only one patient in the high-dose pUDK-HGF group had an amputation in the left thigh during the trial.
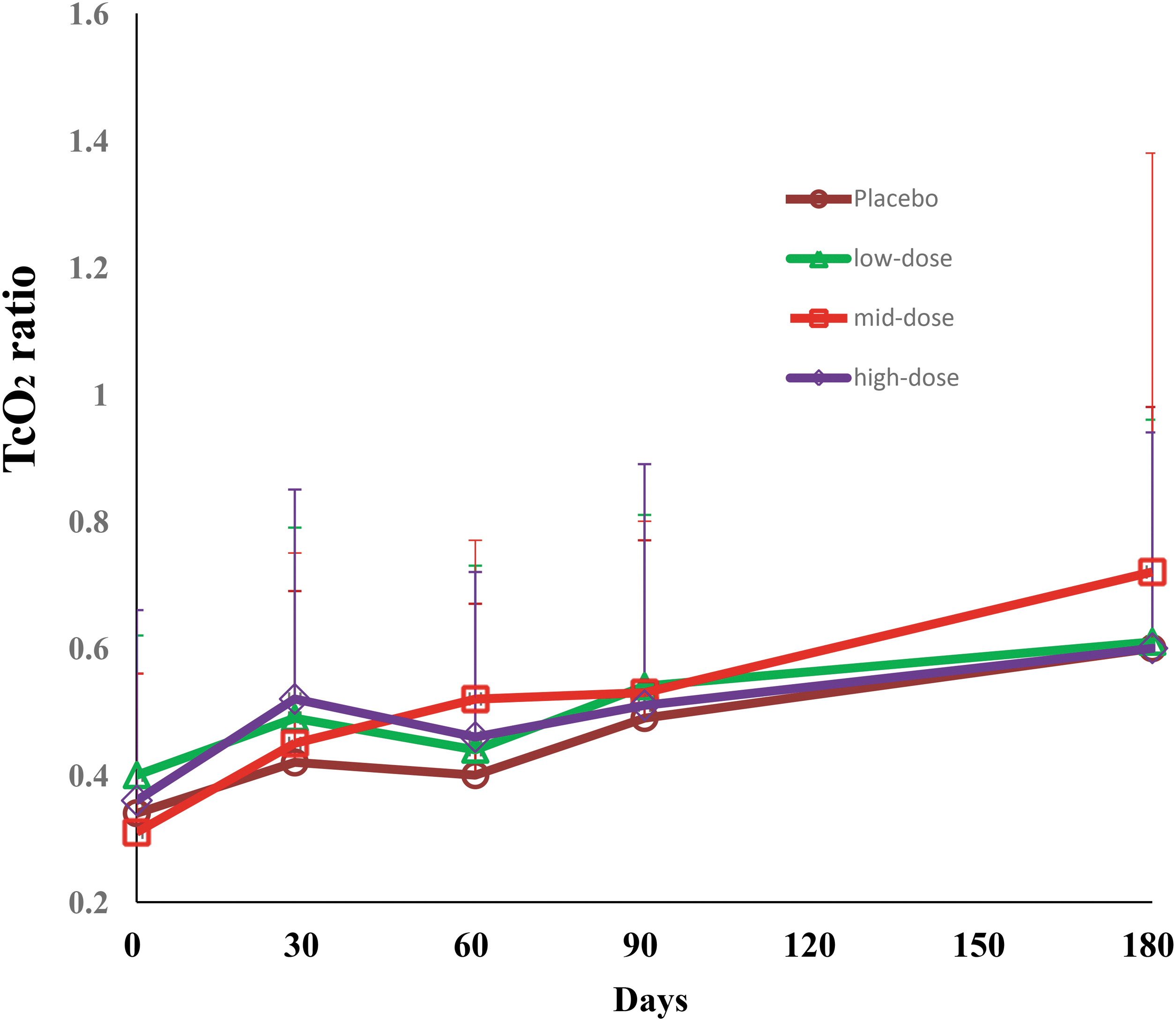
The changes of TcPO2 ratio from baseline over the 6-month follow-up in the resting pain patient cohort. The TcPO2 ratio in each group significantly increased from baseline onward, but no statistical difference by Fisher's exact test among groups at any visiting time points. TcPO2, transcutaneous oxygen pressure. Color images are available online.
Efficacy endpoints in ulcer patients
In the cohort of ulcer patients, pUDK-HGF treatment tended to be superior to the placebo in reduction of the ulcer area with ulcers <1,000 mm2 at baseline, as shown in Table 4 and Fig. 5.
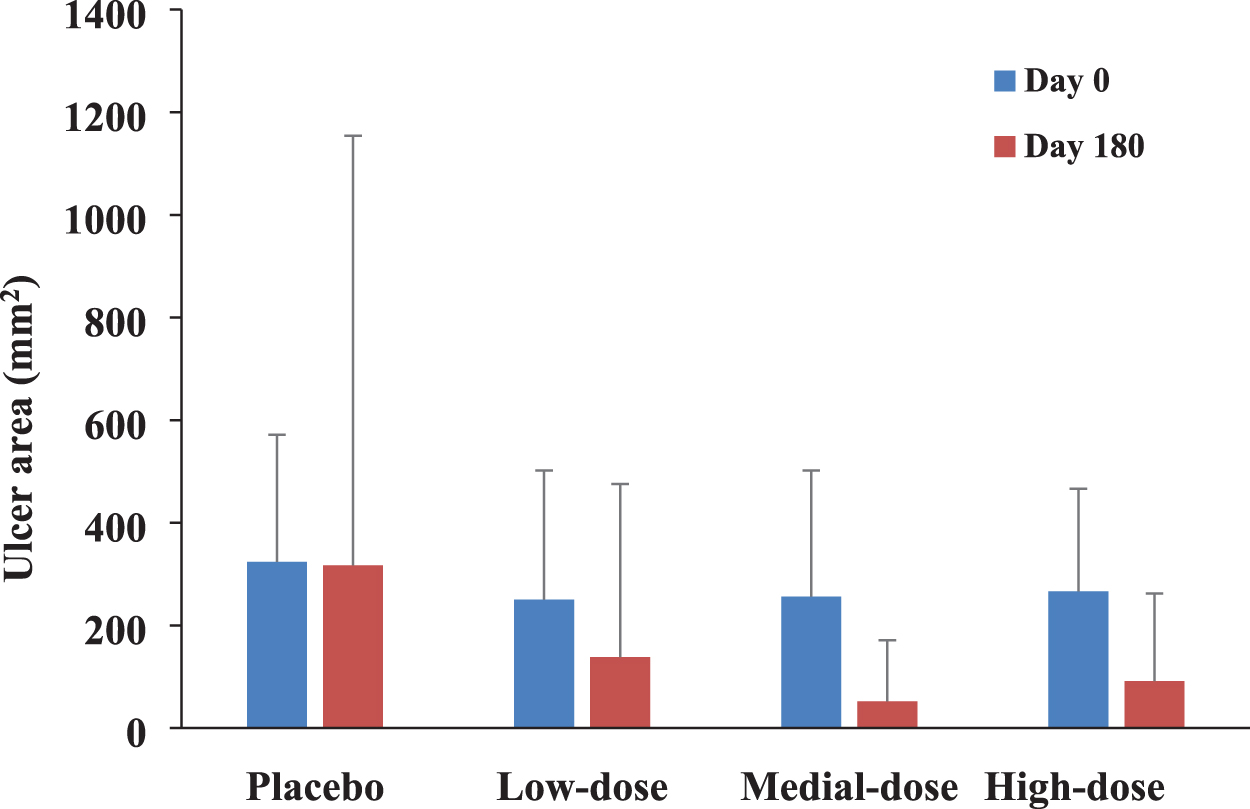
The ulcer area changes from baseline to 6 months in patients with ulcer <1,000 mm2 at baseline. pUDK-HGF treatment tended to be superior to the placebo in promoting ulcer healing. No statistical difference among groups on day 180. Color images are available online.
The ulcer size changes and healing of ulcer patients on day 180
The primary endpoint was the percentage of patients with complete ulcer closure on day 180. More patients with ulcers smaller than 1,000 mm2 at baseline in all the pUDK-HGF groups reached this primary endpoint compared with the placebo group (44.4% in the placebo group vs. 55.6%, 75.0%, and 61.1% in the high-dose, mid-dose, or low-dose groups, p > 0.05) on day 180, but these differences were not statistically significant. Only one ulcer (10 ulcers in total) larger than 1,000 mm2 on baseline had completely healed on day 180 in the high-dose pUDK-HGF treatment group.
The second endpoint was the percentage of patients displaying a >50% reduction in their ulcer size from baseline (defined as responding patients). More responding patients were observed in the mid-dose pUDK-HGF treatment (83.3%) than in the placebo group (72.2%) on day 180.
The VAS score was also evaluated in all the ulcer patients as the second endpoint, and the mid-dose pUDK-HGF treatment produced the greatest reduction in the VAS pain score. Twelve patients (12/25, 48.0%) who received the mid-dose pUDK-HGF treatment reported complete pain relief on day 180, whereas only three reported this effect in the placebo group (3/23, 13.0%, p = 0.0820). Altogether, 23 patients (23/25, 92.0%) in the group who received the mid-dose pUDK-HGF treatment reported a >50% reduction in the VAS pain score compared with the placebo group (14/23, 60.87%, p = 0.0010).
Although the ABI, TBI, and TcPO2 measurements in all the pUDK-HGF treatment groups showed obvious increases from baseline to day 180, no statistical differences were apparent for any of the time points during the follow-up visits.
The pooled efficacy analysis
As the efficacy differences among the three doses of pUDK-HGF treatment are not significant, the pooled data analysis of three pUDK-HGF doses versus placebo was performed to investigate the overall efficacy of pUDK-HGF, as shown in Table 5. The pooled analysis demonstrated that a very significant high proportion of patients in pUDK-HGF treatment (pUDK-HGF group: 47 of 71 patients, 66.20% vs. placebo group: 9 of 29 patients, 31.03%, p = 0.0013) achieved complete pain relief compared with the placebo group in the resting pain patient cohort on day 180. And overall, pUDK-HGF treatment also exhibited significant improvement in complete ulcer healing (pUDK-HGF group: 40 of 66 patients, 60.61% vs. placebo group: 8 of 23 patients, 34.78%, p = 0.0324) in the ulcer patient cohort.
The pooled analysis of complete pain relief and complete ulcer healing
Combined data of low-, mid-, or high-dose pUDK-HGF on day 180.
Safety and tolerability
A safety assessment was also conducted on the 240 patients who received at least one injection of pUDK-HGF or placebo in the two cohorts. Intramuscular injections of pUDK-HGF were well tolerated, and no unexpected SAEs were observed during the 6-month study period. There were no statistically significant differences in the occurrence of AEs, SAEs, adverse reactions, or injection-site reactions among the three pUDK-HGF and placebo groups, as shown in Fig. 6.
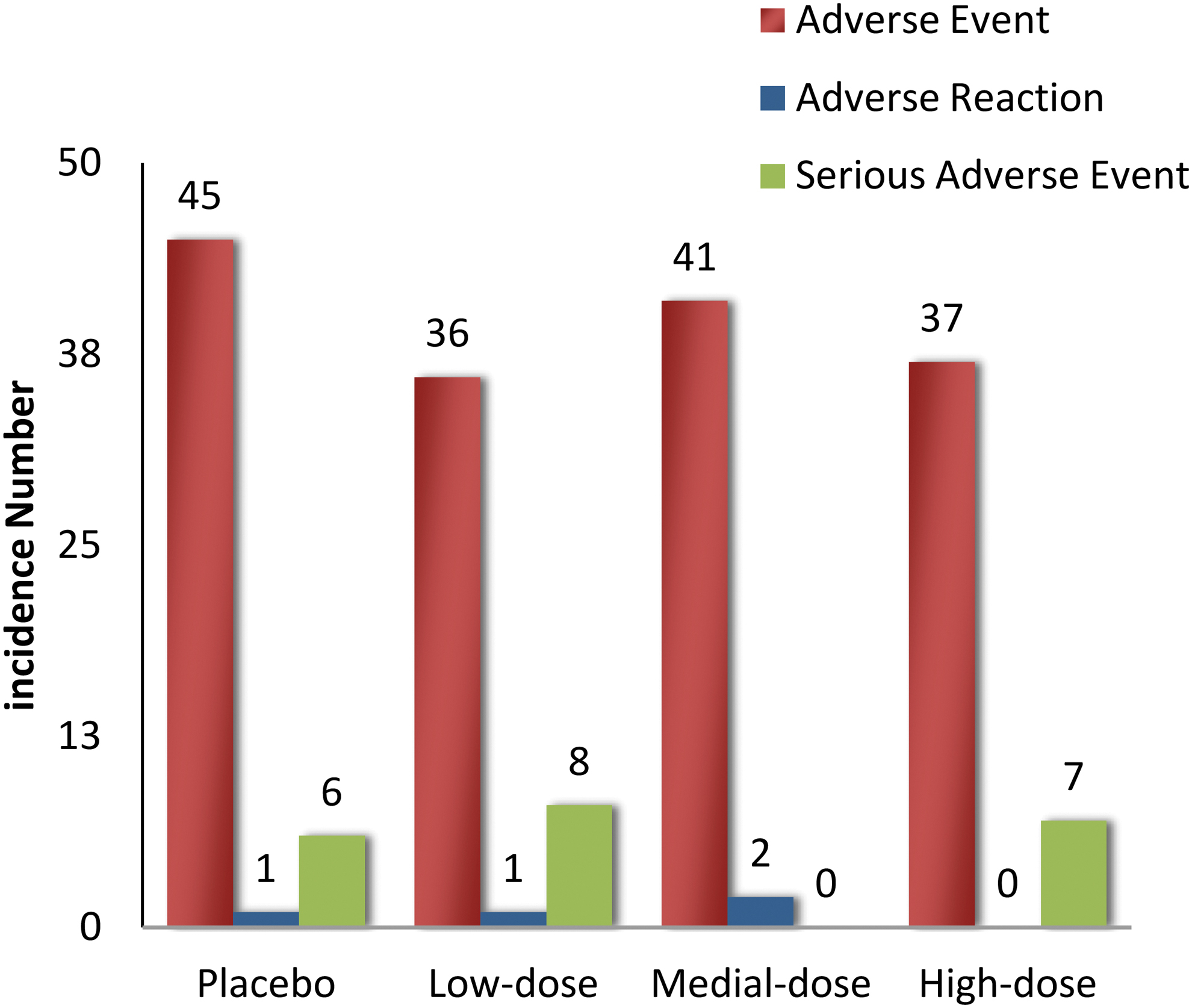
The incidence of AEs, SAEs, and adverse reaction in placebo and low-, mid-, or high-dose pUDK-HGF groups. Intramuscular injections of pUDK-HGF were well tolerated. There were no statistically significant differences in the occurrence of AEs, SAEs, adverse reactions, or injection-site reactions among the three pUDK-HGF and placebo groups. AEs, adverse events; SAEs, serious adverse events. Color images are available online.
In total, 159 AEs (159/240, 66.25%) occurred in all patients. These AEs, which included headache, dizziness, upper respiratory tract infection, hypertension, diarrhea, and abnormal laboratory indexes, were unrelated to the treatment and did not require any significant interventions during the study.
Twenty-one SAEs (21/240, 8.75%) occurred in patients over the course of the trial, with 7 of them (7/58, 12.07%) occurring in the high-dose patients, none (0/60, 0%) occurring in the mid-dose patients, 8 occurring (8/58, 13.79%) in the low-dose patients, and 6 (6/64, 9.38%) occurring in the placebo group. The SAEs were pulmonary infection, thromboembolic events, heart failure, double leg pain, acute cerebral embolism, foot infection and necrosis, gastric perforation, lower extremity arteriosclerosis progression, ulcer progression, and death; they were classified as being unrelated to pUDK-HGF or placebo treatments.
Three patients died of cerebral embolism (in the high-dose pUDK-HGF group), asthma (in the high-dose group) and CLI progression (in the low-dose group), and these events were considered to be independent of pUDK-HGF treatment. There was no progression of diabetic retinopathy, and no developing malignancy over the study period.
Altogether, five adverse reactions occurred over the course of the study, with two patients displaying limb edema (one in the placebo group and one in the low-dose group), while one in the mid-dose group had pruritus, one in the mid-dose group had limb pain, and one in the mid-dose group had an increased platelet count. These reactions, which were considered to be possibly related to pUDK-HGF administration or the placebo treatment, were alleviated without medical treatment during the follow-up period. Eight patients (placebo, 2; low-dose, 3; mid-dose, 2; and high-dose, 1) experienced mild injection-site reactions, including pain, itching, swelling, and bruising. The reactions were not treated and were alleviated during the follow-up period.
Discussion
Chronic CLI reflects the presence of significant atherosclerotic disease of the lower extremity vessels in patients. Resting pain and ulcerative lesions are two clinical manifestations of this disease state, and the extent to which the different clinical stages advance varies according to the degree of ischemia and the patient's response to medical therapy. The primary goals in treating CLI are to relieve ischemic pain, heal ischemic ulcers, prevent limb loss, improve the patients' functionality and quality of life, and prolong their survival. In this phase II clinical trial, patients with resting pain and skin ulcers were enrolled as two separate cohorts to obtain more accurate efficacy and safety of intramuscular injection with naked plasmid pUDK-HGF. Complete relief of resting pain and complete ulcer closure were ethically selected as the primary endpoints for this study, according to the guidelines for the clinical investigation of medicinal products for the treatment of peripheral arterial occlusive disease from the European Medicines Agency.
In the resting pain cohort, significantly more patients in the pUDK-HGF groups achieved the primary endpoint on day 180. The numbers of responding patients with >50% pain relief in the pUDK-HGF groups were also significantly higher than those in the placebo groups on day 180. After the first injection of pUDK-HGF on day 0, the VAS score reduced and showed a statistically significant difference between the placebo and pUDK-HGF groups on days 60, 90, and 180. This indicated that the patients who underwent pUDK-HGF treatment experienced long-term pain relief, rather than the temporary relief provided by analgesics.
In the cohort of ulcer patients, pUDK-HGF treatment in every dosage tended to be superior to the placebo in the higher percentage of patients displaying complete ulcer healing. And overall, the pUDK-HGF treatment exhibited significant improvement in complete ulcer healing in the pooled analysis. The most completely healed ulcer had a baseline area <1,000 mm2, the average ulcer area was 250.17–266.15 mm2. Only one ulcer larger than 1,000 mm2 at baseline (1,829.06 mm2) showed complete healing in the high-dose pUDK-HGF group. These results suggest that the present treatment process promotes ulcer healing with an area less than 300 mm2, and the frequency of pUDK-HGF treatment via local injections should be increased or combined with topical application of pUDK-HGF.
Our preclinical study indicated that angiogenesis is the mechanism underlying the analgesic action and promoting wound healing of pUDK-HGF, through which the establishment of collateral circulation occurs. Our recent work indicated that intramuscular injections of pUDK-HGF promoted blood flow and proliferation of satellite cells, and inhibited inflammatory cell recruitment, collagen accumulation, and the expression of pronociceptive factors in a rat skin/muscle incision and retraction model. 18,19 Intrathecal injection of pUDK-HGF in SMIR rats inhibited spinal glial cell activation and reduced the cytotoxicity associated with the expression of interleukin (IL)-1β, IL-6, tumor necrosis factor α, and inducible NO synthase, as well as decreasing nitric oxide production from activated glial cells. These factors might also play a part in the pain relief mechanism of pUDK-HGF. pUDK-HGF also significantly accelerated reepithelialization in skin wound healing of diabetic rats in a manner related to the β1-integrin/integrin-linked kinase (ILK) pathway. 20
In this study, the minimum value of the TcPO2 ratio for the anterior calf/chest, posterior calf/chest, and dorsal foot/chest was used to evaluate the microcirculation status of the limbs to obviate the effect of environmental factors such as temperature, humidity, and atmospheric pressure, among others. 21 This ratio was considered more reliable and comparable than direct TcPO2 measurements. The TcPO2 ratio in the pUDK-HGF treatment group increased significantly on 180 days when compared with the baseline in both the pain relief cohort and the ulcer cohort; however, there were no statistical differences at any of the follow-up time points.
Altogether, 240 patients representing the two cohorts were analyzed for treatment safety. Most AEs and SAEs were classified as being unrelated to the treatment received, and were most likely caused by CLI or comorbid conditions. All pUDK-HGF doses were well tolerated by the patients.
Another important goal of this trial was to determine the appropriate pUDK-HGF dose for a phase III trial. In three pUDK-HGF treatment groups, the mid-dose pUDK-HGF (6 mg) provided the most effective pain relief in the resting pain cohort and the most effective ulcer healing in the cohort of ulcer patients. Therefore, the mid-dose pUDK-HGF appears to be the most appropriate dose for a phase III clinical trial, although the efficacy differences among three doses are small, possibly due to not large pharmacological dosage differences (4.0, 6.0, or 8.0 mg), or a biphasic dose–response effect. HGF protein and HGF plasmid displayed the biphasic dose–response effect in some previous studies, 8,22,23 with a dose–response curve at lower doses and a bell-shaped dose–response effect at a high dose.
The present study is different from that of GU previous report 24 in two different plasmids, pUDK-HGF and NL003, although with a similar clinical trial design. pUDK-HGF is a simple plasmid expressing only one HGF(728 amino acids) isoform, while NL003 expressed two isoforms of HGF(723 amino acids and 728 amino acids). Our current study indicates that the plasmid expressing only one isoform also significantly reduces resting pain severity and improves ulcer healing. Furthermore, in our study, the resting pain and ulcer patients were enrolled as two separate cohorts to obtain an accurate efficacy of intramuscular injection of pUDK-HGF.
Our present study is also different from Morishita report in the injection strategies, although both plasmids (pVAX1HGF and pUDK-HGF) express a single HGF (728 amino acids) isoform. 25 pVAX1HGF was delivered into the calf or distal thigh muscles of the ischemic limb under echosonographic guidance. The injection volume was determined according to the angiographic findings and the available muscle mass. The patients were treated with pVAX1HG two or three times, 4 weeks apart. pVAX1HGF was conditional and time-limited approved in Japan to treat CLI patients in March 2019 based on four clinical studies. Our present study was designed to increase the number of injection sites as the dosage increased. Multiple-site injections of pUDK-HGF were easily given to the muscle from starting of stenosis to lower extremity following the blood vessel direction. The patients were treated with pUDK-HGF three times, 2 weeks apart.
The limitation of our present study is dividing the limited number of patients into three pUDK-HGF dose groups and placebo group, although 120 patients were enrolled in each cohort. This resulted in a small baseline imbalance in the patient characteristics and a trend outcome in each dosage group, such as that seen with wound healing and TcPO2, but the pooled overall pUDK-HGF treatment exhibited significant improvement in complete ulcer healing. In a phase III clinical trial, the mid-dose of pUDK-HGF (6 mg) will be fixed and administered to an enlarged sample size to provide a clear efficacy result.
Above all, this phase II clinical study has demonstrated that intramuscular injection of pUDK-HGF is safe, can significantly reduce resting pain in CLI patients, and promote better ulcer healing in CLI patients than the placebo. The mid-dose pUDK-HGF is recommended as an appropriate dose for a larger size phase III clinical trial of CLI therapy. The phase III clinical trial was approved by the China Food and Drug Administration (CFDA) in September 2017.
Footnotes
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by the National Science and Technology Major Project, China, No. 2014ZX09101-045-002.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.