
Abstract

Hematopoietic stem cells (
Ex vivo HSC-based gene therapy trials mainly used/use polytropic vesicular stomatitis virus G (VSVG) pseudotyped lentiviral vectors (LVs). Features such as serum instability and others shown in Fig. 1 indicate that VSVG-LVs are not the best candidates for in vivo gene therapy. In addition, the low-density lipoprotein receptor, the receptor for VSVG-LVs, is only present on CD34+ stem and progenitor cells after 2–3 days stimulation in culture. 1 This receptor will not be readily available on quiescent HSCs in vivo, so other candidate viral glycoproteins are needed to achieve high-level in vivo HSC gene delivery.
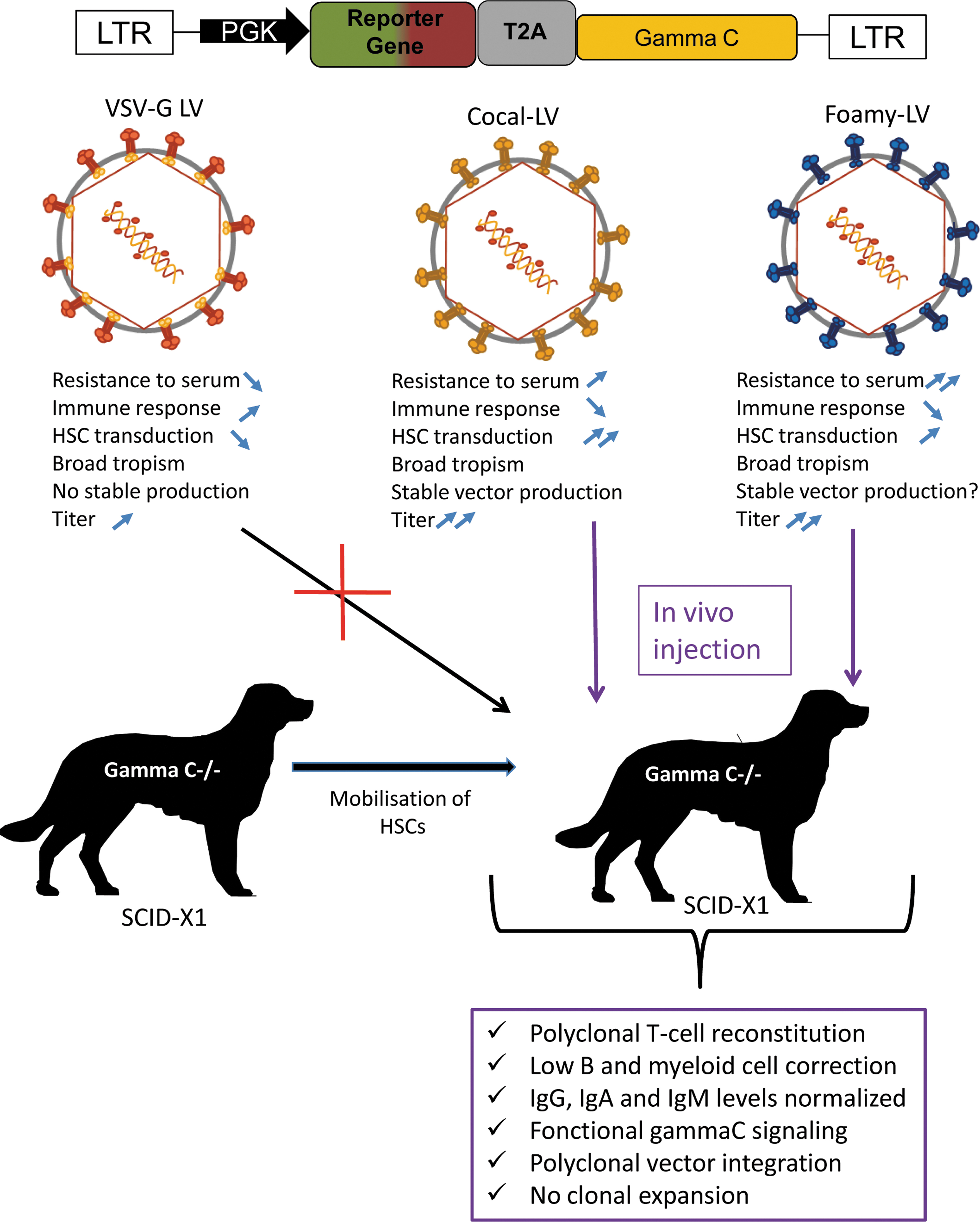
Comparison of lentiviral vectors for in vivo HSC gene therapy in an SCID-X1 preclinical dog model. HSC, hematopoietic stem cell; SCID, severe combined immune deficient.
The study by Rajawat et al. in this issue evaluated in vivo gene therapy with cocal viral (CV) envelope pseudotyped LVs to treat severe combined immune deficient (SCID)-X1 dogs with no prior conditioning regimen. The cocal virus envelope belongs to the same viral family as VSVG but it is distinct in the sense that it is more resistant to complement-mediated inactivation by serum. In contrast to VSVG-LVs, CV glycoprotein pseudotyped LVs can be produced at higher titers than VSVG-LVs, and CV envelope allows to generate stable packaging cell lines. Moreover, it has a broad tropism for cells from different species: human, nonhuman primate, murine, and canine HSCs are readily gene modified by CV-LVs (Fig. 1).
To make human stem and progenitor cells (HSPCs) accessible for transduction into the blood stream, these authors used a combinatory regimen to mobilize the CD34+ cells from the bone marrow. They efficiently mobilized these cells as indicated by an approximately eightfold increase of CD34+ HSPCs in peripheral blood. These authors present follow-up data from a previous publication, in which they obtained for >45 months a long-term therapeutic benefit of foamy virus (FV) glycoprotein enveloped vectors injected in vivo to rescue dogs with the SCID-X1 disease phenotype. 2 More specifically, Rajawat et al. compared these FV-LV pseudotypes with CV-LVs to obtain immune correction in SCID-X1 dogs. A single injection of CV-LVs expressing a functional copy of gamma C cDNA corrected the clinical phenotype of canine SCID-X1 immunodeficiency. In these treated dogs, a rapid reconstitution of T cells (quicker than for the foamy vector used previously) was achieved that showed a naive (CD45RA+) phenotype and polyclonal T cell repertoire. Gene marking in myeloid and B cells remained low. Nevertheless, the authors reported IgG and IgA production and normal IgM levels, suggesting isotype switching as seen in FV-LV–treated SCID-X1 dogs. Overall the safety profile was favorable for both FV and CV envelope pseudotyped vectors as shown by vector insertion analysis and the fact that no clonal dominance was detected (Fig. 1).
This is a first report on in vivo HSC-based gene therapy in a preclinical large animal model for SCID-X1 and is thus an important step to bring the application of in vivo HSC correction closer to the clinic. This in vivo strategy might also be attractive for treatment of bone marrow failures such as Fanconi anemia (FA), for which it is a challenge to isolate sufficient bone marrow HSCs and maintain the gene-corrected HSCs ex vivo for efficient long-term correction. 3 Moreover, in FA the corrected stem cells have a selective advantage, so only a small fraction of the HSCs might need to be corrected cells in vivo to alleviate disease. FA might, therefore, be one of the target diseases that would mostly benefit from in vivo HSC correction. But this will also pave the way to in vivo cure of many inherited blood diseases.
Nevertheless, prudence is warranted since vectors for in vivo HSC transduction ideally should be specific for the target cell to avoid vector spreading and transduction of antigen presenting cells, which carries the risk of eliciting a transgene-specific immune response leading to the elimination of transduced cells and loss of therapeutic benefit. Since CV-LVs and FV-LVs are polytropic, a careful long-term evaluation is required for off-target cell transduction, causing severe health risks and adverse effects. To address this safety concern, another team engineered recently novel targeting vectors highly specific for T cell gene delivery, which allowed to generate chimeric antigen receptor T cells directly in vivo. 4,5
In conclusion, in vivo gene therapy as proposed by Rajawat et al. might in the future consist of a single injection of this vectorized medicine into the blood stream, thus making this therapy more broadly available to patients while reducing costs, risks for the patients (elimination of conditioning), and potentially improving therapeutic outcomes through conservation of stem cell properties.